
INTRODUCTION
Modern genomic technologies have allowed for comprehensive characterization of somatic gene mutations found in tumor cells. In cohorts of newly-diagnosed meningioma, mutations in AKT1, SMO, KLF4, PIK3CA, and TRAF7 have been identified, in addition to the long-established NF2 gene inactivation that is characteristic of this disease [1]. More recently, we and others have identified additional genomic alterations that are specifically associated with progressive higher-grade meningiomas, including BAP1 inactivation [2] and telomerase reverse transcriptase promoter (TERTp) mutations [3].
Telomere maintenance is considered a hallmark of neoplasia [4]. Over 90% of human tumors express the enzyme telomerase, which actively counteracts telomere shortening [5]. TERTp mutations, initially discovered in melanomas [6], are among the most common recurrent alterations in human cancer. In addition to TERTp mutations, aberrant telomerase expression can be caused by TERT rearrangements [7], DNA amplifications or transcript fusions [5]. Telomerase-independent cancers maintain their telomeres through a homologous recombination-dependent process known as alternative lengthening of telomeres (ALT), which is associated with genomic alterations in the genes DAXX or ATRX [5].
Telomerase activation has been reported as a frequent phenomenon in meningiomas, with an association between activation and World Health Organization (WHO) grade [3]. Intriguingly, TERTp mutations were reported to be rare in WHO grades I and II meningiomas [8–10], the vast majority of which are cured by surgical resection, but were found at higher frequencies (up to 28%) in recurrent tumors and those with malignant histopathological appearance, which is predictive of a higher recurrence rate [3, 8].
However, a longitudinal genomic analysis of telomerase gene alterations across the spectrum of initial and recurrent disease remains incomplete. It is possible that other known cancer-specific mechanisms of telomere maintenance such as ATRX/DAXX mutations or TERT rearrangements could be present in meningiomas. With this in mind, we characterized a large cohort of patients with progressive/higher-grade meningiomas at both initial diagnosis and recurrence. To investigate the selective pressures that manifest in genomic differences during meningioma progression, we also sequenced temporally and spatially distinct sites of the meningiomas in our cohort to assess for intratumoral heterogeneity.
RESULTS
Somatic TERTp mutations are frequent in recurrent meningiomas
TERTp was sequenced in all 64 tumor samples derived from 26 patients with progressive higher-grade meningiomas, across the temporal spectrum of disease from the initial pre-treatment specimen to the final recurrent tumor. Canonical TERTp mutations were detected in nine samples (9/64= 14%) from six patients (three females and three males, 6/26 = 23%). Three TERTp mutations were located at 146 bp (referred C250T) and three were located 124 bp (referred C228T) upstream of the translation start (Table 1). We screened DNA from matched blood samples available in four positive cases and did not detect TERTp mutations, confirming that this is a somatic (non-germline) alteration, as has previously been reported in other cancers [11].
Table 1: Patients’ characteristics
Gender |
Age at first diagnosis |
Initial WHO grading |
Tumor location |
TERT promoter status |
Nr. of surgeries |
RTx |
CTx |
|
|---|---|---|---|---|---|---|---|---|
Pat01 |
f |
62 |
II |
Convexity |
wild-type |
4 |
Yes |
No |
Pat02 |
m |
73 |
III |
Intraventricular |
wild-type |
2 |
Yes |
No |
Pat03 |
m |
48 |
III |
Convexity |
wild-type |
1 |
Yes |
