
INTRODUCTION
Fabry disease (FD, OMIM #301500) is a rare X-linked recessive disorder characterized by the absence or reduced activity of α-galactosidase A (α-GalA). This enzyme deficiency leads to deposition of globotriaosylceramide (Gb3) in body fluids and in the vascular endothelium of many organs [1]. The initial signs and symptoms appear in childhood or adolescence and include angiokeratoma, acroparasthesia, corneal opacities, hypohidrosis and gastrointestinal symptoms [2–5].
Vascular dysfunction is the main manifestation of later disease progression observed in FD patients, who typically manifest abnormalities of renal function, cardiac defects and cerebrovascular complications, resulting in early demise, typically in the fourth or fifth decade of life [6]. Enzyme replacement therapy (ERT) with recombinant α-GalA has been widely used for the treatment of FD patients. Clinical trials using agalsidase alfa (Replagal® Shire HGT) and agalsidase beta (Fabrazyme® Genzyme Corp) have shown that ERT is safe and well tolerated and is able to remove Gb3 inclusions from smooth muscle, epithelial cells, myocardium and kidney [7–12].
However, it has become evident that the removal of stored Gb3 from endothelial cells does not prevent progression of vascular disease in all patients [13], specifically in advanced stage with renal impairment, suggesting a limited success in treatment. Involvement of modulators in the vascular pathophysiology of FD, unrelated to α-GalA and Gb3 accumulation, such as genetic and environmental factors, has been largely investigated. Several studies reported the association of single nucleotide polymorphisms (SNPs) or mutations in inflammatory and coagulation factor genes, such as interleukin 6 (c.−174G>C), endothelial nitric oxide synthase (p.Glu298Asp), the factor V (p.Arg506Gln), and the gene encoding the vitamin-K-dependent protein Z (c.−13A>G, IVS6 + 79G>A), with an increased risk of cerebral lesions and stroke in patients with FD [14–16]. In the present study, we investigated whether, in addition to α-GalA, genetic variants in genes encoding drug absorption, distribution, metabolism, and excretion (ADME) proteins exert some effect on response variability to ERT in a group of 37 FD patients. We compared the genetic profiling of 1936 variants across 231 genes in 28 responders versus 9 non-responders using the Affymetrix Drug Metabolizing Enzyme and Transporters (DMET) Plus platform.
RESULTS
Response to ERT
According to Mains Severity Score Index (MSSI) twenty-eight patients were classified as responders and nine as non-responders. At baseline total MSSI score in responders ranged from 3 to 45 (median 24). Twenty-one, six and one patients showed mild, moderate and severe involvement respectively. In non-responders total MSSI score ranged from 14 to 36 (median 25). Six non-responders showed mild while three moderate involvement. Figure 1 shows significant changes in total MSSI score between responders and non-responders after at least 1 years of ERT (median duration of treatment 5 years, range 1–10 years) (p = 0.0003). Five responders showed an improvement in total MSSI score (median change −1.8, ranged from −1 to −3), twelve worsened (median change +2.8, ranged from +1 to +8), and ten remained stable. In non-responders the total MSSI score increased with a median change of 9.5, ranged from +4 to +27. When we evaluated MSSI score for single clinical parameters (general, cardiovascular, neurological and renal), we observed a significant difference between the two groups in cardiovascular baseline MSSI score (p = 0.001), also after Bonferroni correction (p = 0.006). At follow-up non-responders showed an increase in MSSI score for general (p = 0.002) and renal (p = 0.004) parameters in addition to cardiovascular (p = 0.0004) (Figure 2). No correlation has been observed between response to ERT and age, sex and treatment duration.
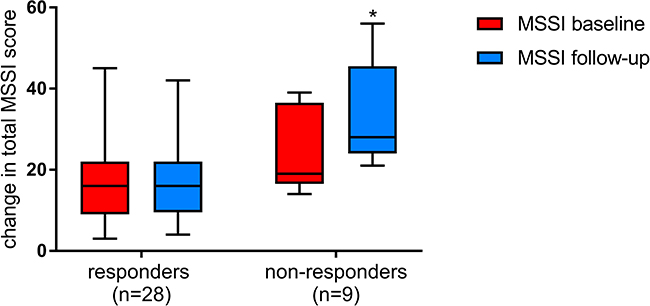
Figure 1: Changes in total MSSI scores among responders and non-responders at baseline (red) and after at least 1 years of ERT (blue). The box plots show, median (rule), interquartile range (box), and minimum and maximum values (whiskers) (GraphPad Prism v.7). *p ≤ 0.05.
