
INTRODUCTION
The tumor suppressor genes BRCA1 and BRCA2 maintain genomic stability through their involvement in homologous recombination (HR) double-stranded break DNA repair among other processes [1].
Carrying a mutation in the BRCA1 or BRCA2 genes increases a woman’s lifetime risk of developing breast and ovarian cancer, although there are considerable differences in disease manifestation. At the age of 80, cumulative cancer risk for BRCA1 and BRCA2 mutation carriers ranges from 72% to 69% for breast cancer development, and from 44% to 17% for ovarian cancer [2]. This high variability may be explained by other genetic modifiers and/or environmental factors.
Given the relation of synthetic lethality that exists between one of the components of the Base Excision Repair (BER) pathway, PARP1 (poly[ADP-ribose] polymerase 1), and both BRCA1 and BRCA2 genes [3], it is likely that other members of the BER pathway exhibit a similar behavior. We hypothesized that common genetic variants in genes involved in BER might modify a woman’s lifetime risk of developing breast and ovarian cancer if she is a BRCA1 or BRCA2 mutation carrier. In particular, two Single Nucleotide Polymorphisms (SNPs) in the OGG1 and NEIL2 genes were identified as cancer risk modifiers for BRCA1 and BRCA2 mutation carriers, respectively [4]. Although the molecular mechanism underlying these associations is not clear yet, both SNPS were in transcriptional regulatory regions of genes encoding DNA glycosylase enzymes which play an important role in the first steps of the pathway.
The BER pathway corrects base lesions from deamination, oxidation or methylation [5, 6] which represent the majority of endogenous DNA damage due to chemical reactions during cellular metabolism [7]. There are 11 DNA glycosylases which have the ability of recognizing a wide variety of lesions thanks to a DNA binding domain, the helix-hairpin helix DNA binding motif (like OGG1) [8] and the helix-2turn-helix domain (like NEIL2) [9]. In bi-functional DNA glycosylases, like OGG1 or NEIL2, base lesions are excised from the DNA thanks to its glycosylase activity and AP lyase activity, although they may have different DNA-structure/substrate affinities. For example, the OGG1 incises DNA at 8-oxoG residues, and is active only on duplex DNAs [10]. In contrast, NEIL2 shows preferential activity on bubble DNA or single-stranded DNA regions [11] and present high incising activity for several cytosine-derived lesions with robust activity for 5-hydroxyuracil and weaker activity for dihydrouracil, 5-hydroxycytosine, thymine glycol and 8-oxoG [10] .
If they are not repaired, these lesions may evolve into mutation (C:G→T transversions [12] or DNA single-strand [7] or double-strand breaks (DSBs) [13, 14], which are the principal source of genomic instability [15, 16].
Certain SNPs in DNA glycosylase genes could affect negatively to the general performance of the BER pathway and contribute by increasing the levels of genome instability and hence to a higher cancer risk, especially in presence of a defective BRCA1 or BRCA2 background. As an example, we previously identified that the single nucleotide polimorphism “rs2304277”, located 1.8Kb downstream the 3’-untranslated region (UTR) of OGG1 gene, was associated with an increased ovarian cancer risk for BRCA1 mutation carriers [4]. We tried to explain this cancer association at a molecular level and we discovered that the SNP was associated with a constitutive OGG1 transcriptional down-regulation, which contributed to a higher genome and telomere instability, especially in those individuals harboring mutations in BRCA1 [17].
Similarly, the SNP rs804271, localized within the NEIL2 promoter region, is associated with increased breast cancer risk for BRCA2 mutation carriers [4]. This SNP forms part of several transcription-factor binding motifs that are responsive to oxidative stress [18]. It has previously been reported that SNPs 5´- UTR upstream the coding region of the NEIL2 gene influence gene transcription levels and alter levels of genetic damage [19]. In this study, we have explored in two independent set of samples with different BRCA status the role of this SNP at transcriptional level and its possible implication on DNA damage and genome instability to explain its cancer risk modifier effect.
RESULTS
SNP frequency in FBOC series
We genotyped the rs804271 in FBOC (familial breast and ovarian cancer) individuals, and we found a SNP allelic frequency of 0.39, similar as reported for European population 0.41 in Ensembl data base (http://www.ensembl.org). No significant differences in the genotypic frequencies were detected among the different BRCA and control groups (Supplementary Table 1).
NEIL2 mRNA levels are activated by rs804271 SNP: In silico studies (HaploReg and GTEX public data), FBOC series and LCLs
The SNP rs804271 is located at the 5′- UTR region of the NEIL2 gene, within a transcriptional regulatory domain at Transcriptional Start Site (TSS) of the gene. We explored the possible phenotypic effects of this SNP by using HaploReg Database web server [20] and we found that 18 proteins are predicted to interact within TSS and 3 binding motifs for transcription factors TFs (E2F1, SIN3A and YY1) are predicted to be altered in the presence of this specific SNP (rs804271), (Supplementary Table 2).