No |
Pat04 |
m |
42 |
III |
Convexity |
wild-type |
1 |
Yes |
No |
Pat05 |
f |
52 |
III |
Convexity |
wild-type |
1 |
No |
No |
Pat06 |
m |
75 |
III |
Convexity |
C228T mutation |
1 |
Yes |
No |
Pat07 |
m |
75 |
II |
Frontobasal |
C228T mutation |
5 |
Yes |
No |
Pat08 |
m |
65 |
II |
Convexity |
C250T mutation |
6 |
Yes |
No |
Pat09 |
f |
64 |
II |
Frontobasal |
wild-type |
3 |
Yes |
No |
Pat10 |
m |
50 |
II |
pertoclival |
wild-type |
3 |
Yes |
Imatinib |
Pat11 |
m |
54 |
II |
Frontobasal |
wild-type |
4 |
Yes |
No |
Pat12 |
f |
49 |
II |
Infratentorial |
wild-type |
4 |
Yes |
No |
Pat13 |
m |
65 |
II |
Convexity |
wild-type/ LPCAT1-TERT fusion |
8 |
Yes |
No |
Pat14 |
m |
61 |
III |
Infratentorial |
wild-type |
3 |
Yes |
No |
Pat15 |
m |
18 |
I |
Frontobasal |
wild-type |
4 |
Yes |
Temozolomide |
Pat16 |
f |
6 |
II |
Temporobasal |
wild-type |
4 |
Yes |
No |
Pat17 |
f |
47 |
III |
Convexity |
wild-type |
1 |
No |
No |
Pat18 |
m |
50 |
III |
Unknown |
wild-type |
3 |
No |
No |
Pat19 |
m |
75 |
III |
Unknown |
wild-type |
1 |
No |
No |
Pat20 |
f |
62 |
III |
Frontobasal |
wild-type |
1 |
Yes |
Hydroxyurea |
Pat21 |
f |
60 |
II |
Temporobasal |
wild-type |
4 |
Yes |
No |
Pat22 |
m |
71 |
I |
Frontobasal |
wild-type |
2 |
Yes |
No |
Pat23 |
m |
57 |
II |
Convexity |
wild-type |
3 |
Yes |
No |
Pat24 |
f |
74 |
II |
Frontobasal |
C250T mutation |
2 |
No |
No |
Pat25 |
f |
45 |
II |
Convexity |
C228T mutation |
2 |
Yes |
No |
Pat26 |
f |
47 |
I |
Convexity |
C250T mutation |
2 |
Yes* |
No |
M: male, f: female, RTx: radiotherapy, CTx: chemotherapy. WHO: World Health Organization. * upfront radiotherapy.
Somatic TERTp mutations are acquired during progression in recurrent meningiomas
A matched comparison of primary meningiomas and their recurrences is often challenging due to the long time lapse between initial tumor and recurrence. Our cohort had 19 matched pairs, allowing the determination of timing of acquired mutations during progression. To investigate whether the TERTp mutation was acquired at recurrence in our cohort, we examined TERTp status longitudinally in samples. Interestingly, out of five TERTp-mutant cases with the initial specimen available, four somatic TERTp mutations were absent in the initial, grade II tumor, emerging only later in the recurrent tumors (Table 2, Figure 1). On inspection of clinical demographic factors (Table 1), we could not identify differences in the initial presentation between patients who ultimately developed TERTp mutations compared to those with TERTp wild-type meningiomas. The median age of the patient at first diagnosis was comparable in both groups and we could not discern a correlation between the tumor location and TERTp mutation status. These findings highlight the temporal dynamics of evolution in recurrent meningioma and, ultimately, could provide insight into natural history. These results suggest that TERTp mutations arise later in tumor evolution in progressive meningiomas, consistent with similar observations in other cancers like melanomas or hepatocellular carcinoma [12–14].
Table 2: Emergence of TERT promoter mutations in high-grade meningioma
Initial WHO grading |
TERT promoter status |
Recurrent tumor WHO grading |
TERT promoter status |
Number of recurrences |
|
|---|---|---|---|---|---|
Pat06 |
III |
C228T mutation |
No recurrence |
n.a. |
0 |
Pat07 |
II |
wild-type |
III |
C228T mutation |
5 |
Pat08 |
II |
wild-type |
III |
C250T mutation |
6 |
Pat24 |
II |
wild-type |
III |
C250T mutation |
1 |
Pat25 |
II |
wild-type |
III |
C228T mutation |
1 |
Pat26 |
I |
Unknown |
II |
C250T mutation |
3 |
f: female m: male, n.a.: not applicable, WHO: World Health Organization.
