
INTRODUCTION
Gynecologic cancers, including cervical, endometrial, and ovarian cancer, constitute one of the main causes of death from cancer worldwide [1]. Patients with gynecologic cancers are often treated with combination platinum and taxane-based chemotherapy, but many patients develop recurrent disease with poor survival as a result of acquired or intrinsic drug resistance [2–4]. In particular, Cisplatin resistance is a critical issue that must be overcome when considering the treatment of gynecologic cancers, as well as several other cancers that display Cisplatin resistance.
Statins are well-known agents that inhibit HMGCR and are commonly used to prevent hypercholesterolemia [5]. Recently, statins have received increasing attention because of emerging evidence of their antitumor effects [6, 7]. Furthermore, the importance of MVA pathway metabolites and enzymes in the survival of cancer cells has been highlighted by the latest studies [8, 9].
KLFs are a subfamily of the zinc-finger class of DNA-binding transcriptional regulators and are involved in the regulation of differentiation, development, cellular proliferation, growth-related signal transduction, and apoptosis [10, 11]. It was reported that KLF2 and KLF6 act as tumor suppressor genes, and downregulation of KLF2 or KLF6 was associated with poor survival in several cancers [12–17]. RHOB is one of the Rho family of small GTPases, signaling molecules that regulate many cellular processes including cytoskeletal dynamics, cell motility, cell adhesion, cell division, and transcription [18]. The Rho GTPases thereby contribute to wound healing, inflammation, and cancer progression [18]. RHOB is also known as a tumor suppressor that promotes growth inhibition and induces apoptosis in cancer cells [19, 20].
In this study we found that statins preferentially led to viability reduction of Cisplatin-resistant cells compared with Cisplatin-sensitive cells, and that expression of KLF2, KLF6, and RHOB was induced in response to Lovastatin. We investigated the involvement of these tumor suppressor genes and MVA pathway-associated genes in Cisplatin resistance.
RESULTS
Lovastatin sensitized Cisplatin-resistant cells
We evaluated the effects of Cisplatin and Lovastatin on cell viability of Cisplatin-resistant HCP4, PCDP5 cells and parental Hela, PC3 cells, respectively, by cell proliferation assay. The IC50 of Cisplatin and statins for Hela, HCP4, PC3 and PCDP5 cells were calculated with CalcuSyn software. HCP4 and PCDP5 cells were 37-fold and 18-fold more resistant to Cisplatin than their parental cells, respectively (Figure 1 and Table 1). In contrast, HCP4 and PCDP5 cells were 13-fold and 7-fold more sensitive to Lovastatin than their parental cells, respectively (Figure 1 and Table 2). HCP4 and PCDP5 cells were also more sensitive than their parental cells to other statin-related agents, including Simvastatin, Pravastatin, Compactin, Fluvastatin, Atorvastatin, Pitavastatin, and Pravastatin (Figure 1 and Table 2). We also evaluated the effects of Lovastatin on Cisplatin-resistant DDP10 cells, oxaliplatin-resistant OX2 cells and Mithramycin-resistant MM4 cells derived from T24 cells (Supplementary Table 1). DDP10, OX2 and MM4 cells were 7.1-fold, 15.6-fold and >270-fold more resistant to Cisplatin, Oxaliplatin and Mithramycin, respectively, when compared with parental T24 cells. DDP10 and OX2 cells were 1.3-fold and 2.2-fold more sensitive to Lovastatin, respectively, while MM4 cells were not sensitive to this compound.
