
INTRODUCTION
Rheumatoid arthritis (RA) is the most common form of systemic autoimmune arthritis with unknown etiology, characterized by systemic inflammation and persistent poly-joint synovitis, principally leading to injury of the flexible joints, often with symptoms of joint pain and swelling, stiffness, bone destruction and fatigue, as well as implications of extra articular organs [1, 2]. The prevalence of RA varies largely in different populations, from 0.25% in Eastern Asians to 0.75% in European ancestry, and to as high as 6% in American Indians [3]. It remains largely unknown whether genetics, cultural, or environmental factors contribute to these differences. During the past years, an increasing list of genetic associations with RA has emerged from genome wide association studies (GWAS), which attributes great relevance to immune system contributed by profound sources of genetic variation with a panel of surface and intracellular signaling molecules as well as cytokines [4, 5]. GWAS has also revealed a complex picture of both shared and population-specific genetic susceptibility loci to this autoimmune disease in comparison of Asian and European populations [6, 7]. Generally, GWASs are designed to capture common genetic variation, and to date, a large portion of the heritability of complex traits has not been explained [8], which has prompted us to explore other potential sources of genetic susceptibility to RA, such as rare variants.
Whole-exome sequencing (WES) has become a popular and powerful technique for the identification of rare variants that alter protein functions, which may contribute to disease pathogenesis [9-11]. Protein-coding variants are more straightforward to annotate biological functions and pinpoint causative genes. To our knowledge, there is no comprehensive WES on RA in Han Chinese population in the literatures yet. As nowadays most RA-associated polymorphisms identified by GWASs are in non-coding region (Supplementary Table 6), in our study, we performed WES of 124 subjects in Chinese ancestry to investigate both rare and common variants predicted to be deleterious. In addition, we further characterized their cellular biological function categories of multiple risk variants based on the analysis of ingenuity pathways. Using this strategy, we have for the first time demonstrated potential causal variants resulted from the established RA risk loci as well as from novel candidate genes and pathways associated with RA in Chinese population.
RESULTS
Deleterious variants in novel RA candidate genes
WES data were generated from 58 RA patients with a median coverage of 76-fold on targeted exome regions (Supplementary Figure 1A). An average of 96% of all targeted regions was covered by at least 20-fold. The healthy controls had a median coverage of 68-fold on targeted exome regions, and an average of 94% of those regions was covered by at least 20-fold (Supplementary Figure 1B). A total of 3,537,952 variants were identified from 58 RA samples (Figure 1). After applying the quality filters by removing synonymous variants, we found 72,024 exonic and splicing variants, including nonsynonymous substitutions and a small number of stop-gain, stop-loss, frameshift and non-frameshift indels (Supplementary Table 1). Of these, 135 variants were identified as deleterious based on the “pathogenic” annotation in ClinVar, and an additional 21,755 variants predicted to be deleterious were identified using an ensemble logistic regression score.
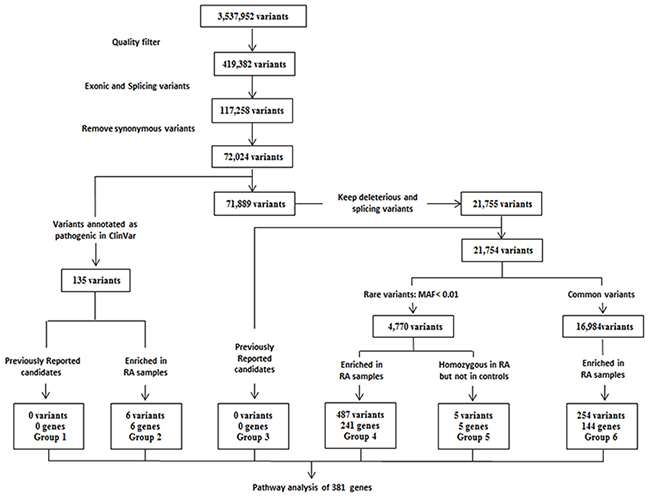
Figure 1: Experimental workflow of whole-exome sequencing was shown to detect and prioritize variants conferring susceptibility to rheumatoid arthritis (RA) using variant filtration and gene burden analysis. The variant list for all groups can be found in Supplementary Table 3. MAF = minor allele frequency in the 1000 Genomes Southern Han Chinese (phase III) population. Pathway analysis in candidate genes identified from 58 RA patients was performed using DAVID 6.8 (https://david.ncifcrf.gov/summary.jsp).
It was initial surprising that our identified genes were not found in previously reported candidate risk variants GWAS data (group 1 and group 3 in Figure 1; Supplementary Table 3 and 6), such as HLA-associated genes. However, after reviewing the location of the reported RA-associated variants, we found that only 9 of over 200 variants are located in the exome area, including CTLA4 (rs231775), FCGR2A (rs1801274), IL6R (rs2228145), OLIG3 (rs2230926), PTPN22 (rs2476601), RTKN2 (rs3125734), SH2B3 (rs3184504), TNFAIP3 (rs223092) and TYK2 (rs34536443). Our data has applied WES technique focusing on exome region, thus explaining the reason why we have identified a new set of RA-associated genes. Interestingly, two novel risk variant loci were identified associated with TGFβ1 (transforming growth factor β1) and FOXP3 (forkhead box P3) genes (group 4 and group 6 in Figure 1; Supplementary Table 3) whereas other known variations of these two genes were previously found to be involved in the risk to RA [12-14].