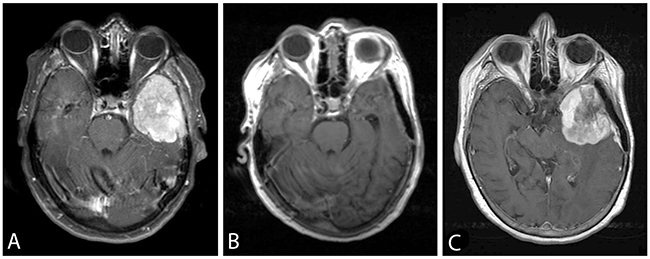
Figure 1: Axial post-gadolinium T1-weighted images showing the TERTp wild-type sphenoid wing meningioma WHO grade II (Case Pat24) (A) pre- and (B) postoperatively. An adjuvant treatment was not pursued after initial resection. (C) 22 months later the patient showed a local recurrent tumor. At that time point, the tumor was histologically progressive to grade III and carried a newly detected TERTp mutation. The patient had a rapidly progressive subsequent course, and died 6 months after the second surgery.
Spatial intratumoral heterogeneity in TERTp-mutant meningiomas
From the perspective of tumor cell phylogeny, later emergence of a subclonal mutational event typically manifests as spatial heterogeneity within a bulk tumor mass. For instance, increased cytogenetic abnormality in recurrent meningioma has recently been demonstrated using array comparative genomic hybridization [15]. However, to date there has been no targeted assessment of intratumoral spatial heterogeneity of meningioma driver genes in progressive samples, due to the fact that most analyses were performed on single tissue samples from individual patients. We therefore scored TERTp status at spatially distinct sites in selected recurrent meningiomas in our cohort.
In two patients who underwent surgery for recurrent tumors at distinct locations, we detected the TERTp mutation in only one of two sites (Figure 2). To further explore intratumoral heterogeneity, we sequenced the TERTp in further 8 separate specimens of geographically distinct regions taken from both meningiomas. TERTp mutations were present in 5 of the 8 additional samples (Figure 3). Along with the observation that not all recurrent tumors have TERTp mutations, this finding suggests ongoing subclonal selection during the evolution of progressive meningiomas, resulting in at least two distinct meningioma cell subpopulations (TERTp wild-type and TERTp-mutant).
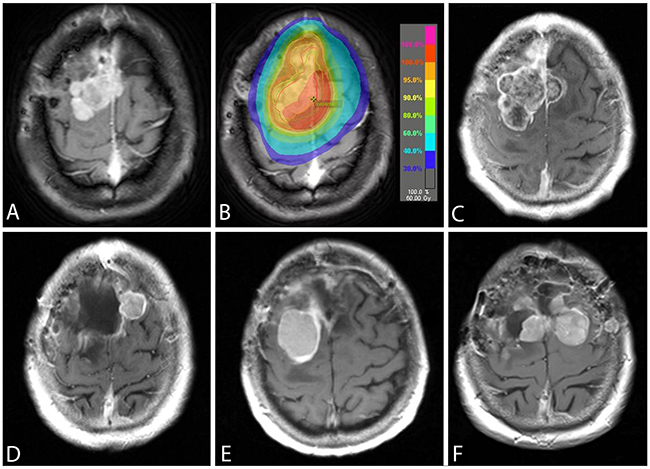
Figure 2: An axial post-gadolinium T1-weighted image showing the TERTp wild-typeconvexity WHO grade II meningioma (case Pat08) (A). A radiation therapy with 60 Gy was applied after a subtotal tumor resection (B) and the patient was progression-free for 12 months. The follow-up MRI showed a progressive tumor (C) and the pathological diagnosis was consistent with an anaplastic meningioma WHO grade III with a new developed TERTp mutation. While the recurrence in (D) was proven to be TERTp wild-type, the subsequent recurrence (E) was TERTp-mutant. At the time of last surgery (F), the TERTp mutation was detected in the right-sided, but not in the left-sided meningioma. The patient passed away 22 months after the first emergence of TERTp mutation due to progressive disease.
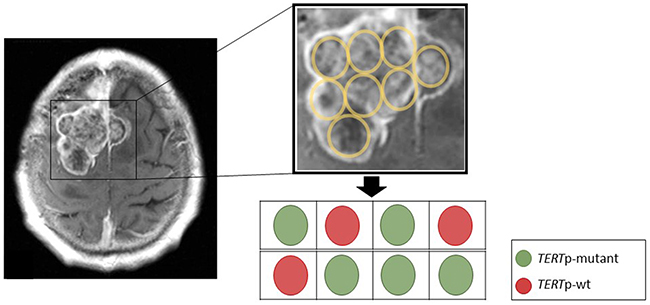
Figure 3: Spatial representation of the tumor from Figure 2C for TERTp mutation status. The sequencing of TERTp mutation on DNA extracted from 8 different paraffin embedded tumor blocks from the surgical resection specimen to provide a widely spread and independent geographical separation for the sampling. Although we are not able to directly localize these block specimens with the MRI scan, sequencing revealed a mutation in 5 out of 8 samples, providing evidence for spatial intratumoral heterogeneity in TERTp mutant meningiomas.