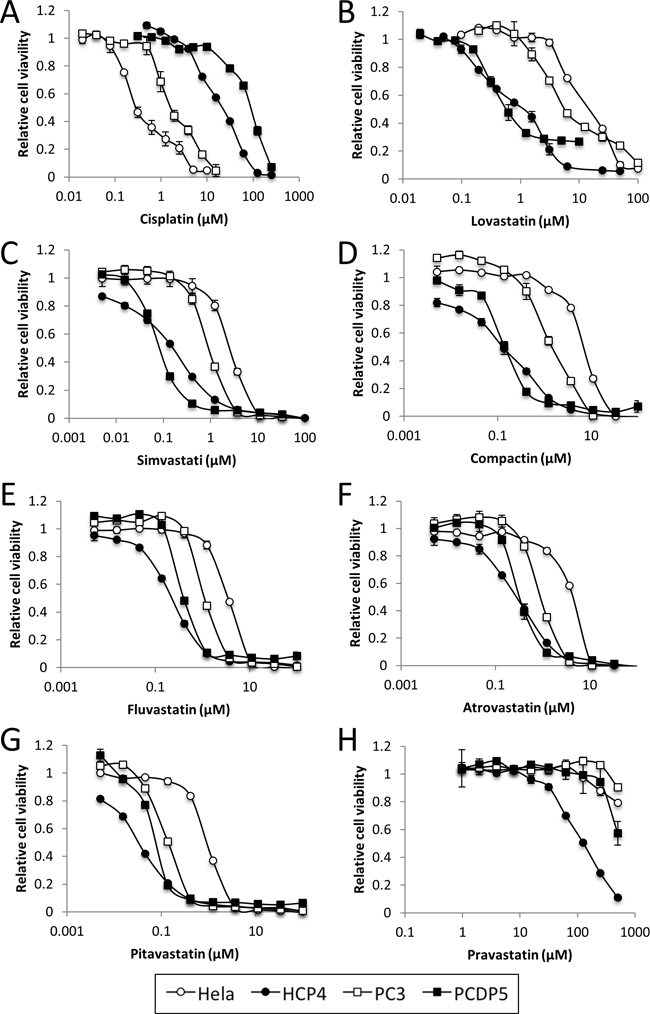
Figure 1: Statins sensitized Cisplatin-resistant cells. Hela, HCP4, PC3 and PCDP5 cells were treated with serial dilutions of Cisplatin or seven kinds of statin. After 72 h, the surviving cells were stained with TetraColor ONE for 2–3 h. All values represent the mean of at least two independent experiments.
Table 1: Evaluation of IC50
Hela |
HCP4 |
Ratio (HCP4/Hela) |
PC3 |
PCDP5 |
Ratio (PCDP5/PC3) |
|
|---|---|---|---|---|---|---|
Cisplatin |
0.530 |
19.564 |
36.884 |
2.877 |
53.139 |
18.468 |
(μM) |
±0.042 |
±1.271 |
±0.209 |
±0.673 |
Table 2: Evaluation of IC50
Hela |
HCP4 |
Ratio (Hela/HCP4) |
PC3 |
PCDP5 |
Ratio (PC3/PCDP5) |
|
|---|---|---|---|---|---|---|
Lovastatin |
12.499 |
0.927 |
13.48 |
10.719 |
1.508 |
7.11 |
(μM) |
±0.859 |
±0.019 |
±0.131 |
±0.078 |
||
Simvastatin |
1.961 |
0.111 |
17.67 |
1.052 |
0.216 |
4.87 |
(μM) |
±0.958 |
±0.007 |
±0.147 |
±0.098 |
||
Compactin |
5.671 |
0.083 |
68.33 |
1.382 |
0.291 |
4.75 |
(μM) |
±1.589 |
±0.012 |
±0.136 |
±0.046 |
||
Fluvastatin |
2.486 |
0.245 |
10.15 |
2.450 |
0.878 |
2.79 |
(μM) |
±0.943 |
±0.056 |
±1.120 |
±1.227 |
||
Atorvastatin |
1.815 |
0.188 |
9.65 |
0.916 |
0.593 |
1.54 |
(μM) |
±0.069 |
±0.021 |
±0.046 |
±0.201 |
||
Pitavastatin |
0.662 |
0.028 |
23.64 |
0.106 |
0.104 |
1.02 |
(μM) |
±0.065 |
±0.001 |
±0.008 |
±0.032 |
||
Pravastatin |
>500 |
128.637 |
N.C. |
>500 |
>500 |
N.C. |
(μM) |
±12.731 |
N.C. indicates “not calculated”
Lovastatin induced apoptosis of Cisplatin-resistant cells
To determine the mechanism of viability reduction of Lovastatin for HCP4 cells we performed cell cycle analysis after treatment with Lovastatin. We used Lovastatin at 1 μM and 10 μM concentrations because 10 μM Lovastatin strongly reduce the viability of both Hela and HCP4 cells (by approximately 70% and 5%, respectively), while 1 μM Lovastatin could reduce the viability of HCP4 cells (by approximately 50%) but could not of Hela cells (Figure 1). As shown in Figure 2A and 2B, 10 μM Lovastatin significantly increased the sub-G1 population of HCP4 cells, but not Hela cells, after 48 h. These results suggested that 10 μM Lovastatin preferencally induced apoptosis of HCP4 cells. Next, we performed western blot analysis to examine the expression of the apoptosis-related proteins cleaved caspase 3, cleaved caspase 9, and PARP-1. As shown in Figure 2C, cleaved caspase 3, 9 and PARP-1 proteins were strongly induced by Lovastatin in a time-dependent manner in HCP4 cells. These proteins were also induced by Lovastatin in Hela cells, but the expression levels were low compared with HCP4 cells. These results indicated that Lovastatin induced apoptosis in Cisplatin-resistant HCP4 cells more rapidly and to a greater extent than in the parental Hela cells.