Because transcriptional changes could be expected due to the modifications by this SNP at the TSS, we used the GTEx eQTL web server [21] (http://www.gtexportal.org) to test whether rs804271 was associated with changes on NEIL2 mRNA levels in different tissues. Overall, we found significant increased NEIL2 mRNA levels for 30 tissues, including breast (p = 1*10−4), ovary (p = 1.4 * 10−14), and blood (p = 6.6 * 10−13), Supplementary Table 3 although in some of them, such as “Cells - EBV-transformed lymphocytes (LCLs)” the effect was “moderated” (Supplementary Figure 1).
In parallel, we measured NEIL2 mRNA expression levels in FBOC series considering both, the BRCA mutational status and the presence or absence of the NEIL2- variant to stratify and compare expression values among groups. We found no significant differences in the NEIL2 mRNA levels between BRCA1 and/or BRCA2 mutation carriers compared to controls (Figure 1A). In contrast, when we stratified by the presence of the SNP we detected a common NEIL2 mRNA up-regulation pattern that was similar for each BRCA mutational group (Figure 1B). We performed linear regression analysis to confirm that the rs804271 was associated with significant higher NEIL2 mRNA levels (β = 0.24; p = 0.01) among the FBOC individuals.
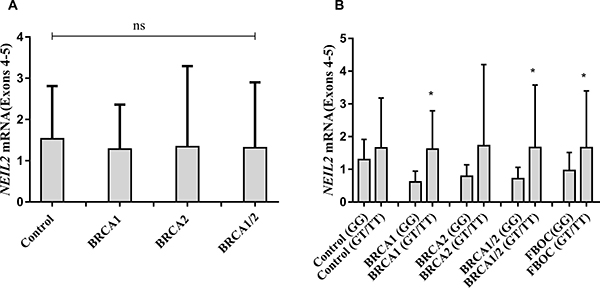
Figure 1: (A) Comparative analysis of NEIL2 mRNA expression according BRCA mutational status in FBOC series (BRCA1 and BRCA2 mutation carriers are compared with Controls). (B) Comparative analysis of NEIL2 mRNA expression according the SNP status ((Carriers (GT/TT) Vs Non-carriers (GG)) among the different FBOC groups (BRCA1, BRCA2 mutation carriers and BRCA1/BRCA2 non-carrier Controls). Bars represent the mean and the standard deviation for each group. Unpaired student t test was used to test for potential significant differences between means. (*p < 0.05).
Finally, we measured NEIL2 mRNA basal levels among the 20 LCLs considering the BRCA and SNP status. Although we detected higher NEIL2 mRNA levels for those LCLs harboring the SNP, these differences were not significant (Supplementary Figure 2). This result confirmed the tissue variability previously observed in the data provided by GTEX (Supplementary Figure 1).
NEIL2 mRNA and protein levels are correlated and both predict NEIL2-derived DNA damage
Because protein sample from FBOC series was not available, we decided to use the LCL panel (n = 20) to test NEIL2 protein levels (Supplementary Figure 3). Spearman correlation analysis confirmed that NEIL2 mRNA and protein levels were significantly correlated among LCLs in basal conditions (r = 0.51; p = 0.02), Figure 2A.
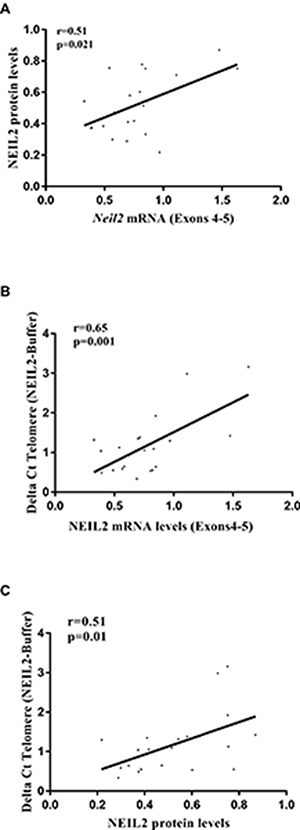
Figure 2: (A) Correlation analysis between NEIL2 mRNA and protein levels. (B) Correlation analysis between NEIL2 mRNA levels and the relative amount of “NEIL2-lesions”. (C) Correlation analysis between the NEIL2 protein levels and the relative amount of “NEIL2-lesions”. Spearman test, was used to test whether correlation is significant. significant p-value when (p < 0.05).
In parallel, we measured in the DNA extracted from the same LCLs (n = 20), the amount of base lesions that are recognized and processed by NEIL2 (NEIL2-lesions) at telomeres (detailed information in the material and methods section). We selected this region because NEIL-protein family members have been described to be active at telomeres [22]. Then, we performed a correlation analysis between the NEIL2 mRNA/ protein levels and the relative number of “NEIL2-lesions” detected, independently of the BRCA or the SNP status. We found that both NEIL2 mRNA and NEIL2 protein levels were significantly correlated with the relative number of telomeric “NEIL2-lesions¨ (r = 0.65; p = 0.001 and r = 0.51; p = 0.01, respectively), (Figure 2B and 2C).