Presence of detectable mutant TERTp in progressive meningiomas confers a poor prognosis
In a recently published study, Sahm et al. found a strong association between TERTp mutations and time-to-recurrence [8], but were not able to evaluate overall survival. With the comprehensive follow-up in our patient cohort, we found that patients with emergence of TERTp-mutant meningiomas was associated with a significantly shorter OS than their TERTp wild-type counterparts, when measured from the time of initial diagnosis (2.7 years, 95% CI 0.9 – 4.5 versus 10.8 years, 95% CI 7.8 -12.8, p=0.003) (Figure 4). In addition, patients with a TERTp mutation had a significantly shorter progression-free survival (1.1 years, 95% CI 0.8 - 1.4) in comparison with TERTp wild-type patients (3.6 years, 95% CI 0.3 - 9.6, p= 0.002) (Figure 4). In a multivariate Cox regression model, TERTp mutation emergence showed significant independent association with poor prognosis (hazard ratio 12.74, 95% CI: 1.77 – 91.8, p= 0.012) (Table 3). Other clinical parameters such as age, gender, tumor localization, initial WHO grade, number of recurrences or adjuvant therapy were not independent prognostic factors in our cohort, although it is possible that there are other clinical variables which may account for the differences in outcome.
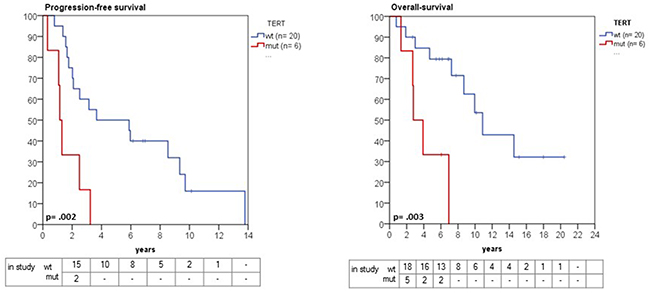
Figure 4: Kaplan-Meier estimates of progression and overall survival in grade II and III meningioma in relation to TERTp mutation status. Patients with a TERTp mutation had a significantly shorter progression-free survival (1.1 years, 95% CI 0.8 - 1.4 versus 3.6 years, 95% CI 0.3 - 9.6, p= 0.002). Likewise, overall survival was significantly shorter in TERTp-mutant compared with wild-type patients (2.7 years, 95% CI 0.9 – 4.5 versus 10.8 years, 95% CI 7.8 -12.8, p=0.003).
Table 3: Stepwise backward logistic regression for overall survival
Hazard ratio (HR) |
95% CI |
p-value |
|
|---|---|---|---|
Age |
0.96 |
0.95 -1.03 |
0.79 |
Gender |
1.31 |
0.36 -4.73 |
0.68 |
Tumor localization (skull base/ non-skull base) |
1.18 |
0.32 – 4.33 |
0.96 |
Initial WHO grade (I/II vs. III) |
4.34 |
0.38 – 48.6 |
0.23 |
TERTp mutation |
12.74 |
1.77 – 91.8 |
0.012 |
Number of recurrences |
1.34 |
0.2 - 6.2 |
0.71 |
Adjuvant radio- or chemotherapy |
1.02 |
0.057 – 18.52 |
0.98 |
Frequency of TERTp mutations in a subset of radiation-associated meningiomas
Five of 6 patients with TERTp mutation had received radiation before surgical resection of the specimen demonstrating the mutation, raising the possibility of a causal association between receipt of radiation and emergence of TERTp mutation. To test the hypothesis that TERTp mutations might be enriched in post-radiation meningiomas, we performed a TERTp mutation analysis in a separate cohort consisting of 19 patients with post-radiation meningiomas (n=19 patients, total of 24 samples). On initial diagnoses, these meningiomas spanned all WHO grades (4 grade I, 12 grade II and 3 grade III). However, TERTp mutation was detected only in two patients (10.5%). Both had received radiation in childhood, one patient for an orbital tumor, and a second for a pilocytic astrocytoma. Thus, we did not detect a markedly elevated rate of TERTp mutation in meningiomas arising after receipt of radiation.