In order to identify novel genes and pathways that could enhance understanding of RA pathogenesis, we performed a gene burden analysis to identify genes for which deleterious variants were enriched in our Han Chinese RA samples compared to healthy control and public control samples. Six such genes were identified (group 2 in Figure 1; Supplementary Table 2 and 3). Of these, a missense variant of SAA1 (Serum Amyloid A1) was found in 3 RA patients but not present in healthy controls. SAA1 is highly expressed in response to inflammation and tissue injury, and strongly associated with activity of the disease and risk of cardiovascular and renal involvement in RA patients [15-17], suggesting that this novel deleterious variant may potentially contribute to RA disease risk through its interference with proinflammatory effectors. Additional pathogenic variant of OXCT1 (3-Oxoacid CoA-Transferase 1) was predicted to be damaging (disease-related, D) in our RA patients, encoding Succinyl-CoA:3-ketoacid coenzyme A transferase 1 (SCOT1), which is a key enzyme for synthesis and degradation of ketone bodies involved in cardiovascular disease [18, 19].
Rare variants are more likely to predict a significant impact on protein function and result in clinically relevant consequences than common ones [20]. Thus, we grouped variants that were indicated to be deleterious into rare (minor allele frequency <1%) and common variants, which did not overlap with previously reported candidates (Supplementary Table 6). Performing a gene burden analysis for variants within each of these groups, we identified 241 genes (group 4 in Figure 1; Supplementary Table 3) with rare, deleterious variants specifically enriched in our Chinese RA samples compared to healthy control and public control samples. Notably, since the functional impact of rare and deleterious variants is likely to be greater when present as homozygote, 5 rare and deleterious homozygous variants (NCR3LG1, RAP1GAP, CHCHD5, HIPK2 and DIAPH2) were identified in the Chinese RA samples and absent in the controls (group 5 in Figure 1; Supplementary Table 3). Finally, we also identified 144 genes with common and deleterious variants in RA patients (group 6 in Figure 1; Supplementary Table 3).
Pathway discovery
Using our methodology, we identified a total of 381 genes as candidates for increased risk of RA (Supplementary Table 3). In order to identify the associated biologic pathways, we performed the functional enrichment analysis using DAVID 6.8 and identified the pathways of the extracellular matrix (ECM)-receptor interaction, protein digestion and absorption, focal adhesion and glycerophospholipid metabolism as significantly overrepresented (Table 1), which were reported to be relevant in pathogenesis of arthritis [21-24].
Table 1: Pathway analysis for candidate genes conferring susceptibility to RA
|
Pathway |
P value |
Genes |
|---|---|---|
|
Based on genes identified in comparison of RA patients and controls |
||
|
ECM-receptor interaction |
2.1 × 10−3 |
COL4A4, COL6A5, COL11A1, COL11A2, HSPG2, ITGB5, LAMC1, THBS1 |
|
Protein digestion and absorption |
2.3 × 10−3 |
ATP1A1, ATP1A4, COL4A4, COL6A5, COL11A1, COL11A2, MME, PRCP |
|
Focal adhesion |
2.8 × 10−2 |
RASGRF1, COL4A4, COL6A5, COL11A1, COL11A2, FLNB, ITGB5, LAMC1, MYL5, THBS1 |
|
Glycerophospholipid metabolism |
4.8 × 10−2 |
CHAT, GPAT4, LPIN3, LPCAT1, MBOAT1, PTDSS2 |
|
Based on genes only identified in disease duration comparison of RA patients |
||
|
Olfactory transduction |
1.2 × 10−2 |
OR14C36, OR4A15, OR52N4, OR6C74, OR6C75, OR7G3, OR9K2 |
In order to identify variants that might predispose RA patients to disease duration, we repeated the variant filtration and gene burden analysis on Chinese RA samples with the disease duration ≥3-year compared to the disease duration ≤1-year. A total of 277 genes were identified (Supplementary Table 4) compared to the 381 genes identified in the case–control comparison (Supplementary Table 3). Of these, 87 genes were unique to disease duration with exonic variants (Supplementary Table 5). Pathway analysis performed on the 87 genes identified olfactory transduction pathway as significantly overrepresented (Table 1), including OR14C36, OR4A15, OR52N4, OR6C74, OR6C75, OR7G3 and OR9K2.
Structural analysis and function change prediction of mutant proteins
In order to gain structure insights of the protein mutation with pathogenic variants into the clinical conditions of RA patients, we derived a three-dimensionally structure model of SAA1 Gly90Asp (rs79681911) and SCOT1 Thr58Met (rs75134564) by combining homology modeling with point mutation in MOE 2015.09 package. The crystal structures of human SAA1 protein (PDB code: 4IP8.A) and SCOT1 protein (PDB code: 3K6M.C) were selected to be used as templates due to their optimal identity with the target sequences of SAA1 (Protein RefSeq: NP_000322.2) and SCOT1 (Protein RefSeq: NP_000427.1), 83.6% and 83.5%, respectively (Supplementary Figure 3). The SAA1 and SCOT1 models with the best packing quality function and full energy minimization were assessed by Ramachandran plots, indicating that the phi and psi backbone dihedral angles in the models were reasonable (Supplementary Figure 4).