The rs804271 is associated to higher levels of NEIL2-lesions at telomeres in FBOC series
When considering the BRCA status and NEIL2 genotypes, we found significantly higher amount of “NEIL2-lesions” in BRCA1 and BRCA2 mutation carriers compared with controls (p = 0.01 and p < 0.0001 respectively), (Figure 3A). Moreover, when we considered the presence of the SNP (rs804271) we found that those individuals presenting both genetic events (BRCA mutation together with the SNP) presented significantly higher levels of “NEIL2-lesions”, compared to their BRCA1/ BRCA2 counterparts without the SNP or controls (p < 0.05), (Figure 3B).
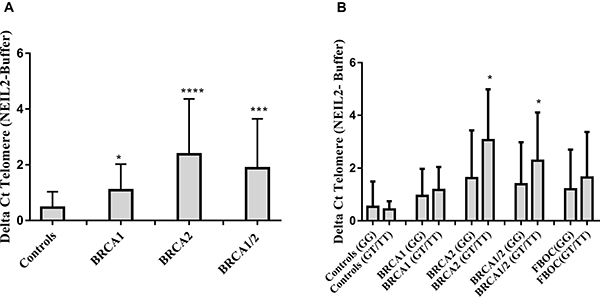
Figure 3: (A) Comparative analysis of the relative number of NEIL2-lesions found at telomeres according BRCA mutational status in FBOC series (BRCA1 and BRCA2 mutation carriers are compared with Controls). (B) Comparative analysis of the relative amount of “NEIL2-lesions” found at telomeres according the SNP status ((Carriers (GT/TT) Vs Non-carriers (GG)) among the different BRCA mutational groups in FBOC series (BRCA1, BRCA2 mutation carriers and BRCA1/BRCA2 non-carrier Controls). Unpaired student t test was used to test for potential significant differences. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Because FPG (formamidopyrimidine [fapy]-DNA glycosylase) (e. coli) recognizes specifically oxidative purines lesions (8-oxoG/methylFapyG) [23], we measured in the DNA from our FBOC individuals the relative amount of “FPG-lesions”. Then, we performed correlation analysis between (“FPG-lesions” and “NEIL2-lesions”) and we detected a significant correlation between both type of lesions (r = 0.40; p = 0,03), (Supplementary Figure 4A), which suggest that that from the wide range of lesions that NEIL2 can recognize [10], the presence of the SNP among BRCA1 and BRCA2 mutation carriers lead preferentially to the accumulation of purine lesions (8-oxoG or methylFapyG).
Because telomeres are susceptible to uracil miss incorporation which is primarily recognized and removed by the uracil DNA glycosylase (UNG) [24], we have measured the relative amount of uracil miss incorporation at the telomere region as NEIL2 is not able to recognize/process this type of lesions. We performed correlation analysis between “Uracil-lesions” and “NEIL2-lesions” and we found no significant correlation between them (Supplementary Figure 4B)
NEIL2-derived DNA damage correlates with γH2AX intensity signal
We measured the γH2AX signal intensity in the cell nucleus of the 20 LCLs (as a marker of DSBs) at basal conditions. We found a direct correlation between the relative amount of “NEIL2-lesions¨ and the nuclear γH2AX intensity signal independently of the BRCA or SNP status (r = 0.31; p = 0.09), (Figure 4).
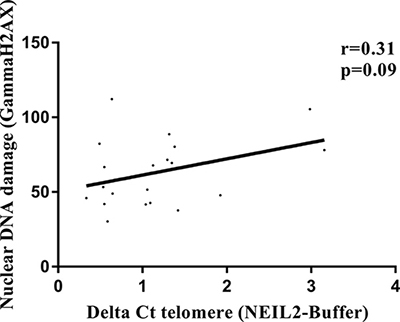
Figure 4: Correlation analysis between relative amount of “NEIL2-lesions” and the γH2AX nuclear intensity signal (DSBs). Spearman test, was used to test whether correlation is significant. significant p-value when (p < 0.05).
DISCUSSION
In the present study, we have tried to gain molecular insights into a common genetic variant (rs804271) previously reported by our group to be associated with increased breast cancer risk in BRCA2 mutation carriers [4]. For that, we have used two independent set of samples to test the SNP effect on NEIL2 transcriptional regulation and its possible implication on genome instability.
This SNP is localized within the TSS of NEIL2 gene. Previous characterization of the NEIL2 promoter region showed that NEIL2 transcription is influenced by certain SNPs located 5′ upstream of the start site [19]. Indeed, in silico analysis predicted that this polymorphism is located within a binding motif for several transcription factors (Supplementary Table 2), and transcriptional modifications due to this SNP may be expected.
Data from Gtex confirmed that the presence of rs804271 was associated with a significant mRNA upregulation in 30 tissues including breast (p = 0.00001), ovary (p = 1.4 * 10−14), and blood (p = 6.6 * 10−13), (Supplementary Table 3). However, for some tissues, such as “Cells - EBV-transformed lymphocytes (LCLs)”, this effect was “moderate” (Supplementary Figure 1), suggesting that the intensity of the SNP effect may be tissue specific. We validated these results in our FBOC series and we found, independently of the BRCA status, significantly increased NEIL2 mRNA levels in the blood from FBOC individuals harboring the SNP (β = 0.24; p = 0.01), suggesting that it is associated per se with transcriptional activation of the NEIL2 gene. In contrast, we were not able to detect a significant NEIL2 mRNA upregulation associated to the SNP in the 20 LCL analyzed, confirming the tissue specificity found in the GTEX data. All these results suggest that rs804271 is indeed associated with constitutive transcriptional activation of the NEIL2 gene.