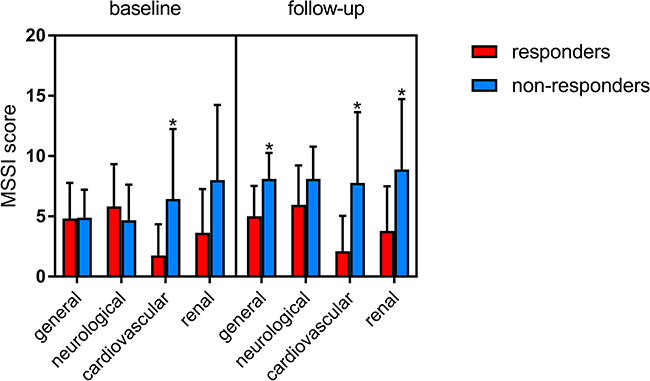
Figure 2: Changes of clinical parameters included in the MSSI score (general, neurological, cardiovascular, renal) among responders and non-responders at baseline and after at least 1 years of ERT (GraphPad Prism v.7). *p ≤ 0.05.
Genetic and statistical results
All 37 patients passed QC metrics and produced useable genotypes with an average call rate >95%. Among the 1936 SNPs included in the DMET assay, we used genotyping data from 993 polymorphic SNPs for statistical analysis. The rs953062 in CYP39A1 failed to meet Hardy-Weinberg equilibrium (HWE) and thus was excluded from further analysis. Three SNPs in ADH4 gene (rs1126670, rs1126671, rs2032349) and one in ADH5 (rs2602836) resulted significantly associated with response to ERT (Table 1). The heterozygous genotypes GT (rs1126670), AG (rs1126671) and CT (rs2602836) resulted more frequent in non-responders compared with responders, while the homozygous genotypes CC (rs2032349 and rs2602836) were more frequent in responders. All related genotypes were confirmed with TaqMan SNP genotyping assays.
Table 1: Genotypic distribution of SNPs in ADH4 and ADH5 among responders and non-responders
SNP IDa |
Gene |
Chr |
Alleles |
Genotype(no.) |
P-value |
OR (95% C.I.) |
|
|---|---|---|---|---|---|---|---|
Responders (N = 28) |
Non-responders (N = 9) |
||||||
rs2602836 |
ADH5 |
4 |
C/T* |
CC=15 |
CC=0 |
0.0052 |
0.046 (0.0024 to 0.8636) |
CT=9 |
CT=8 |
0.0052 |
16.89 (1.8251 to 156.2884) |
||||
TT=4 |
TT=1 |
||||||
rs1126670 |
ADH4 |
4 |
G/T* |
GG=3 |
GG=0 |
0.0002 |
45.82 (2.3885 to 879.1319 |
GT=8 |
GT=9 |
0.0015 |
0.035 (0.0018 to 0.6540) |
||||
TT=17 |
TT=0 |
||||||
rs1126671 |
ADH4 |
4 |
A/G* |
AA=4 |
AA=0 |
0.0001 |
54.47 (2.8144 to 1054.0945) |
AG=7 |
AG=9 |
0.0015 |
0.035 (0.0018 to 0.6540) |
||||
GG=17 |
GG=0 |
||||||
rs2032349 |
ADH4 |
4 |
C/T* |
CC=27 |
CC=6 |
||
CT=1 |
CT=3 |
0.0375 |
0.074 (0.0065 to 0.8414) |
||||
TT=0 |
TT=0 |
||||||
SNPs were tested for association using Fisher’s exact test. SNPs and genes indicated were limited to those in which p < 0.05. SNP ID* is the SNP identifier based on NCBI dbSNP. *genotypes reported as reverse strand.