Structural analysis of SAA1 Gly90Asp (Figure 2A and 2B) revealed that the substitution of glycine with aspartic acid induced the formation of two pairs of hydrogen bonds with two threonine residues (Ala91 and Asp93), exhibiting more stable structure of loop region and promoting the polar interaction. Moreover, this mutation shortened the length of α helix 4, which may affect the stability of SAA1. Structural changes in SAA1 protein caused the surface of Asp90 to be exposed in solvent environment, leading to the increased hydrophilic region. In addition, the construction of 3D models in SCOT1 and its mutant Thr58Met revealed that this substitution resulted in the disappearance of the hydrogen bond between Thr58 and Asp206, the reduction of intramolecular polar interactions and the expansion of hydrophobic region (Figure 2C and 2D), suggesting its potential function alteration in RA pathogenesis.
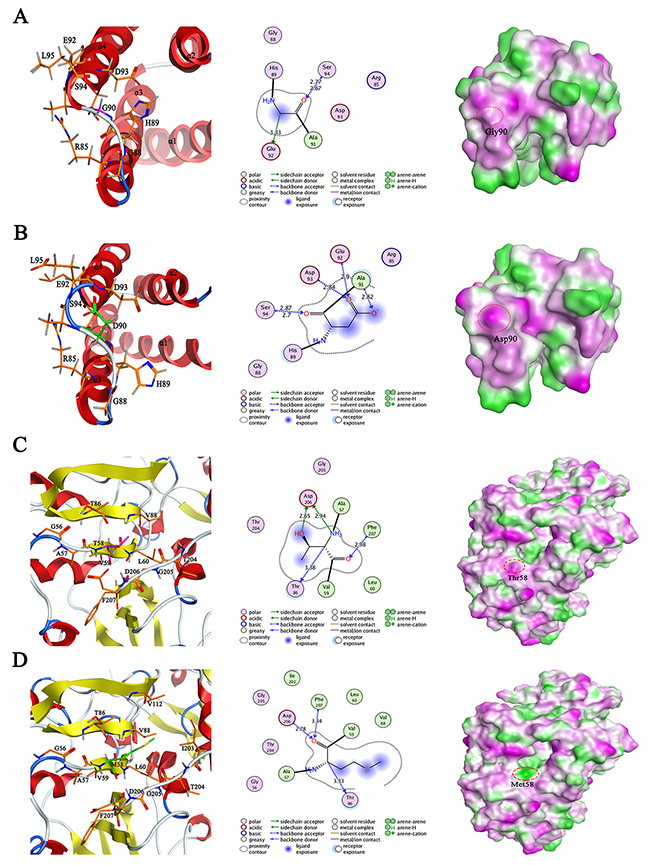
Figure 2: The modeled 3D structure comparison of human wild type SAA1 (A) and its mutant G90D (B), as well as wild type SCOT1 (C) and its mutant T58M (D). Left panel: the ribbon secondary structure diagram with α helices in red and β sheets in yellow; middle panel: the proposed interactions between mutated residue and its surrounding residues, distances are not represented to scale; right panel: the lipophilic surface representation by showing hydrophilic (magenta), neutral (green) and lipophilic (white).
DISCUSSION
In this study, we performed perspective WES aiming to identify potentially causal variants in a cohort of Chinese RA patients. We focused on investigating the occurrence frequency of variants in genes previously associated with RA as well as novel genes. We generated a previously reported GWAS candidate risk loci list in Supplementary Table 6, including Caucasians and Black Americans. It was initial surprising that our identified genes were not found in this list of GWAS data (group 1 and group 3 in Figure 1), such as HLA-associated genes. However, after reviewing the location of the reported RA-associated variants, we found that very few variants are located in the exome area, including CTLA4 (rs231775), FCGR2A (rs1801274), IL6R (rs2228145), OLIG3 (rs2230926), PTPN22 (rs2476601), RTKN2 (rs3125734), SH2B3 (rs3184504), TNFAIP3 (rs223092) and TYK2 (rs34536443). Notably, most common variants identified by GWAS analysis are located in the non-coding region of the genome, while WES is a powerful technique for the identification of protein-coding variants, which are more straightforward to annotate biological functions and pinpoint causative genes. Our current work may be treated as a pilot study, and it is significantly valuable for our future works by expending samples of RA patients in Han Chinese population, even in comparison with different ethnicities.
Despite known variants of TGFβ1 and FOXP3 genes associated with increased RA risk [12-14], two novel risk variant loci in these two genes were for the first time identified to be implicated in the RA risk (group 4 and group 6 in Supplementary Table 3). A novel splicing variant (rs199982059) of TGFβ1 was found to be significantly enriched in 4 RA patients, but absent in healthy controls. TGFβ1 is a pivotal protein in the pathogenesis of a number of autoimmune disorders and its dysregulation is also increasingly implicated in the risk of developing RA [25-27]. RNA splicing is a focal point on connection between genetic variations and complex disorders [28, 29], and this novel splicing variant of TGFβ1 might provide new insights into the genetic determinants of RA disease. In addition, a novel missense variant (chrX:49114808) of FOXP3 was observed in 8 RA patients. FOXP3 is a unique regulatory T cell (Treg)-specific marker and important in the development of RA-derived Treg cells as a transcriptional factor [30, 31]. In spite of the other known variants in TGFβ1 and FOXP3 genes associated with RA, these two newly-identified variants in our Chinese RA patients may offer the novel genetic contributions to the RA risk.