TERT rearrangements in malignant meningiomas
TERT rearrangements have been described in several neoplasms and are usually associated with increased TERT expression [5]. To assess whether TERT gene rearrangements were present in progressive higher-grade meningiomas, we performed targeted RNA fusion gene analysis in TERTp wild-type meningioma cases (n=28) using Anchored Multiplex PCR (AMP). We identified a novel chromosomal rearrangement involving TERT in a recurrent WHO grade III meningioma sample (patient 13, Table 1). The fusion involved the juxtaposition of exon 11 of LPCAT1 and exon 2 of TERT within chromosome 5p15.33 (Figure 5). The remaining samples did not have detectable TERT exon 2 fusion transcripts.
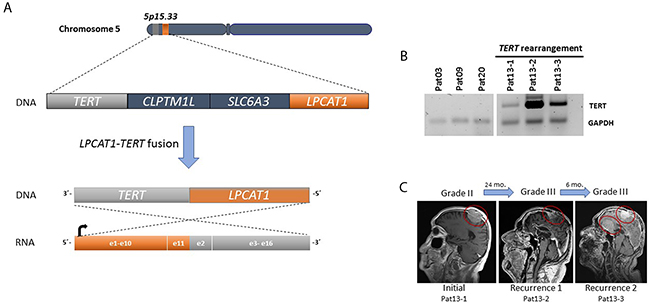
Figure 5: (A) A fusion was detected between exon 11 of LPCAT1 (orange) and exon 2 of TERT (gray) within chromosome 5p15. 33. (B) TERT expression (reverse transcriptase PCR) in three subsequent samples of patient 13 with TERT rearrangements compared to three samples from patients without TERT alterations. TERT was amplified in all consecutive samples. GAPDH was used as a control. (C) Representative MRIs for tumor growth history for patient 13.
Progressive meningiomas lack ATRX and DAXX mutations
As an alternative to upregulation of TERT expression, cancers can also use the ALT pathway for telomere maintenance, associated with inactivating mutations in either the ATRX or DAXX genes [16]. Therefore, we screened 18 samples of our TERTp-wild-type (n=15) and TERTp-mutant (n=3) malignant meningioma for mutations in both genes using targeted sequencing. None of the samples showed ATRX, DAXX mutations or deletions. In addition, we performed ATRX immunohistochemistry from meningiomas in our cohort at different stages during progression (n= 42). In one case, ATRX expression was lost by IHC, however, targeted analysis by SNaPshot NGS did not reveal mutations or deletions in the ATRX gene. Together with a recent report of no loss of ATRX or DAXX expression by IHC in 58 grade II and III meningiomas [16], these results provide evidence that the ALT pathway is not a frequent mechanism for telomere maintenance in progressive higher-grade meningiomas.
DISCUSSION
We demonstrate that mutations of TERTp are highly-prevalent in progressive higher-grade meningiomas, found in up to 25% of cases. These findings support similar observations reported by Goutagny et al., who found an increased incidence of TERTp mutations in patients with meningiomas that underwent malignant progression [3]. However, Goutagny et al. showed in all patients with multiple specimens that the TERTp mutation was present in the initial low-grade tumor [3]. In contrast, we reveal the emergence of TERTp mutations during the progression interval, providing evidence for the outgrowth of subpopulations of tumor cells during the evolution of these tumors.
Similarly, a recent integrated genomic analysis detected TERTp mutations in 4/31 recurrent atypical meningioma samples, but not in any of 110 first-diagnosed, non-recurrent grades I and II meningiomas [9]. The authors speculated that acquisition of an activating TERTp mutation could be an essential step in formation of atypical meningiomas [9]. As a proof of this later clonal emergence, our study demonstrates TERTp mutations were heterogeneously detected in geographically distinct sites within recurrent bulk tumor mass, providing critical evidence in support of this model. Further consistent with this model of accelerated tumor progression, we show that TERTp-mutant meningiomas are associated with a significantly shorter overall survival than their TERTp wild-type counterparts.