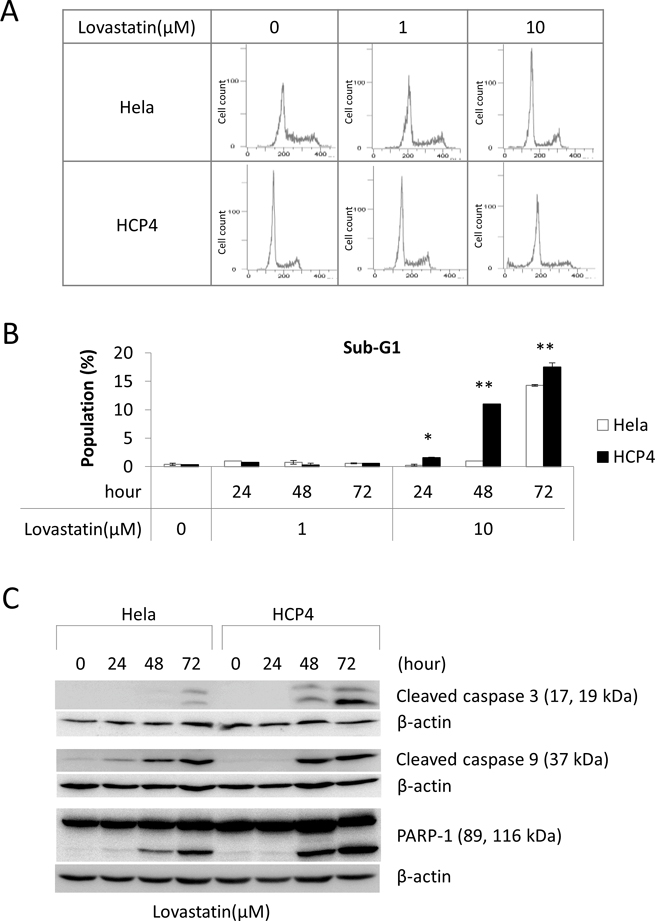
Figure 2: Lovastatin induced apoptosis of Cisplatin-resistant cells. (A) Hela and HCP4 cells were treated with the indicated concentration of Lovastatin for 48 h. Cell cycle analysis was performed by flow cytometry. (B) The sub-G1 population was calculated form the results of (A). All values represent the mean of at least three independent experiments. Significant differences are compared for each cell line under the same condition; * and ** indicate P < 0.05 and P < 0.01, respectively. (C) Hela and HCP4 cells were treated with 1 μM Lovastatin for the indicated time. Lysates (50 μg) were subjected to western blot analysis with the indicated antibodies.
HMGCS1 and HMGCR were upregulated in Cisplatin-resistant HCP4 cells
To clarify the mechanism underlying the sensitivity of Cisplatin-resistant HCP4 cells to Lovastatin we examined the MVA cascade. Western blot analysis revealed that cellular expression levels of HMGCS1 and HMGCR in HCP4 cells were 2.6-fold and 2.9-fold higher than those in Hela cells, respectively (Figure 3A). Real-time PCR analysis showed that the mRNAs of these genes were also upregulated in HCP4 cells (Figure 3B). Next, we performed metabolome analysis for Hela and HCP4 cells and found that the ratio of the amount of HMG-CoA in Hela cells to HCP4 cells was 1.1 (data not shown). These results suggested that the MVA cascade was activated in HCP4 cells compared with Hela cells, but the metabolized HMG-CoA was not accumulated in HCP4 cells.