We have also identified six novel and deleterious genes that are classified as pathogenic in ClinVar database (Supplementary Table 2). Of these, a missense variant (rs79681911) of SAA1, initially characterized by serum amyloid a variant (OMIM 104750) and required for the amyloidosis disease process, was identified in our RA patients. SAA1 has been reported to play a pathogenic role in the pro-inflammatory cascades in RA, therefore, this novel deleterious variant may be implicated in RA risk as a sensitive indicator of inflammatory activity [32]. Additional pathogenic variant (rs75134564) of OXCT1 was predicted to be disease-related in 4 RA patients based on LR score, which previously implicated in Succinyl-CoA acetoacetate transferase deficiency (OMIM 601424) in clinic. OXCT1 encoding enzyme SCOT1 is essential for ketone body metabolism and involved in cardiovascular disease, which are shown to be strongly associated with the course of RA [33-35], suggesting this enzyme may potentially contribute to RA prognosis. Importantly, the 3D structural analysis of these two mutants revealed that the substitution of mutation points may be involved in the functional alteration of the proteins and further impact on RA disease progression (Figure 2).
We sought to identify novel genes or biological candidate pathways fundamental to the risk of RA disease, including both rare and common variants. To elucidate additive effects of polygenic variants that affect the same gene or pathway, we performed gene burden test and pathway analysis. Notably, the biological impact of rare and deleterious variants is likely to be greater when present as two copies. In our study, 5 homozygous variants (group 5 in Figure 1; Supplementary Table 3) were detected in our RA patients but not in healthy controls. Intriguingly, a frameshift indel variant (rs61406813) of NCR3LG1 (natural cytotoxicity triggering receptor 3 ligand 1) was identified in our RA patients as homozygote. NCR3LG1 could be detected on monocytes and neutrophils after application of inflammatory stimuli [36], and it was initially described as a tumor cell–expressed ligand of NKp30, which is found to be implicated in RA-associated inflammation [37]. Additionally, a missense variant (rs61014678) of RAP1GAP (RAP1 GTPase Activating Protein) was identified as damaging (disease-related, D) by determinant of LR model in our RA patients. RAP1GAP regulates the activity of the ras-related RAP1 protein, which involves in induction of apoptotic pathway in synovial fibroblasts and plays a critical role in oxidative stress and T cell behavior in RA synovial tissues [38, 39]. Thus, these two homozygous variants may perform stronger functions in RA pathogenic mechanisms.
Our WES analysis totally identified 381 genes that may partially contribute to RA pathogenesis and disease progression, including 3 genes (TGFβ1, FOXP3 and SAA1) previously implicated in RA and 378 novel candidate genes. Biologic pathway analysis might help us to deeply understand RA pathogenesis, and previously biological pathways have been identified from genes in large-scale association analysis of GWAS data (Supplementary Table 6), such as autoimmune thyroid disease, natural killer cell mediated cytotoxicity and T cell receptor signaling pathways. We deciphered enrichment of our identified deleterious genes within additional pathways of ECM-receptor interaction, protein digestion and absorption, focal adhesion and glycerophospholipid metabolism based on our WES data (Table 1), which have been implicated in the autoimmune conditions or pathogenesis of RA [21-24]. We also sought to identify potential deleterious variants associated with disease duration among RA patients. Our pathway analysis focusing on variants enriched among RA patients with disease duration ≥3-year highlighted seven novel genes in olfactory transduction pathway (Table 1), which has been previously reported to be implicated in regulating inflammatory responses [40].
Pathogenesis of RA is complicated and includes both environmental and genetic factors. Recently, gut microbiota has been evident of being implicated in RA pathogenesis and treatment responses as a critical environmental factor that influences metabolic and immune homeostasis [41], involvement of protein digestion and absorption, glycerophospholipid metabolism and olfactory transduction pathway [42, 43], which were also enriched by novel candidate genes identified in our Chinese RA patients (Table 1). In addition, the homozygous variant NCR3LG1 (group 5 in Supplementary Table 3) may mediate autoimmune and microbial infection-induced inflammation by associating with the ligand of NKp30 [37]. Therefore, these involved novel deleterious genes might be convincingly considered genetic contributions to microbial alteration in relation to the pathogenesis and development of RA.
Genetic factors on the × chromosome always contribute to the increased risk of developing autoimmune disorders in females compared with males, such as RA [44]. Here, four novel and deleterious variants were investigated to be associated with sex bias in 58 Chinese RA patients, including OTC (Ornithine Transcarbamylase) (rs72554348), DIAPH2 (Diaphanous Related Formin 2) (rs363755), ARSE (Arylsulfatase E) (rs56393981) and FOXP3 (chrX:49114808) (Supplementary Table 7). Notably, OTC, ARSE and FOXP3 were previously reported to be implicated in x-linked diseases [45-49], in which these three novel variants identified in our study are also associated with female, supporting that the association of variants on × chromosome and RA may further provide molecular evidence as a risk factor contributing to increased susceptibility in Chinese female RA patients.