DISCUSSION
ERT has been widely used for the treatment of FD. However, it is still unclear why some patients under ERT progress to renal, cardiovascular and cerebrovascular dysfunctions. Growing evidence is supporting the notion that Gb3 acts as a second messenger inducing oxidative stress and inflammation in FD vasculopathy [17–19]. In this context, Biancini et al. [20] found decreased levels of antioxidant defenses in FD patients while compared to controls: reduced glutathione and glutathione peroxidase activity and increased superoxide dismutase/catalase ratio in erythrocytes. Also, authors reported increased plasma levels of malondialdehyde and protein carbonyl groups in FD patients, as a consequence of higher lipid peroxidation and protein damage, compared to controls. However, the underlined molecular mechanisms that lead to cell and tissue damage in FD vasculopathy can be only partially explained by Gb3 accumulation, as demonstrating by lack of ERT responsiveness in FD patients despite Gb3 removal.
In this study, we investigated the impact of ADME gene variants on treatment failure in a cohort of 37 FD patients under ERT. We found three SNPs in ADH4 gene (rs1126670, rs1126671, rs2032349) and one in ADH5 gene (rs2602836) associated with disease progression. Both genes belong to the human alcohol dehydrogenase (ADH) family clustered on chromosome 4q22-23 [21]. Active ADH enzyme is formed by a dimerization interaction between two of nine possible different subunits, each encoded by a unique gene. The ADH4 gene encodes the human π subunit and contributes to the metabolization of a wide variety of substrates, including ethanol, retinol, other aliphatic alcohols, hydroxysteroids, and lipid peroxidation products. This gene is expressed primarily in the liver and at lower levels in the gastrointestinal tract and spleen. The ADH5 gene encodes the χ subunit and is involved in the metabolism of alcohols and aldehydes [22]. Unlike other members of the ADH family, ADH5 is ubiquitously expressed. The ADH4 rs1126671 at exon 7 results in the amino acid substitutions Val-Ile and might affects the function of the π subunit. The rs1126670 at exon 6 and the rs2032349 at exon 3 are synonymous changes, respectively of Pro-Pro and Ser-Ser. The rs2602836 in ADH5 gene is located at intergenic level and could be important for gene expression level. In previous study, ADH variants have been implicated in the risk for alcohol and drug dependence [23], schizophrenia and autism [24], cancer [25, 26]. ADH genes are also involved in the metabolism of 4-hydroxynonenal (4-HNE) to produce alcohol 1,4-dihydroxy-2-nonene (DHN). 4-HNE is an aldehyde which can be formed as secondary product during lipid peroxidation and its levels increase significantly in plasma and tissues in disease associated with oxidative stress, such as atherosclerosis and diabetes, and neurological disorders [27]. Studies have found abundance of 4-HNE in the vascular endothelial and smooth muscle cells. Levels of 4-HNE in vasculature are not only dependent on the rate of lipid peroxidation and 4-HNE synthesis, but also on the removal of 4-HNE adducts by phase II metabolic pathways. Because at high level 4-HNE can react with protein and DNA to form adducts, with consequent toxicity, it is evident that mutations and/or functional SNPs in genes involved in 4-HNE metabolism, such as ADH genes, could reduce the rapid intracellular metabolism of this compound and could be crucial for cell survival in a compromised oxidative stress system. ADH4 is highly active in the reduction of 4-HNE, supporting a defense role for cells [28].
We suggest that SNPs in ADH4/ADH5 genes, observed in this study, could be considered a risk factor, linked to oxidative stress state, for disease progression in FD patients despite enzyme replacement therapy. However, for the absence of functional analysis and the small size of population our study should be considered a hypothesis generating study. Validation of our results has been planned in a larger and independent cohort to evaluate the correlation between 4-HNE plasmatic levels, ADH4/ADH5 genotypes and MSSI score index. In conclusion, our results provide a basic tool and framework in the light of personalized medicine for FD.
MATERIALS AND METHODS
Patients
From January 2004 to May 2014, 37 patients with a clinical and molecular diagnosis of FD have been regularly monitored at the Pediatric Unit of Magna Graecia University of Catanzaro and at the Department of Nephrology of Federico II University of Naples. Of these, 33 patients received infusions of agalsidase alfa 0.2 mg/kg every other week, the remaining 4 were treated with agalsidase beta every 2 weeks at a dose of 1 mg/ kg. ERT with algasidase alfa was initiated 1–6 years (median:2.3) after diagnosis (median age 35.7 years, range 15–62 years), while ERT with algasidase beta 1–4 years (median: 3.2 years) after diagnosis (median age 41.7 years, range 25–57 years).