A recent work in which NEIL1 and NEIL2 (Neil1 −/− /Neil2 −/−) double and NEIL1, NEIL2 and NEIL3 (Neil1 −/− /Neil2 −/− /Neil3 −/−) triple knock-out mouse models have been characterized, no accumulation of oxidative DNA damage, no changes in the mutation frequencies under normal physiological conditions and more importantly, no cancer predisposition for these mice has been observed [25]. This would agree with our results in which it is NEIL2 “excess” and not its “absence” that may be deleterious and responsible for the increased risk effect of this SNP in BRCA2 mutation carriers.
In the line of this hypothesis, it has been previously described that NEIL2 gene is frequently amplified in esophageal adenocarcinoma and that tumors with copy number gains of NEIL2 gene present significant poor prognosis [26, 27]. In addition, we have observed that NEIL2 gene is frequently upregulated in several tumor types (Supplementary Figure 5A), and more importantly that NEIL2 mRNA upregulation or copy number amplification has prognostic value for some of those tumors (Supplementary Figure 5B).
The molecular mechanism by which NEIL2 mRNA upregulation could be deleterious for BRCA2 mutation carriers is unclear. However, high expression levels of BER related enzymes have been associated with tissue oxidative DNA base damage [12]. In addition, it was described that rs804271 (previously ss74800505) was associated with both NEIL2 transcriptional modifications and significantly increased mutagen-induced genetic damage [19]. In fact, in LCLs we found a significant positive correlation between the amount of NEIL2 mRNA or protein levels and “NEIL2-lesions” (r = 0.65; p = 0.001 and r = 0.51; p = 0.02, respectively) (Figure 2B). Moreover, in FBOC the SNP was also associated with higher amount of “NEIL2-lesions” compared to their counterparts without the SNP, although it was only significant for BRCA1 and BRCA2 mutation carriers (p = 0.03) (Figure 3B).
A possible explanation for this result, could be that the NEIL2 enzyme “excess” as consequence of the SNP could lead to the recognition and binding to DNA lesions for which normally it presents low excision activity, like 8-oxoG [10]. Indeed, we found a significant correlation between “NEIL2-lesions” and “FPG-lesions” (r = 0.40; p = 0.003) (Supplementary Figure 4A), which mostly correspond to purine bases lesions (8-oxoG/methylFapyG). This could lead to a delay in the repair and to the accumulation of “NEIL2-lesions” in the DNA.
In the context of BRCA1 and BRCA2 deficiency, this accumulation of base lesions would be deleterious since both enzymes are involved in transcription-coupled repair of 8-OxoG [28] and protect against oxidative DNA damage converted into DSBs [14]. Indeed, our results in the LCLs confirmed that the relative number of “NEIL2-lesions” at the telomere was correlated with nuclear γH2AX intensity signal (a marker for DSBs) independently of the BRCA or SNP status (r = 0.31; p = 0.09) (Figure 4).
In summary, our hypothesis would be that this SNP activates at transcriptional level NEIL2 gene expression leading to a cascade of events that converge in the accumulation of unresolved “NEIL2-lesions” that may be converted into DSBs. In a system with a defective HR DNA repair, as it is the case for BRCA2 mutation carriers, this SNP would contribute to higher genome instability and finally to a higher cancer risk for this specific group of patients.
MATERIALS AND METHODS
Familial breast and ovarian cancer (FBOC) series
We studied a group composed of 166 individuals belonging to 51 families meeting high-risk criteria, and screened for deleterious mutations in the BRCA1 and BRCA2 genes, as reported previously [29]. Of these families, 25 carried a deleterious mutation in the BRCA1 gene, 25 in BRCA2.
Eighty individuals were used as non-carrier controls: they were relatives of BRCA1/2 mutation carriers who did not have personal cancer antecedents and did not harbor the corresponding familial mutation in the BRCA1 or BRCA2 genes.
All cases and controls signed an appropriate informed consent form and the ethics committee of the hospital involved (Fuenlabrada University Hospital) approved the proposal.
We used this set of samples to calculate the SNP frequency, to quantify NEIL2 mRNA levels in peripheral blood and to measure the accumulation of oxidative DNA damage at telomeres (NEIL2-lesions, FPG-lesions, uracil accumulation) in blood DNA. (Table 1).
Table 1: FBOC series information
Families (n) |
Healthy carriers (n) |
Cancer cases (n) |
rs804271 genotyped (n) |
NEIL2 mRNA (n) |
NEIL2-lesions |
FPG-lesions |
uracil-lesions |
||
|---|---|---|---|---|---|---|---|---|---|
BRCA1 |
25 |
21 |
19 |
40 |
24 |
25 |
14 |
14 |
|
BRCA2 |
25 |
23 |
23 |
46 |
30 |
35 |
18 |
18 |
|
Controls |
na |
0 |
0 |
80 |
29 |
25 |
20 |
20 |
|
Information regarding number of healthy BRCA1 and BRCA2 mutation carriers or cancer cases and the sample size for each experimental section.