In summary, we have performed WES to present support and advance our understanding of associations with genetic variants that may be involved in the development of RA in the Chinese population. The variants highlighted include previously implicated genes as well as novel genes and pathways, involved in regulation of adaptive immune response, transmission of nerve impulse and chromosome organization. This study significantly extends the work of GWAS and provides new insight into fundamental etiologic mechanisms in this common autoimmune disease. While further experiments are required to validate our results and define the underlying biological mechanisms of these novel variants, these findings can serve as a starting point to elucidate the pathogenesis and potential impact on RA through genetics to functional insights.
MATERIALS AND METHODS
Patients
58 patients diagnosed as having RA were unrelated individuals of Han Chinese descent recruited from hospitals in Southern and Eastern China (Guangzhou and Changzhou) using 2010 Rheumatoid Arthritis Classification Criteria established by American College of Rheumatology and European League Against Rheumatism Collaborative Initiative (2010 ACR/EULAR) [50]. In addition, 66 healthy and unrelated blood donors of Han Chinese ancestry from Medical Center for Physical Examination and Health Assessment, were included as controls. Detailed descriptions of sequenced individuals and clinical characteristics of the enrolled patients are provided in Table 2 and 3. Written informed consent was obtained from all of the participants, and the study was registered in Chinese Clinical Trial Registry (ChiCTR-ROC-17010351) and approved by the local ethics committees of Macau University of Science and Technology (Macau, China).
Table 2: Description of the 124 sequenced individuals
|
Parameter |
Cases |
Controls |
|---|---|---|
|
N |
58 |
66 |
|
Sex |
43 female, 15 male |
41 female, 25 male |
|
Age |
48.48±14.08 |
35.23±10.73 |
Table 3: Demographic and clinical characteristics of the 58 patients with rheumatoid arthritis
|
Women/men, no. (%) |
43(25.9%)/ 15(74.1%) |
|
Age at diagnosis, mean±SD years |
|
|
All |
45.62±14.19 |
|
Women |
44.02±12.60 |
|
Men |
50.20±17.68 |
|
Disease duration (years), mean±SD years |
3.32±4.23 |
|
≤1-Year, no |
26 |
|
≥3-Year, no |
23 |
|
Rheumatoid factor +/−, no.(%) |
45 (83.3%) / 9 (16.7%); 4 Not Test |
|
No. of tender joints, mean±SD years |
6.66±7.37 |
|
No. of swollen joints, mean±SD years |
3.26±4.60 |
Ancestry composition analysis
We verified the population ethnicity information of the RA and healthy control samples by ancestry composition analysis (Supplementary Figure 2) using admixture v1.3.0 [51] (https://www.genetics.ucla.edu/software/admixture) and multidimensional scaling in PLINK v1.07 (http://zzz.bwh.harvard.edu//plink/) [52]. Three ethnic populations were used as reference samples from 1000 Genome Project Phase III data (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ALL.chr9.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz), including Utah Residents with Northern and Western European Ancestry (EUR-CEU), Yoruba in Ibadan, Nigeria (AFR-YRI) and Southern Han Chinese (EAS-CHS).
Generation of candidate gene list
A list of 159 candidate genetic variants reported by previous studies with the P value threshold of P < 1 × 10−5 (Supplementary Table 6) was prepared based on Rheumatoid Arthritis associated genes in the NHGRI GWAS Catalog [53] and literatures [6, 7, 54, 55].
Library preparation and WES
Blood samples were collected according to protocols approved by local institutional review boards. Genomic DNA was extracted from peripheral blood mononuclear cells (PBMCs) using PureLink® Genomic DNA Mini Kit (Invitrogen, USA) according to the manufacturer’s protocol. 500 ng of double-stranded DNA was determined by Qubit (Invitrogen, USA) and randomly fragmented to 150-200bp with Covaris cracker (Covaris, USA). Fragments with specific indexes were hybridized with probes. After PCR amplification and quality control, libraries were sequenced by next-generation sequencing. Agilent liquid phase hybridization was applied to efficiently enrich whole exons which would be sequenced on Illumina platform. Agilent SureSelect Human All ExonV5/V6 (Agilent Technologies, USA) with reagents were used for sequencing libraries and capture, which was recommended by the instruction manual and followed by optimized experimental procedures.
Sequencing was performed on an Illumina HiSeq × sequencer with a paired-end read length of 150bp in the Genomics Core Facility at Novogene (Genome Sequencing Company, Beijing, China). Data generated in this study will be submitted to the National Center for Biotechnology Information (NCBI) BioProject.
WES data analysis
To analyze the entire cohort of samples for genotype calls, variant analysis and joint genotyping were performed according to the pipeline recommended by the Genome Analysis Toolkit software and the GATK Best Practices procedures on RA patients and healthy controls [56-59]. Briefly, Burrows-Wheeler Aligner (BWA) [56] software is utilized to align the raw sequencing reads in FASTQ formats to the 1000 Genomes (GRCh37 + decoy) human genome reference. The BWA alignment files were converted to BAM files with SAMtools v1.1 [57], which is used for sorting the BAM files. Duplicate reads were marked for BAM files with Picard MarkDuplicates (https://sourceforge.net/projects/picard/). The coverage and depth were computed based on the final BAM file. Local realignment, base quality recalibration, variant calling, joint genotyping, and variant quality score recalibration and filtration were applied using with GATK v3.7 (https://software.broadinstitute.org/gatk/). Default settings were used for BWA, SAMtools, Picard and GATK tools.