Clinical characteristics of patients are summarized in Table 2. Study protocols were approved by institutional ethics committee and written informed consent was obtained from all participants.
Table 2: Patient characteristics
Total |
Male |
Female |
|
|---|---|---|---|
Total patients (no.) |
37 |
17 |
20 |
Age (y; median SD) |
40.5 ± 13.3 |
40.1 ± 16.8 |
40.9 ± 7.6 |
ERT |
|||
with Agalsidase alfa |
33 |
17 |
16 |
with Agalsidase beta |
4 |
0 |
4 |
The characteristics of 37 patients with a clinical and molecular diagnosis of FD enrolled in the study are indicated. Patients have been monitored regularly at the Pediatric Unit of Magna Graecia University of Catanzaro and at the Department of Nephrology of Federico II University of Naples and received ERT with algasidase alfa or beta.
Treatment outcome
Disease progression and treatment effects in individual patients were assessed longitudinally as changes from baseline using MSSI [29]. The MSSI scoring system consists of four sections that include general, neurological, cardiovascular and renal signs and symptoms [30]. For each component, a single rating was assigned, and the corresponding points were summed to produce a total score. Individual scores were then combined to calculate the total score MSSI. MSSI was evaluated as mild for values between 0–20, moderate for values between 20–40, and severe for values higher than 40. Baseline MSSI values were obtained before treatment, follow up values after at least 1 year of ERT. Patients were classified as non-responders if MSSI values changes from mild (at baseline) to moderate or from moderate to severe.
Methods
Genomic DNA was extracted from peripheral blood using Perfect Pure DNA Blood kit (5 Prime) and analyzed using the DMET Plus assay (Affymetrix, Santa Clara, CA), as previously described [31–35]. DMET Console version 1.1 (Affymetrix, Santa Clara, CA) was used to perform genotype calls from intensity array data using the Dynamic Genotype Boundaries algorithm. We applied a call rate less than 95% as exclusion criteria from further analysis. Association analysis was performed using DMET-Analyzer Tool software [36]. The observed genotype frequencies for each SNP were tested for HWE in both groups, responders and non-responders, using χ2 test. Odds ratios (ORs) and corresponding 95% confidence intervals (CIs) were calculated for 2 × 2 table using Med Calc v12.3.0. Unpaired t-test and Bonferroni correction were done in GraphPad Prism 7 (GraphPad Software, Inc).
All genotypes of interest were validated using pre-designed TaqMan SNP genotyping assays (Assay ID: C_519458_40, C_11941799_30, C_11941805_40 and C_9523470_10, Applied Biosystem). PCR amplification and endpoint plate read were carried out on a ViiA7TM Real-Time PCR System (Applied Biosystem). All reactions were performed in duplicate in a final volume of 10 μL accordingly to the manufacturer’s recommendations. Mismatched genotypes, which constituted < 0.5% of the total number of duplicate genotypes performed, were discarded.
Abbreviations
ADME: Adsorption, distribution, metabolizing enzyme; α-GalA: α-galactosidase A; DMET: Drug metabolizing enzyme and transporters; ERT: Enzyme replacement therapy; FD: Fabry disease; Gb3: Globotriaosylceramide; 4-HNE: 4-hydroxynonenal ; HWE: Hardy–Weinberg equilibrium; PCR: Polymerase chain reaction (PCR); SNP: Single nucleotide polymorphism.
Author contributions
F.S. and M.T.D. designed and executed the experiments, F.S., M.T.D and L.P. wrote the manuscript, L.P. and D.C. read and approved the manuscript, S.S., A.N., F.F., K.R., A.P. and E.R. collected patients samples and information, M.A. and P.H.G. supported DMET data analysis
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Desnick RJ, Wasserstein MP, Banikazemi M. Fabry disease (alpha-galactosidase A deficiency): renal involvement and enzyme replacement therapy. Contrib Nephrol. 2001; 136:174–92.
2. Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008; 372:1427–35.
3. Pensabene L, Sestito S, Nicoletti A, Graziano F, Strisciuglio P, Concolino D. Gastrointestinal Symptoms of Patients with Fabry Disease. Gastroenterol Res Pract. 2016; 2016:9712831.