In addition to the TERTp mutations, we identified for the first time in meningioma a fusion that involves the TERT gene (LPCAT1:TERT). In this case, the patient (pat13) had a meningioma grade II that progressed to grade III within two years (Figure 5). Interestingly, LPCAT1-TERT fusions has been described in lung adenocarcinoma [17] and in a pediatric hepatocellular carcinoma [18]. TERT rearrangements are a common genetic alteration in high-risk neuroblastomas and are associated with increased TERT expression and a poor prognosis [19].
Taken together, these findings indicate that meningiomas likely give rise to more malignant clones during an ongoing evolutionary process, underscoring the crucial role of genetic diversity in tumor cell evolution. Furthermore, this heterogeneity might explain the variable detection frequency of TERTp mutation that has been reported in atypical meningiomas [9]. Indeed, the high treatment-failure rate of progressive meningiomas may be due to this increased heterogeneity allowing subclonal escape from adverse selection pressures such as hypoxia, metabolic stress, chemotherapy, or radiotherapy [20].
Our findings have implications for understanding the biology of a subset of progressive higher-grade meningiomas, providing important insight into the complexity of intratumor heterogeneity. Distinct molecular etiologies drive progressive higher-grade meningiomas, overlaid on the known genomic alterations in newly-diagnosed benign tumors. These differences highlight the potential role of TERT alterations as a clinical biomarker allowing for the stratification of patients in the recurrent setting for clinical trials in progressive higher-grade meningioma. Sensitive techniques for the detection of TERTp-mutant subclones may allow for the early diagnosis of clinically-aggressive meningioma, a long-sought goal of molecular analyses of this disease.
MATERIALS AND METHODS
Patient and tumor characteristics
Twenty-six patients with treatment-resistant meningiomas formed the study cohort, with specimens available from initial diagnosis and sequential recurrences (n=64 tumors). Patient demographic characteristics are shown (Table 1). Tumor locations included 11 convexity, 9 skull base, two infratentorial and one intraventricular meningiomas; MRI imaging at time of initial diagnosis was not available for two patients. Three patients presented initially with a WHO grade I meningioma, while 13 patients had a grade II meningioma at initial diagnosis, and 10 patients had a de novo grade III meningioma at first surgery. Most patients (n= 19) in this study had recurrent surgery in addition to the initial diagnostic procedure, during the median follow-up of 11.3 years (range 4.4 – 16.8 years), undergoing an average of 3 surgeries (range 1-8). In one case upfront radiotherapy was performed, whereas the majority (21/26, 80.7%) received radiotherapy during the course of treatment after the initial surgery. In two cases, the initial surgical specimen was unavailable, as that surgery had been performed at an outside hospital. Overall, 19 patients had one or more recurrent meningioma specimens available, with the initial, pre-treatment tumor sample available for these cases, except for patient Pat26.
Tumor and blood samples from patients were obtained from the Departments of Neurosurgery at the University Hospital Dresden and the Massachusetts General Hospital in Boston under established Institutional Review Board approvals for genetic studies. When fresh tumor tissue from surgical specimens was available, it was immediately frozen at −80 °C. When frozen tissue was not available, formalin-fixed, paraffin-embedded tissues were used for DNA extraction. Control slides stained with hematoxylin and eosin were reviewed by a neuropathologist to assure a tumor cell content of at least 80% for nucleic acid extraction. DNA isolation was performed using the QIAmp DNA Mini Kit 50 (Qiagen, Hilden, Germany).
TERT promoter mutation
The TERTp was assessed either by amplification using Sanger sequencing performed using ABI Prism 3730 DNA Analyzer or using the fluorescence PCR technique previously described [21]. Mutations between the residues of −124 (1295228) to −146 (1295250) bp from the ATG start site in the TERTp were scored, as described previously by Killela et al. [22].
SNaPshot Next Generation Sequencing Archer® FusionPlex®
Specimens were subjected to additional analysis utilizing SNaPshot, a hybrid capture based method for single nucleotide variant (SNV) and insertion/deletion (indel) detection in genomic DNA, and Archer® FusionPlex®, an anchored multiplex polymerase chain reaction (AMP) technique by ArcherDx (Boulder, CO) for fusion detection in RNA [23]. SNaPshot targets 108 genetic loci frequently mutated in 15 cancer genes, including TERTp, ATRX, and DAXX. AMP using the Archer® FusionPlex® Solid Tumor Kit detects gene rearrangements involving over 50 genes, including unknown fusion gene partners such as novel TERT exon 2 fusion transcripts [23].