Further filtration for the joint genotyped variants was performed using Variant Tools [58]. We applied the following filters to generate a list of preliminary variants by removing false-positive variants through Variant Quality Score Recalibration with tranche truth sensitivity threshold <9.9.00, as well as variants with low read depth (DP) <10 and poor genotyping quality (GQ) <20, keeping exonic or splicing variants based on University of California, Santa Cruz (UCSC) genome browser build 37 human Reference Sequence Gene annotation, and removing synonymous variants. From the preliminary variant list, variants annotated as “pathogenic” in ClinVar and deleterious variants were identified, respectively, including those candidate genes that overlapped with previous studies or passed the case-control gene burden test threshold. Deleterious variants were predicted to be damaging (disease-related, D) or benign/neutral (tolerated, T) based on LR score determined by logistic regression (LR) model [59]. The novel deleterious variants were divided into the rare and common variant groups, which were distinguished by minor allele frequency (MAF) in Chinese Southern population from the 1000 Genomes Project phase III study.
Analysis of burden association signal
Case–control gene burden analysis was assessed on both rare and common deleterious variants to investigate causal genes using RA patients with > 80% Chinese ancestry as cases and two types of controls: 105 southern Chinese samples from the 1000 Genomes Project phase III study and 66 healthy controls with >80% Chinese ancestry. Regardless of DP or GQ, all available genotype calls contributed to the number of allele count across the retained deleterious variants in each individual gene. The gene burden ratio was calculated by dividing the allele frequency in cases by the allele frequency in controls. We identified an enrichment of deleterious variants in a gene according to the gene burden ratio >1.5-fold with both types of controls, or the deleterious alleles in the gene with at least 3 RA cases if zero allele frequency in the controls. We further identified genes with rare variants that were homozygous in RA cases but not present in controls, which were considered greater contribution to functional impact.
Pathway analysis
To discover enriched functional-related gene groups, pathway analysis was performed using DAVID Bioinformatics Resource 6.8 program (DAVID 6.8) (https://david.ncifcrf.gov/summary.jsp) with a Modified Fisher Exact P value less than 0.05 as the significance threshold and strong enrichment in the annotation categories.
Structural analysis of proteins
Homology modeling is one of the best and reliable ways to construct the three dimensional (3D) structure of protein [60]. Firstly, protein sequence was imported into the Molecular Operating Environment (MOE) 2015.09 software (Chemical Computing Group Inc., Montreal, Canada) to search an optimal template. The top ranked structure based on the Z score towards the target sequence was selected as the template. Target protein sequence and its corresponding crystal structure coordinates of template were separately loaded and aligned. A series of protein models were independently constructed by using a Boltzmann-weighted randomized procedure [61]. Amber force field [62, 63] was applied in the process of construction and energy minimization. Finally, the model with the best packing quality function was selected for further full energy minimization, and the stereochemical qualities of protein model was assessed by means of Ramachandran plots.
To analyze the effect on the point mutation in the 3D structure of the protein, the mutant protein were carried out in Residue Scan module of MOE 2015.09 software based on the 3D structure of homology modeling. In addition, we further analyze the hydrogen bonds, solvent interactions, metal ligation and nonbonded interaction between the target mutant residue and its surrounding key amino acid residues.
Author contributions
All the authors contributed to the manuscript and involved in drafting the article or revising it critically for important intellectual content, and all authors contributed to study conception and design, acquisition of data or analysis and interpretation of data, and approved the final version to be published.
ACKNOWLEDGMENTS
We wish to thank the Novogene Genomics Core Facility for generating the exome sequencing data for this project. We would like to thank Prof. David C. Ward for his valuable comments and suggestion on the data as well as advice on writing the manuscript.
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
FUNDING
This work was supported by grant from Macao Science and Technology Development Fund (grant 077/2014/A2).
Ethics approval
Local ethical committees of Macau University of Science and Technology (Macau, China). This study was registered in Chinese Clinical Trial Registry (ChiCTR-ROC-17010351).
REFERENCES
1. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010; 376: 1094-108.
2. Turesson C, Jacobsson L, Bergström U. Extra-articular rheumatoid arthritis: prevalence and mortality. Rheumatology. 1999; 38: 668-74.
3. Oliver JE, Worthington J, Silman AJ. Genetic epidemiology of rheumatoid arthritis. Curr Opin Rheumatol. 2006; 18: 141-6.
4. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010; 42: 508-14.
5. Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, Todd JA, Donnelly P, Barrett JC, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007; 447: 661-78.
6. Freudenberg J, Lee HS, Han BG, Shin HD, Kang YM, Sung YK, Shim SC, Choi CB, Lee AT, Gregersen PK, Bae SC. Genome-wide association study of rheumatoid arthritis in Koreans: population-specific loci as well as overlap with European susceptibility loci. Arthritis Rheum. 2011; 63: 884-93.
7. Jiang L, Yin J, Ye L, Yang J, Hemani G, Liu AJ, Zou H, He D, Sun L, Zeng X, Li Z, Zheng Y, Lin Y, et al. Novel risk loci for rheumatoid arthritis in Han Chinese and congruence with risk variants in Europeans. Arthritis Rheumatol. 2014; 66: 1121-32.
8. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, et al. Finding the missing heritability of complex diseases. Nature. 2009; 461: 747-53.
9. Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010; 42: 30-5.
10. Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS, Stevens CR, Schubert CR, Collaboration AA, Greenberg ME, Gabriel SB, Walsh CA. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012; 8: e1002635.
11. Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012; 485: 237-41.
12. Zhou TB, Zhao HL, Fang SL, Drummen GP. Association of transforming growth factor-beta1 T869C, G915C, and C509T gene polymorphisms with rheumatoid arthritis risk. J Recept Signal Transduct Res. 2014; 34: 469-75.
13. Saad MN, Mabrouk MS, Eldeib AM, Shaker OG. Genetic case-control study for eight polymorphisms associated with rheumatoid arthritis. PLoS One. 2015; 10: e0131960.
14. Paradowska-Gorycka A, Jurkowska M, Felis-Giemza A, Romanowska-Prochnicka K, Manczak M, Maslinski S, Olesinska M. Genetic polymorphisms of Foxp3 in patients with rheumatoid arthritis. J Rheumatol. 2015; 42: 170-80.
15. Upragarin N, Landman WJ, Gaastra W, Gruys E. Extrahepatic production of acute phase serum amyloid A. Histol Histopathol. 2005; 20: 1295-307.
16. Carty CL, Heagerty P, Heckbert SR, Enquobahrie DA, Jarvik GP, Davis S, Tracy RP, Reiner AP. Association of genetic variation in serum amyloid-A with cardiovascular disease and interactions with IL6, IL1RN, IL1beta and TNF genes in the Cardiovascular Health Study. J Atheroscler Thromb. 2009; 16: 419-30.
17. Targonska-Stepniak B, Majdan M. Serum amyloid A as a marker of persistent inflammation and an indicator of cardiovascular and renal involvement in patients with rheumatoid arthritis. Mediators Inflamm. 2014; 2014: 793628.
18. Wentz AE, d’Avignon DA, Weber ML, Cotter DG, Doherty JM, Kerns R, Nagarajan R, Reddy N, Sambandam N, Crawford PA. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J Biol Chem. 2010; 285: 24447-56.
19. Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017; 25: 262-84.
20. Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, Warren L, Aponte J, Zawistowski M, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012; 337: 100-4.
21. Lv W, Wang Q, Chen H, Jiang Y, Zheng J, Shi M, Xu Y, Han J, Li C, Zhang R. Prioritization of rheumatoid arthritis risk subpathways based on global immune subpathway interaction network and random walk strategy. Mol Biosyst. 2015; 11: 2986-97.
22. Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis. 2013; 72: 110-7.
23. Choe JY, Hun Kim J, Park KY, Choi CH, Kim SK. Activation of dickkopf-1 and focal adhesion kinase pathway by tumour necrosis factor alpha induces enhanced migration of fibroblast-like synoviocytes in rheumatoid arthritis. Rheumatology (Oxford). 2016; 55: 928-38.
24. Yang Z, Matteson EL, Goronzy JJ, Weyand CM. T-cell metabolism in autoimmune disease. Arthritis Res Ther. 2015; 17: 29.
25. Padyukov L, Lampa J, Heimburger M, Ernestam S, Cederholm T, Lundkvist I, Andersson P, Hermansson Y, Harju A, Klareskog L, Bratt J. Genetic markers for the efficacy of tumour necrosis factor blocking therapy in rheumatoid arthritis. Ann Rheum Dis. 2003; 62: 526-9.
26. Mattey DL, Nixon N, Dawes PT, Kerr J. Association of polymorphism in the transforming growth factor {beta}1 gene with disease outcome and mortality in rheumatoid arthritis. Ann Rheum Dis. 2005; 64: 1190-4.
27. Sugiura Y, Niimi T, Sato S, Yoshinouchi T, Banno S, Naniwa T, Maeda H, Shimizu S, Ueda R. Transforming growth factor β1 gene polymorphism in rheumatoid arthritis. Annals of the rheumatic diseases. 2002; 61: 826-28.
28. Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, Morris Q, Barash Y, Krainer AR, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015; 347: 1254806.
29. Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK. RNA splicing is a primary link between genetic variation and disease. Science. 2016; 352: 600-04.
30. Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007; 446: 685-9.
31. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, Liu X, Xiao L, Chen X, Wan B, Chin YE, Zhang JZ. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med. 2013; 19: 322-8.
32. Connolly M, Mullan RH, McCormick J, Matthews C, Sullivan O, Kennedy A, FitzGerald O, Poole AR, Bresnihan B, Veale DJ, Fearon U. Acute-phase serum amyloid A regulates tumor necrosis factor alpha and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum. 2012; 64: 1035-45.
33. Fraser DA, Thoen J, Bondhus S, Haugen M, Reseland JE, Djoseland O, Forre O, Kjeldsen-Kragh J. Reduction in serum leptin and IGF-1 but preserved T-lymphocyte numbers and activation after a ketogenic diet in rheumatoid arthritis patients. Clin Exp Rheumatol. 2000; 18: 209-14.
34. Chodara AM, Wattiaux A, Bartels CM. Managing cardiovascular disease risk in rheumatoid arthritis: clinical updates and three strategic approaches. Curr Rheumatol Rep. 2017; 19: 16.