4. Rigoldi M, Concolino D, Morrone A, Pieruzzi F, Ravaglia R, Furlan F, Santus F, Strisciuglio P, Torti G, Parini R. Intrafamilial phenotypic variability in four families with Anderson-Fabry disease. Clin Genet. 2014; 86:258–63.
5. Sestito S, Ceravolo F, Concolino D. Anderson-Fabry disease in children. Curr Pharm Des. 2013; 19:6037–45.
6. Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, Sorensen SA, Wilcox WR, Desnick RJ. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009; 24:2102–11.
7. Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, Finkel R, Packman S, Bichet DG, Warnock DG, Desnick RJ, Fabry Disease Clinical Trial Study Group. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007; 146:77–86.
8. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ, International Collaborative Fabry Disease Study Group. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001; 345:9–16.
9. Kim JH, Lee BH, Hyang Cho J, Kang E, Choi JH, Kim GH, Yoo HW. Long-term enzyme replacement therapy for Fabry disease: efficacy and unmet needs in cardiac and renal outcomes. J Hum Genet. 2016; 61:923–9.
10. Mehta A, Beck M, Elliott P, Giugliani R, Linhart A, Sunder-Plassmann G, Schiffmann R, Barbey F, Ries M, Clarke JT, Fabry Outcome Survey Investigators. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009; 374:1986–96.
11. Ortiz A, Abiose A, Bichet DG, Cabrera G, Charrow J, Germain DP, Hopkin RJ, Jovanovic A, Linhart A, Maruti SS, Mauer M, Oliveira JP, Patel MR, et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase beta: data from the Fabry Registry. J Med Genet. 2016; 53:495–502.
12. Concolino D, Amico L, Cappellini MD, Cassinerio E, Conti M, Donati MA, Falvo F, Fiumara A, Maccarone M, Manna R, Matucci A, Musumeci MB, Nicoletti A, et al. Home infusion program with enzyme replacement therapy for Fabry disease: The experience of a large Italian collaborative group. Mol Genet Metab Rep. 2017; 12:85–91.
13. Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, Desnick RJ, Germain DP, International Fabry Disease Study Group. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004; 75:65–74.
14. Altarescu G, Chicco G, Whybra C, Delgado-Sanchez S, Sharon N, Beck M, Elstein D. Correlation between interleukin-6 promoter and C-reactive protein (CRP) polymorphisms and CRP levels with the Mainz Severity Score Index for Fabry disease. J Inherit Metab Dis. 2008; 31:117–23.
15. Altarescu G, Moore DF, Schiffmann R. Effect of genetic modifiers on cerebral lesions in Fabry disease. Neurology. 2005; 64:2148–50.
16. Heltianu C, Costache G, Azibi K, Poenaru L, Simionescu M. Endothelial nitric oxide synthase gene polymorphisms in Fabry’s disease. Clin Genet. 2002; 61:423–9.
17. De Francesco PN, Mucci JM, Ceci R, Fossati CA, Rozenfeld PA. Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide. Mol Genet Metab. 2013; 109:93–9.
18. Shen JS, Meng XL, Moore DF, Quirk JM, Shayman JA, Schiffmann R, Kaneski CR. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol Genet Metab. 2008; 95:163–8.
19. Simoncini C, Chico L, Concolino D, Sestito S, Fancellu L, Boadu W, Sechi GP, Feliciani C, Gnarra M, Zampetti A, Salviati A, Scarpelli M, Orsucci D, et al. Mitochondrial DNA haplogroups may influence Fabry disease phenotype. Neurosci Lett. 2016; 629:58–61.
20. Biancini GB, Vanzin CS, Rodrigues DB, Deon M, Ribas GS, Barschak AG, Manfredini V, Netto CB, Jardim LB, Giugliani R, Vargas CR. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim Biophys Acta. 2012; 1822:226–32.
21. Duester G, Hatfield GW, Smith M. Molecular genetic analysis of human alcohol dehydrogenase. Alcohol. 1985; 2:53–6.
22. Edenberg HJ. Regulation of the mammalian alcohol dehydrogenase genes. Prog Nucleic Acid Res Mol Biol. 2000; 64:295–341.
23. Birley AJ, James MR, Dickson PA, Montgomery GW, Heath AC, Martin NG, Whitfield JB. ADH single nucleotide polymorphism associations with alcohol metabolism in vivo. Hum Mol Genet. 2009; 18:1533–42.