Lymphoblastoid cell lines
A second set of 20 LCLs was established by Epstein-Barr virus transformation of peripheral blood lymphocytes from thirteen healthy women carrying heterozygous mutations in BRCA1 and seven non-carrier relatives used as controls. Mutational analysis had been performed by Sanger sequencing (BRCA status) or Taqman probe (rs804271) (Supplementary Table 4). None of the women included in the study had personal antecedents of cancer. This LCL panel has been previously described by our group [30]. Cell lines were cultured in RPMI-1640 media (Sigma-Aldrich) supplemented with non-heat-inactivated 20% fetal bovine serum (Sigma-Aldrich), penicillin-streptomycin (Gibco) and Fungizone (Gibco). The cultures were carried out in 25 cm2 flasks (Corning) at 37°C in 5% CO2 atmosphere and cell lines were maintained in exponential growth by daily dilution to 106 cells/ml of full media.
We used this sample set to analyze the correlation between NEIL2 mRNA – protein levels, the relative number of “NEIL2-lesions” found at DNA, and the relative number of double stranded brakes (DSB) at DNA.
SNP genotyping (rs804271)
Single Nucleotide Polymorphism rs1466785, located in the NEIL2 gene is a cancer risk modifier for BRCA2 mutation carriers [4]. Imputation using 1000 Genomes data showed that there were several SNPs in strong linkage disequilibrium (LD) with rs1466785, the original SNP reported in Osorio et al. [4]. Of these, we considered rs804271 to be the best candidate, given that it showed the most significant associations and that there existed functional data supporting its putative role in cancer [19].
DNA was extracted from peripheral blood of FBOC patients or LCLs using MagNAPure LC 2.0 (Roche Diagnostics, Indianapolis, Indiana) following the manufacturer’s instructions. DNA quantification and quality were assessed by NanoDrop® (ND-1000 V3.7.1). A specific Taqman probe for rs804271 was used to genotype the presence/absence of the polymorphism among the sample collection. Allelic discrimination assays were conducted using the 7900HT Fast Real-Time PCR System (Applied Biosystems). Probe design for rs804271 is (G>T) instead of (C>A). Along the manuscript we refer to the variant as G>T.
NEIL2 mRNA expression analysis
RNA was extracted from peripheral blood cells using TRIzol Reagent (Ambion®, Life Techonogies) according to the manufacturer’s instructions. NanoDrop® (ND-1000 V3.7.1) was used to assess both RNA quantity and quality. Two microliters of cDNA at a final concentration of 10-20 ng/μl were mixed in triplicate with GoTaq® qPCR MasterMix 1x (Promega), NEIL2 cDNA primers (F/R) and GAPDH cDNA primers (F/R) at final concentrations of 500nM. Primers used were: NEIL2 4-5 exons (F: GTCACACCCACCTGTGACAT; R: GCACTCAGGACTGAACCGAG) and GAPDH (F: CCTGCACCACCAACTGCTTA; R: CCATCACGCCACAGTTTCC).All reagents were used following the manufacturer’s instructions. qPCR was done using the QuantStudio S6 system (Applied Biosystems).
NEIL2 protein quantification
The expression level of endogenous NEIL2 protein was analyzed by western blot. Briefly, cell lysates were prepared in RIPA buffer (Sigma) and protease inhibitors cocktail (Roche). Protein content was determined by Lowry analysis (Bio-Rad). Eighty micrograms of proteins were analyzed by SDS-PAGE on polyacrylamide gels and transferred to Immobilon-FL membranes (Millipore). Membranes were blocked in TBS-T (50 mM Tris-HCl, 150 mM NaCl, pH 7.5 plus 0.1% Tween 20) and 5% nonfat milk for 1 hour at RT. Blots were probed with following primary antibodies: rabbit anti-NEIL2 (Atlas Antibibodies, #HPA064460) at 1/1000 dilution or mouse anti-GAPDH (manufactured by the monoclonal antibodies core nit from the Spanish National Cancer Research Centre) at 1/3000 dilution in TBS-T containing 5% nonfat milk. The secondary antibodies were HRP-conjugated (Dako) and the immunoblots were developed using the ECL system (GE Healthcare). ImageLab software version 4.1 (Bio-Rad) was used for image acquisition and images were analyzed using ImageJ software for quantification of signal intensity/area for both proteins.
Oxidative DNA damage studies “NEIL2-lesions”
We used a qPCR-based method to evaluate the oxidative DNA damage within telomeric DNA [32], based on differences in PCR kinetics between DNA template digested by formamidopyrimidine-DNA glycosylase (FPG) and undigested DNA. Quantitative real-time amplification of genomic DNA was performed as described by O’Callaghan et al. [31].