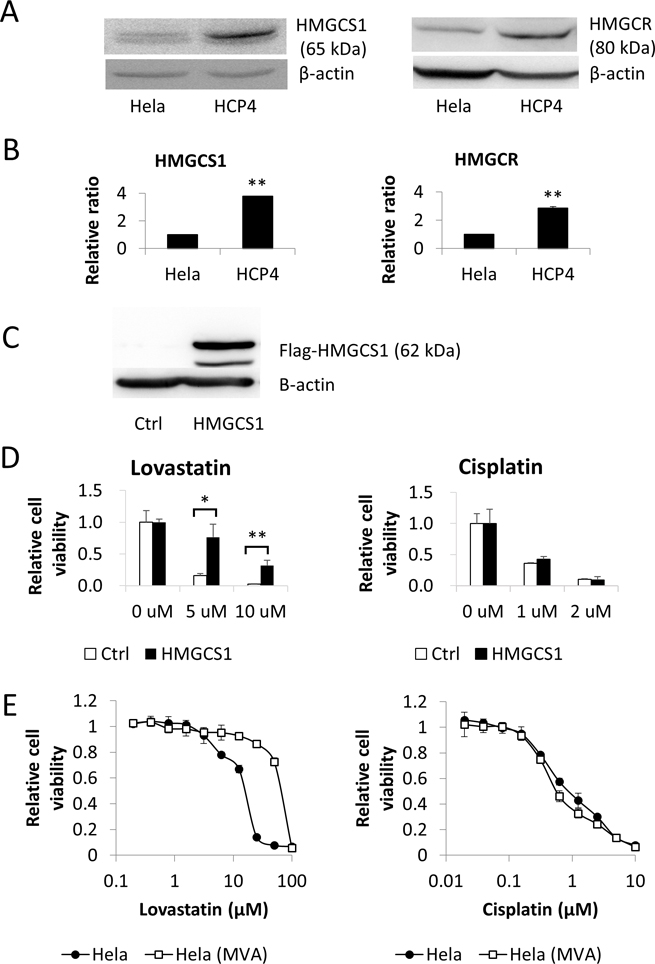
Figure 3: HMGCS1 was upregulated in Cisplatin-resistant HCP4 cells. (A) Lysates (50 μg) of Hela and HCP4 cells were subjected to western blot analysis with the indicated antibodies. (B) Total RNA from each cell line was used for quantitative real-time RT-PCR. All values represent the mean of at least two independent experiments. mRNA expression of Hela cells was set to 1; * and ** indicate P < 0.05 and P < 0.01, respectively. (C) Flag-HMGCS1 and empty vector (Ctrl) were transfected into Hela cells and transfectants were selected with 3 ng/mL Puromycin for 2 weeks. Lysates (50 μg) were subjected to western blot analysis with the indicated antibodies. (D) Transfectants were treated with the indicated concentration of Lovastatin or Cisplatin. After 72 h, cells expressing GFP were quantified with a LUNA-FL™ Dual Fluorescence Cell Counter. All values represent the mean of at least three independent experiments. * and ** indicate P < 0.05 and P < 0.01, respectively. (E) Hela(MVA) indicates Hela cells cultured with 100 μM MVA for 1 month. Hela and Hela(MVA) cells were treated with serial dilutions of Lovastatin (left panel) or Cisplatin (right panel). After 72 h, the surviving cells were stained with TetraColor ONE for 2–3 h. MVA (100 μM ) was added to Hela(MVA) cells until the end of the assay. All values represent the mean of at least two independent experiments.
Mevalonic acid cascade is not involved in Cisplatin resistance
As enhancement of the MVA cascade in Cisplatin-resistant cells was suspected we examined the effect of MVA on Cisplatin sensitivity. First, we performed a cell viability assay with Hela cells overexpressing HMGCS1 and GFP independently (Figure 3C and Supplementary Figure 1). These cells were treated with Cisplatin or Lovastatin at the indicated concentration and GFP-expressing cells were counted. As shown in Figure 3D, there was a marked difference in sensitivity to Lovastatin between HMGCS1 overexpressing Hela cells and control cells but not to Cisplatin. Furthermore, to elucidate whether MVA was involved in Cisplatin resistance, we added Cisplatin to Hela cells that had been cultured for 1 month with 100 μM MVA. As shown in Figure 3E, IC50 ratios of Hela(MVA) for Hela to Lovastatin and Cisplatin were 48.8 and 0.9, respectively. These data indicate that administration of MVA to HeLa cells confers resistance to Lovastatin but not to Cisplatin.
Lovastatin induced KLF2, KLF6, and RHOB expression
To elucidate the mechanism by which Lovastatin sensitized the Cisplatin-resistant cells we performed mRNA microarray analysis of HCP4 and PCDP5 cells treated with or without 1 μM Lovastatin. We found that 65 mRNAs and 54 mRNAs were induced more than 2-fold by Lovastatin treatment, HCP4 (Supplementary Table 2) and PCDP5 (Supplementary Table 3) cells, respectively. Only three genes, KLF2, KLF6, and RHOB, were commonly increased more than 2-fold. Expression levels of these mRNAs were verified by real-time PCR. As shown in Figure 4A, KLF2, KLF6, and RHOB mRNA levels were elevated in HCP4 cells after 24 h treatment with 1 μM Lovastatin. However, in HeLa cells, strongly elevated levels of these mRNAs was only observed after 96 h treatment with 1 μM Lovastatin. Another statin, Compactin, also induced expression of KLF2 and RHOB mRNAs, but not KLF6 mRNA (Supplementary Figure 2). Next, cellular protein levels of RHOB were investigated. As shown in Figure 4B, in the normal culture condition, RHOB protein of Hela cells was higher than that of HCP4 cells. However, Lovastatin induced rapidly and strongly RHOB protein expression in HCP4 cells (Figure 4B). Unfortunately, some antibodies against KLF2 and KLF6 were purchased, but specificities could not be confirmed. To elucidate whether Lovastatin induced the promoter activities of these genes, Hela and HCP4 cells stably transfected with promoter-luciferase constructs were treated with Lovastatin. As shown in Figure 4C, promoter activity of three genes were induced by Lovastatin and these inducible promoter activities were correlated with mRNA expressions levels.