24. Zuo L, Wang K, Zhang XY, Pan X, Wang G, Tan Y, Zhong C, Krystal JH, State M, Zhang H, Luo X. Association between common alcohol dehydrogenase gene (ADH) variants and schizophrenia and autism. Hum Genet. 2013; 132:735–43.
25. Goode EL, White KL, Vierkant RA, Phelan CM, Cunningham JM, Schildkraut JM, Berchuck A, Larson MC, Fridley BL, Olson JE, Webb PM, Chen X, Beesley J, et al, and Ovarian Cancer Association Consortium, and Australian Ovarian Cancer Study Group. Xenobiotic-Metabolizing gene polymorphisms and ovarian cancer risk. Mol Carcinog. 2011; 50:397–402.
26. Wei RR, Zhang MY, Rao HL, Pu HY, Zhang HZ, Wang HY. Identification of ADH4 as a novel and potential prognostic marker in hepatocellular carcinoma. Med Oncol. 2012; 29:2737–43.
27. Dalleau S, Baradat M, Gueraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013; 20:1615–30.
28. Siems W, Grune T. Intracellular metabolism of 4-hydroxynonenal. Mol Aspects Med. 2003; 24:167–75.
29. Parini R, Rigoldi M, Santus F, Furlan F, De Lorenzo P, Valsecchi G, Concolino D, Strisciuglio P, Feriozzi S, Di Vito R, Ravaglia R, Ricci R, Morrone A. Enzyme replacement therapy with agalsidase alfa in a cohort of Italian patients with Anderson-Fabry disease: testing the effects with the Mainz Severity Score Index. Clin Genet. 2008; 74:260–6.
30. Whybra C, Kampmann C, Krummenauer F, Ries M, Mengel E, Miebach E, Baehner F, Kim K, Bajbouj M, Schwarting A, Gal A, Beck M. The Mainz Severity Score Index: a new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin Genet. 2004; 65:299–307.
31. Arbitrio M, Di Martino MT, Barbieri V, Agapito G, Guzzi PH, Botta C, Iuliano E, Scionti F, Altomare E, Codispoti S, Conforti S, Cannataro M, Tassone P, et al. Identification of polymorphic variants associated with erlotinib-related skin toxicity in advanced non-small cell lung cancer patients by DMET microarray analysis. Cancer Chemother Pharmacol. 2016; 77:205–9.
32. Di Martino MT, Arbitrio M, Guzzi PH, Leone E, Baudi F, Piro E, Prantera T, Cucinotto I, Calimeri T, Rossi M, Veltri P, Cannataro M, Tagliaferri P, et al. A peroxisome proliferator-activated receptor gamma (PPARG) polymorphism is associated with zoledronic acid-related osteonecrosis of the jaw in multiple myeloma patients: analysis by DMET microarray profiling. Br J Haematol. 2011; 154:529–33.
33. Di Martino MT, Arbitrio M, Leone E, Guzzi PH, Rotundo MS, Ciliberto D, Tomaino V, Fabiani F, Talarico D, Sperlongano P, Doldo P, Cannataro M, Caraglia M, et al. Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: a DMET microarray profiling study. Cancer Biol Ther. 2011; 12:780–7.
34. Di Martino MT, Scionti F, Sestito S, Nicoletti A, Arbitrio M, Hiram Guzzi P, Talarico V, Altomare F, Sanseviero MT, Agapito G, Pisani A, Riccio E, Borrelli O, et al. Genetic variants associated with gastrointestinal symptoms in Fabry disease. Oncotarget. 2016; 7:85895–85904. https://doi.org/10.18632/oncotarget.13135.
35. Dumaual C, Miao X, Daly TM, Bruckner C, Njau R, Fu DJ, Close-Kirkwood S, Bauer N, Watanabe N, Hardenbol P, Hockett RD. Comprehensive assessment of metabolic enzyme and transporter genes using the Affymetrix Targeted Genotyping System. Pharmacogenomics. 2007; 8:293–305.
36. Guzzi PH, Agapito G, Di Martino MT, Arbitrio M, Tassone P, Tagliaferri P, Cannataro M. DMET-analyzer: automatic analysis of Affymetrix DMET data. BMC Bioinformatics. 2012; 13:258.