Measurement of telomere damage
Oxidative DNA damage within telomeres
We used a qPCR-based method to evaluate the oxidative stress within telomeric DNA. We followed the procedure described by O’Callaghan et al. based on differences in PCR kinetics between DNA template digested by formamidopyrimidine-DNA-glycosylase (FPG) and undigested DNA [32]. Briefly, FPG is a bacterial DNA glycosylase that recognizes and cuts the oxidized bases from DNA, principally 8-oxoG, AP sites that are converted in single-strand breaks (SSBs) by its AP-lyase activity. These SSBs reduce amplification efficiency, thus, the ΔCq after digesting DNA by FPG (Cq digested – Cq undigested) is proportional to the oxidative damage in the amplified region. The incubation and qPCR amplification of genomic DNA was performed as described by O’Callaghan et al. [31].
Quantification of “NEIL2-lesions” accumulation at telomeres
The telomere oxidation protocol previously described can be potentially adapted to quantify the accumulation of different base lesions incubating the DNA with other glycosylases that are sensitive to other specific base lesions. Following this premise, we used NEIL2 enzyme to measure the “NEIL2-lesions” accumulation (5hydroxyuracildihydrouracil, 5-hydroxycytosine, thymine glycol and 8-oxoG) at telomeres [9]. We optimized the protocol using a low NEIL2 concentration, decreasing DNA amount and incubation time. 200 ng of genomic DNA was incubated with 5,6 μM NEIL2 (provided by Dr. Thomas Helleday, Karolinska Institutet, Stockholm, Sweden) or without (replaced with H20) in a buffer (25 mM TrisHcl pH 8.0, 15 mM NaCl, 2 Mm MgCl2 and 0.0025% Tween 20) for 4 hours at 37°C. The reaction was stopped by incubation at 95°C for 5 min. qPCR analysis was performed on 10 ng of digested or undigested genomic DNA following the same reagents and conditions that in the original protocol for FPG [31].
Quantification of uracil accumulation at telomeres
Following this premise, we used UNG to measure the accumulation of uracil at telomeres that is recognized and excised by this enzyme [33]. We optimized the protocol using a low UNG concentration, decreasing DNA amount and incubation time. 180 ng of genomic DNA was incubated with 130 nM UNG (provided by Dr. Thomas Helleday, Karolinska Institutet, Stockholm, Sweden) or without (replaced with H20) in a buffer (25 mM TrisHcl pH 8.0, 15 mM NaCl, 2 Mm MgCl2 and 0.0025% Tween 20) for 30 min at 37°C. The reaction was stopped by incubation at 95°C for 5 min. qPCR analysis was performed on 10 ng of digested or undigested genomic DNA following the same reagents and conditions that in the original protocol for FPG [31].
DNA damage
LCLs were cultured 4 hours before fixation with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, Philadelphia, USA). Two hours before fixation, cells were counted and seeded into a poly-L-lysine-coated (Sigma-Aldrich) μCLEAR bottom 96-well plate (Greiner Bio-One) at a density of 75,000 cells per 100ul full media per well. LCL were then left for 2 hours to attach to the surface of the wells, fixed for 15 min at room temperature, permeabilized in 0.5% Triton X-100 in PBS for 20 minutes at 4°C and stained with primary and secondary antibodies and 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) to visualize nuclei. To detect γ-H2AX we used mouse monoclonal anti-phospho-histone H2AX antibody (Millipore; #05-636). Alexa Fluor 488 from molecular probes (Invitrogen; #A-11034) was used, and fluorescent images were automatically taken for each well of the 96-well plate using an Opera High-Content Screening System (Perkin Elmer). Pictures were taken under non-saturating conditions using a 40x magnification lens to calculate the γ-H2AX nuclear signal intensity.
Statistical analysis
Pearson’s chi-squared test was used to calculate whether differences in the frequency of the SNP among the FBOC groups were significant (Supplementary Table 1).
We performed linear regression analysis to test whether cancer antecedents in BRCA1 and BRCA2 mutation carriers were associated with any of the variables we evaluated in this report, but we did not find significant differences (Significant p-values < 0.05) between healthy BRCA1 and BRCA2 carriers or cancer cases. Hence, we did not stratify for cancer status in these groups (Supplementary Table 5).
We considered heterozygotes and homozygotes (GT/TT) as a single group, to evaluate the effect of the SNP for each of the studied variables, as the cancer modifier effect of rs804271 is dominant for BRCA2 mutation carriers [4].
Significant differences for the different comparative analysis were stablished by unpaired t test analysis (SNP effect on NEIL2 mRNA levels or NEIL2 derived base damage accumulation, Figure 1 and Figure 3, respectively).
Spearman correlation was used to assess for significant correlations between NEIL2 mRNA levels, protein levels and NEIL2 derived base damage accumulation at telomeres (Figure 3). Also, to assess whether NEIL2-lesions correlates with “FPG-lesions”, “UNG-lesions” and γ-H2AX nuclear signal intensity in FBOC and LCLs respectively (Supplementary Figure 4A and Figure 4B).
Statistical calculations were done using SPSS version 18 (SPSS Inc., Chicago, Illinois) and GraphPad Prism 5.03 (San Diego, California); graphs were made using GraphPad Prism 5.03.