35. Pujades-Rodriguez M, Duyx B, Thomas SL, Stogiannis D, Rahman A, Smeeth L, Hemingway H. Rheumatoid arthritis and incidence of twelve initial presentations of cardiovascular disease: a population record-linkage cohort study in England. PLoS One. 2016; 11: e0151245.
36. Schlecker E, Fiegler N, Arnold A, Altevogt P, Rose-John S, Moldenhauer G, Sucker A, Paschen A, von Strandmann EP, Textor S, Cerwenka A. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res. 2014; 74: 3429-40.
37. Mulcahy H, O’Rourke KP, Adams C, Molloy MG, O’Gara F. LST1 and NCR3 expression in autoimmune inflammation and in response to IFN-gamma, LPS and microbial infection. Immunogenetics. 2006; 57: 893-903.
38. Remans PH, Gringhuis SI, van Laar JM, Sanders ME, Papendrecht-van der Voort EA, Zwartkruis FJ, Levarht EW, Rosas M, Coffer PJ, Breedveld FC, Bos JL, Tak PP, Verweij CL, et al. Rap1 signaling is required for suppression of ras-generated reactive oxygen species and protection against oxidative stress in T lymphocytes. J Immunol. 2004; 173: 920-31.
39. Remans PH, Wijbrandts CA, Sanders ME, Toes RE, Breedveld FC, Tak PP, van Laar JM, Reedquist KA. CTLA-4IG suppresses reactive oxygen species by preventing synovial adherent cell-induced inactivation of Rap1, a Ras family GTPASE mediator of oxidative stress in rheumatoid arthritis T cells. Arthritis Rheum. 2006; 54: 3135-43.
40. Al Salihi MO, Kobayashi M, Tamari K, Miyamura T, Takeuchi K. Tumor necrosis factor-alpha antagonist suppresses local inflammatory reaction and facilitates olfactory nerve recovery following injury. Auris Nasus Larynx. 2017; 44: 70-78.
41. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, Lan Z, Chen B, Li Y, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015; 21: 895-905.
42. Ferrebee CB, Dawson PA. Metabolic effects of intestinal absorption and enterohepatic cycling of bile acids. Acta Pharm Sin B. 2015; 5: 129-34.
43. Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan LX, Rey F, Wang T, Firestein SJ, Yanagisawa M, Gordon JI, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013; 110: 4410-5.
44. Khalifa O, Pers YM, Ferreira R, Senechal A, Jorgensen C, Apparailly F, Duroux-Richard I. X-linked miRNAs associated with gender differences in rheumatoid arthritis. Int J Mol Sci. 2016; 17.
45. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature Genet. 2001; 27: 20.
46. Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W, Liu Y, Wang Y, Liu X, Chan MW, Liu JQ, Love R, Liu CG, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell. 2007; 129: 1275-86.
47. Luksan O, Jirsa M, Eberova J, Minks J, Treslova H, Bouckova M, Storkanova G, Vlaskova H, Hrebicek M, Dvorakova L. Disruption of OTC promoter-enhancer interaction in a patient with symptoms of ornithine carbamoyltransferase deficiency. Hum Mutat. 2010; 31: E1294-303.
48. Brunetti-Pierri N, Andreucci MV, Tuzzi R, Vega GR, Gray G, McKeown C, Ballabio A, Andria G, Meroni G, Parenti G. X-linked recessive chondrodysplasia punctata: spectrum of arylsulfatase E gene mutations and expanded clinical variability. Am J Med Genet A. 2003; 117a: 164-8.
49. Jeon GW, Kwon MJ, Lee SJ, Sin JB, Ki CS. Clinical and genetic analysis of a Korean patient with X-linked chondrodysplasia punctata: identification of a novel splicing mutation in the ARSE gene. Ann Clin Lab Sci. 2013; 43: 70-5.
50. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2010; 69: 1580-8.
51. Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009; 19: 1655-64.
52. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81: 559-75.
53. Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014; 42: D1001-D06.
54. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014; 506: 376-81.
55. Diogo D, Kurreeman F, Stahl EA, Liao KP, Gupta N, Greenberg JD, Rivas MA, Hickey B, Flannick J, Thomson B. Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am J Hum Genet. 2013; 92: 15-27.
56. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009; 25: 1754-60.
57. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25: 2078-79.
58. San Lucas FA, Wang G, Scheet P, Peng B. Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics. 2012; 28: 421-2.
59. Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human molecular genetics. 2015; 24: 2125-37.
60. Yamaguchi H, Akitaya T, Yu T, Kidachi Y, Kamiie K, Noshita T, Umetsu H, Ryoyama K. Homology modeling and structural analysis of 11β-hydroxysteroid dehydrogenase type 2. Eur J Med Chem. 2011; 46: 1325-30.
61. Levitt M. Accurate modeling of protein conformation by automatic segment matching. J Mol Biol. 1992; 226: 507-33.
62. Gerber PR, Müller K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J Comput Aided Mol Des. 1995; 9: 251-68.
63. Case D, Darden T, Cheatham T III, Simmerling C, Wang J, Duke R, Luo R, Walker R, Zhang W, Merz K. AMBER 12 Reference Manual; University of California: San Francisco, CA, 2012.