Reverse transcriptase PCR
cDNA was synthesized using 10 μg of total RNA with the High-Capacity cDNA Reverse Transcription Kit according to the manufacturer’s instructions. TERT was amplified using primer sets forward sequence 5’-CTGCAGGCGTACAGGTTTC-3’ and reverse 5’-GTGTCGAGTCAGCTTGAGCA-3’. GAPDH primers were used as control with the forward sequence 5’-GTCAGCCGCATCTTCTTT-3’ and reverse 5’-CGCCCAATACGACCAAAT-3’. The amplified products were separated on a 1.5% agarose gel.
ATRX immunohistochemistry
Immunohistochemistry (IHC) was performed with a polyclonal rabbit antibody against ATRX (dilution 1:100, Abcam, AB97508), using published protocols [24]. Two independent neuro-pathologists evaluated all IHC results (E.A.W. and M.A.). Nuclear staining was considered positive for evaluation. Cases with more than 10% of tumor cells staining positively were considered “retained”. Endothelial cells, cortical neurons and infiltrating inflammatory cells were generally positive and served as internal positive controls.
Clinical data
Demographic, treatment and follow-up data were retrospectively collected. Progression free survival (PFS) was calculated from the initial diagnosis until radiographic tumor progression as determined by the treating physician. Overall survival (OS) was defined as the interval from the day of first surgery until death, with data censored at the last available date of follow-up. All patients’ data were updated on December 10, 2016.
Statistical analysis
The Kaplan-Meier technique was used to estimate PFS and OS and significant differences were analyzed by the log-rank test using the statistical software SPSS. The Mann-Whitney U and Fisher’s exact tests were used to test for association of clinical variables and TERT alterations. P < 0.05 was considered as statistically significant.
Abbreviations
ALT: alternative lengthening of telomeres
CTx: chemotherapy
DAXX: death domain associated protein
OS: overall survival
PFS: progression-free survival
RTx: radiation therapy
TERTp: telomerase reverse transcriptase promoter
WHO: World Health Organization
Author contributions
T.A.J. performed Sanger sequencing, immunohistochemistry, clinical correlations, and sample selection. T.A.J., M.A.K., G.M.S. T.P, and S.S.T. performed Sanger confirmation of detected TERTp mutations for all samples. T.A.J., C.T. and G.M.S analyzed the sequencing data, E.A.W. and M.A. evaluated the ATRX immunohistochemistry. D.D., M.K. and S.A. performed the MR-images and the radiation data. T.P., D.K. K.R. and M.M. assisted with sample collection. S.S. assisted with the statistical analysis, H.E., I.S. and J.C. tested 28 samples for gene fusions and assisted with figure 5. T.A.J wrote the manuscript and made the figures, and G.S., M.K., J.J.M., H.W., P.K.B. and D.P.C. edited the manuscript. M.K., P.K.B. and D.P.C designed and oversaw the project.
CONFLICTS OF INTEREST
All authors have no conflicts of interest to report with regard to this manuscript.
FINANCIAL SUPPORT
This work is supported by U.S. NIH 1R21NS099844 (to D.P. Cahill and P.K. Brastianos), a Burroughs Wellcome Fund Career Award (to D.P. Cahill) and by Max Kade Foundation (to T.A. Juratli). T.A. Juratli received the National Brain Tumor Society Mahaley Clinical Research Award for this study.
REFERENCES
1. Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, Ligon KL, Palescandolo E, Van Hummelen P, Ducar MD, Raza A, Sunkavalli A, Macconaill LE, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013; 45:285-289.
2. Shankar GM, Santagata S. BAP1 mutations in high-grade meningioma: implications for patient care. Neuro Oncol. 2017; 19:1447-1456.
3. Goutagny S, Nault JC, Mallet M, Henin D, Rossi JZ, Kalamarides M. High incidence of activating TERT promoter mutations in meningiomas undergoing malignant progression. Brain Pathol. 2014; 24:184-189.
4. Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
5. Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, Akdemir KC, Seth S, Song X, Wang Q, Lichtenberg T, Hu J, Zhang J, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017; 49:349-357.
6. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA: Highly recurrent TERT promoter mutations in human melanoma. Science. 2013; 339:957-959.
7. Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, Hacker KE, Bhanot G, Gordenin DA, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014; 26:319-330.
8. Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J, Kratz A, Capper D, Schefzyk S, Hielscher T, Wang Q, Sulman EP, Adeberg S, et al. TERT Promoter Mutations and Risk of Recurrence in Meningioma. J Natl Cancer Inst. 2015; 108.
9. Harmancı AS, Youngblood MW, Clark VE, Coşkun S, Henegariu O, Duran D, Erson-Omay EZ, Kaulen LD, Lee TI, Abraham BJ, Simon M, Krischek B, Timmer M, et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun. 2017; 8:14433.
10. Koelsche C, Sahm F, Capper D, Reuss D, Sturm D, Jones DT, Kool M, Northcott PA, Wiestler B, Böhmer K, Meyer J, Mawrin C, Hartmann C, et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013; 126:907-915.
11. Labussière M, Di Stefano AL, Gleize V, Boisselier B, Giry M, Mangesius S, Bruno A, Paterra R, Marie Y, Rahimian A, Finocchiaro G, Houlston RS, Hoang-Xuan K, et al. TERT promoter mutations in gliomas, genetic associations and clinico-pathological correlations. Br J Cancer. 2014; 111:2024-2032.
12. Heidenreich B, Nagore E, Rachakonda PS, Garcia-Casado Z, Requena C, Traves V, Becker J, Soufir N, Hemminki K, Kumar R. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat Commun. 2014; 5:3401.
13. Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, Roncalli M, Zucman-Rossi J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. 2014; 60:1983-1992.
14. Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, Melo M, da Rocha AG, Preto A, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013; 4:2185.
15. Aizer AA, Abedalthagafi M, Bi WL, Horvath MC, Arvold ND, Al-Mefty O, Lee EQ, Nayak L, Rinne ML, Norden AD, Reardon DA, Wen PY, Ligon KL, et al. A prognostic cytogenetic scoring system to guide the adjuvant management of patients with atypical meningioma. Neuro Oncol. 2016; 18:269-274.
16. Fürtjes G, Heß K, Wagner A, Senner V, Brokinkel C, Stummer W, Brokinkel B. Letter: Retention of ATRX and DAXX Expression in Meningiomas. Neurosurgery. 2017; 81:E22-E23.
17. Shi J, Hua X, Zhu B, Ravichandran S, Wang M, Nguyen C, Brodie SA, Palleschi A, Alloisio M, Pariscenti G, Jones K, Zhou W, Bouk AJ, et al. Somatic Genomics and Clinical Features of Lung Adenocarcinoma: A Retrospective Study. PLoS Med. 2016; 13:e1002162.
18. Haines K, Wang L, Sumazin P, Covington KR, Muzny DM, Kumar V, Doddapaneni H, Chao H, Wheeler DA, Tomlinson G, Williams Parsons D, Plon SE, Lopez D. Terrada: Discovery of chimeric transcripts involving APC and TERT in pediatric HCC by RNA sequencing. In AACR Special Conference: Advances in Pediatric Cancer Research: From Mechanisms and Models to Treatment and Survivorship, vol. 76. pp. A33. Fort Lauderdale, Florida: American Association for Cancer Research; 2015:A33.
19. Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, van Noesel MM, George RE, Tytgat GA, Molenaar JJ, Versteeg R. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. 2015; 47:1411-1414.
20. Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer. 2010; 103:1139-1143.
21. Shankar GM, Francis JM, Rinne ML, Ramkissoon SH, Huang FW, Venteicher AS, Akama-Garren EH, Kang YJ, Lelic N, Kim JC, Brown LE, Charbonneau SK, Golby AJ, et al. Rapid Intraoperative Molecular Characterization of Glioma. JAMA Oncol. 2015; 1:662-667.
22. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013; 110:6021-6026.
23. Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, Chen J, Robinson HE, Shim HS, Chmielecki J, Pao W, Engelman JA, Iafrate AJ, Le LP. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014; 20:1479-1484.
24. Heckman LD, Chahrour MH, Zoghbi HY. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. Elife. 2014; 3.