ACKNOWLEDGMENTS
We thank Alicia Barroso her technical assistance. Also to Dr. Thomas Helleday, (Karolinska Institutet, Stockholm, Sweden) that kindly provided NEIL2 and UNG purified enzymes.
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health. Additional funds were provided by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. Donors were enrolled at Biospecimen Source Sites funded by NCI/SAIC-Frederick, Inc. (SAIC-F) subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171), and Science Care, Inc. (X10S172). The Laboratory, Data Analysis, and Coordinating Center (LDACC) were funded through a contract (HHSN268201000029C) to The Broad Institute, Inc. Biorepository operations were funded through an SAIC-F subcontract to the Van Andel Institute (10ST1035). Additional data repository and project management were provided by SAIC-F (HHSN261200800001E). The Brain Bank was supported by supplements to University of Miami grants DA006227 & DA033684 and to contract N01MH000028. Statistical Methods development grants were made to the University of Geneva (MH090941 & MH101814), the University of Chicago (MH090951, MH090937, MH101820, MH101825), the University of North Carolina - Chapel Hill (MH090936 & MH101819), Harvard University (MH090948), Stanford University (MH101782), Washington University St Louis (MH101810), and the University of Pennsylvania (MH101822). The data used for the analyses described in this manuscript were obtained from: [insert, where appropriate] the GTEx Portal on 01/12/2015 and/or dbGaP accession number phs000424.v7.p2 on 01/10/2017.
CONFLICTS OF INTEREST
None.
FUNDING
J.B.’s laboratory is partially funded by the Spanish Ministry of Health PI12/00070, supported by FEDER funds, and the Spanish Research Network on Rare diseases (CIBERER). C.B-B is supported by FIS PI12/00070. J.M.B is supported by grant FPU15/01978 from the Spanish Ministry of Education, Culture and Sport. MU is supported by the Spanish Ministry of Health PI14/00459 with FEDER funds. The study was partially supported by the Spanish Ministry of Economy and Competitiveness (MINECO) SAF2014-57680-R.
REFERENCES
1. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012; 12:68–78. https://doi.org/10.1038/nrc3181.
2. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, Goldgar DE, Terry MB, Rookus MA, et al, and BRCA1 and BRCA2 Cohort Consortium. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017; 317:2402–16. https://doi.org/10.1001/jama.2017.7112.
3. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev. 2008; 8:193–204. https://doi.org/10.1038/nrc2342.
4. Osorio A, Milne RL, Kuchenbaecker K, Vaclová T, Pita G, Alonso R, Peterlongo P, Blanco I, de la Hoya M, Duran M, Díez O, Ramón Y Cajal T, Konstantopoulou I, et al. DNA glycosylases involved in base excision repair may be associated with cancer risk in BRCA1 and BRCA2 mutation carriers. PLoS Genet. 2014; 10:e1004256.
5. Dizdaroglu M, Coskun E, Jaruga P. Repair of oxidatively induced DNA damage by DNA glycosylases: Mechanisms of action, substrate specificities and excision kinetics. Mutat Res. 2017; 771:99–127. https://doi.org/10.1016/j.mrrev.2017.02.001.
6. Dianov GL, Hübscher U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013; 41:3483–90. https://doi.org/10.1093/nar/gkt076.
7. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993; 362:709–15. https://doi.org/10.1038/362709a0.
8. Arai K, Morishita K, Shinmura K, Kohno T, Kim SR, Nohmi T, Taniwaki M, Ohwada S, Yokota J. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene. 1997; 14:2857–61. https://doi.org/10.1038/sj.onc.1201139.
9. Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem. 2002; 277:30417–20. https://doi.org/10.1074/jbc.C200355200.
10. Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: A new perspective. DNA Repair (Amst). 2007; 6:470–80. https://doi.org/10.1016/j.dnarep.2006.10.011.
11. Dou H, Mitra S, Hazra TK. Repair of Oxidized Bases in DNA Bubble Structures by Human DNA Glycosylases NEIL1 and NEIL2. J Biol Chem. 2003; 278:49679–84. https://doi.org/10.1074/jbc.M308658200.
12. Whitaker AM, Schaich MA, Smith MR, Flynn TS, Freudenthal BD. Base excision repair of oxidative DNA damage: from mechanism to disease. Front Biosci (Landmark Ed). 2017; 22:1493–1522. http://dx.doi.org/10.2741/4555.
13. Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 2011; 39:6986–97. https://doi.org/10.1093/nar/gkr331.
14. Fridlich R, Annamalai D, Roy R, Bernheim G, Powell SN. BRCA1 and BRCA2 protect against oxidative DNA damage converted into double-strand breaks during DNA replication. DNA Repair (Amst). 2015; 30:11–20. https://doi.org/10.1016/j.dnarep.2015.03.002.
15. Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008; 9:619–31. https://doi.org/10.1038/nrg2380.
16. Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001; 27:247–54. https://doi.org/10.1038/85798.
17. Benítez-Buelga C, Vaclová T, Ferreira S, Urioste M, Inglada-Perez L, Soberón N, Blasco MA, Osorio A, Benítez J. Molecular insights into the OGG1 gene, a cancer risk modifier in BRCA1 and BRCA2 mutations carriers. Oncotarget. 2016; 7:25815–25. https://doi.org/10.18632/oncotarget.8272.
18. Kinslow CJ, El-Zein RA, Rondelli CM, Hill CE, Wickliffe JK, Abdel-Rahman SZ. Regulatory regions responsive to oxidative stress in the promoter of the human DNA glycosylase gene NEIL2. Mutagenesis. 2010; 25:171–7. https://doi.org/10.1093/mutage/gep058.
19. Kinslow CJ, El-Zein RA, Hill CE, Wickliffe JK, Abdel-Rahman SZ. Single nucleotide polymorphisms 5??? upstream the coding region of the NEIL2 gene influence gene transcription levels and alter levels of genetic damage. Genes Chromosom Cancer. 2008; 47:923–32. https://doi.org/10.1002/gcc.20594.
20. Ward LD, Kellis M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012; 40. https://doi.org/10.1093/nar/gkr917.
21. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013; 45:580–5. https://doi.org/10.1038/ng.2653.
22. Zhou J, Fleming AM, Averill AM, Burrows CJ, Wallace SS. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015; 43:4039–54. https://doi.org/10.1093/nar/gkv252.
23. Tchou J, Bodepudi V, Shibutani S, Antoshechkin I, Miller J, Grollman AP, Johnson F. Substrate Specificity of Fpg Protein: Recognition and cleavage of oxidatively damaged DNA. J Biol Chem. 1994; 269:15318–24. https://doi.org/10.1021/bi1014453.
24. Vallabhaneni H, Zhou F, Maul RW, Sarkar J, Yin J, Lei M, Harrington L, Gearhart PJ, Liu Y. Defective repair of uracil causes telomere defects in mouse hematopoietic cells. J Biol Chem. 2015; 290:5502–11. https://doi.org/10.1074/jbc.M114.607101.
25. Rolseth V, Luna L, Olsen AK, Suganthan R, Scheffler K, Neurauter CG, Esbensen Y, Kuśnierczyk A, Hildrestrand GA, Graupner A, Andersen JM, Slupphaug G, Klungland A, et al. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci Rep. 2017; 7:4384. https://doi.org/10.1038/s41598-017-04472-4.
26. Frankel A, Armour N, Nancarrow D, Krause L, Hayward N, Lampe G, Smithers BM, Barbour A. Genome-wide analysis of esophageal adenocarcinoma yields specific copy number aberrations that correlate with prognosis. Genes Chromosom Cancer. 2014; 53:324–38. https://doi.org/10.1002/gcc.22143.
27. Goh XY, Rees JRE, Paterson AL, Chin SF, Marioni JC, Save V, O’Donovan M, Eijk PP, Alderson D, Ylstra B, Caldas C, Fitzgerald RC. Integrative analysis of array-comparative genomic hybridisation and matched gene expression profiling data reveals novel genes with prognostic significance in oesophageal adenocarcinoma. Gut. 2011; 60:1317–26. https://doi.org/10.1136/gut.2010.234179.
28. Le Page F, Randrianarison V, Marot D, Cabannes J, Perricaudet M, Feunteun J, Sarasin A. BRCA1 and BRCA2 are necessary for the transcription-coupled repair of the oxidative 8-oxoguanine lesion in human cells. Cancer Res. 2000; 60:5548–52.
29. Milne RL, Osorio A, Cajal TRY, Vega A, Llort G, De La Hoya M, Díez O, Carmen Alonso M, Lazaro C, Blanco I, Sánchez-de-Abajo A, Caldés T, Blanco A, et al. The average cumulative risks of breast and ovarian cancer for carriers of mutations in BRCA1 and BRCA2 attending genetic counseling units in Spain. Clin Cancer Res. 2008; 14:2861–9. https://doi.org/10.1158/1078-0432.CCR-07-4436.
30. Vaclová T, Gómez-López G, Setién F, Bueno JM, Macías JA, Barroso A, Urioste M, Esteller M, Benítez J, Osorio A. DNA repair capacity is impaired in healthy BRCA1 heterozygous mutation carriers. Breast Cancer Res Treat. 2015; 152:271–82. https://doi.org/10.1007/s10549-015-3459-3.
31. O’Callaghan NJ, Dhillon VS, Thomas P, Fenech M. A quantitative real-time PCR method for absolute telomere length. Biotechniques. 2008; 44:807–9. https://doi.org/10.2144/000112761.
32. O’Callaghan N, Baack N, Sharif R, Fenech M. A qPCR-based assay to quantify oxidized guanine and other FPG-sensitive base lesions within telomeric DNA. Biotechniques. 2011; 51:403–12. https://doi.org/10.2144/000113788.
33. Doseth B, Visnes T, Wallenius A, Ericsson I, Sarno A, Pettersen HS, Flatberg A, Catterall T, Slupphaug G, Krokan HE, Kavli B. Uracil-DNA glycosylase in base excision repair and adaptive immunity: Species differences between man and mouse. J Biol Chem. 2011; 286:16669–80. https://doi.org/10.1074/jbc.M111.230052.