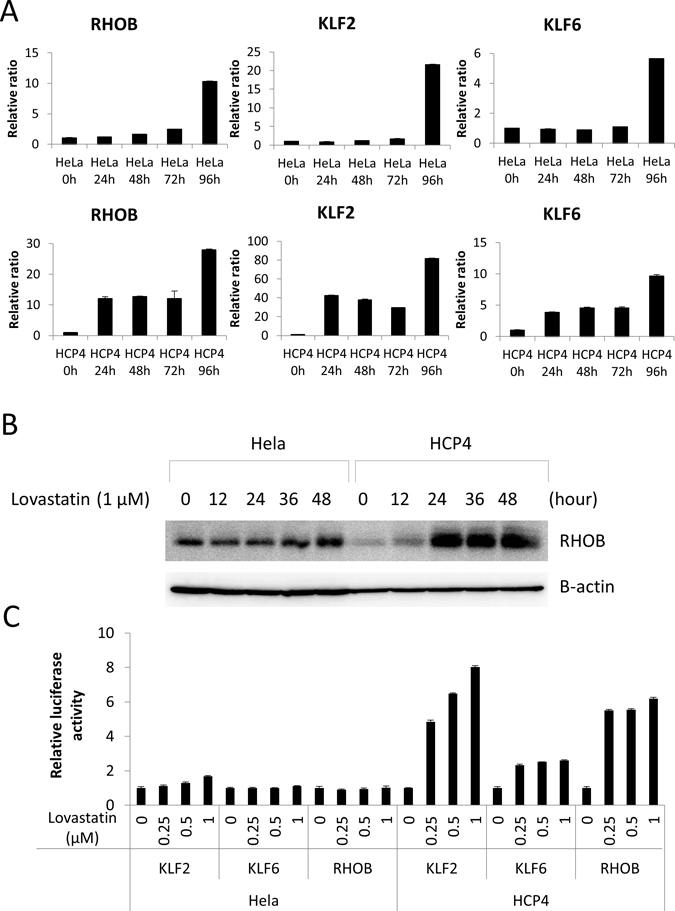
Figure 4: Lovastatin induced KLF2, KLF6, and RHOB expression. (A) Hela and HCP4 cells were treated with 1 μM Lovastatin for indicated time. Total RNA of each cell was used for quantitative real-time RT-PCR. mRNA expression of untreated Hela and HCP4 cells was set to 1. All values represent the mean of at least two independent experiments. (B) Hela and HCP4 cells were treated with 1 μM Lovastatin and collected at indicated time. Lysates (25 μg) were subjected to western blot analysis with the anti-RHOB antibody. (C) Hela and HCP4 cells with integration of KLF2, KLF6, or RHOB promoter–luciferase gene were treated with 0, 0.25, 0.5 1 μM Lovastatin. After 48 h, a luciferase assay was carried out. The results were normalized to protein concentration and are representative of at least three independent experiments.
KLF2, KLF6, and RHOB led to viability reduction
To clarify the mechanism of KLF2, KLF6, and RHOB expressions induced by Lovastatin, we used inducible expression plasmids of the Tet-On system (Supplementary Figure 3). These plasmids contained Flag-tagged KLF2, KLF6, and RHOB driven by the tetracycline response element together with expression sets of rtTA, GFP, and a Puromycin resistance gene (Supplementary Figure 3). Transiently transfected Hela cells and COS1 cells were cultured with 3 ng/mL Puromycin for 2 weeks and selected transfectants were treated with DOX to induce expression of KLF2, KLF6, and RHOB. DOX induced expression of Flag-KLF2, Flag-KLF6, and Flag- RHOB in Hela cells after 24 h. (Figure 5A). At 72 h after DOX treatment, GFP-expressing cells were counted. The number of cells expressing KLF2, KLF6, or RHOB was respectively decreased after DOX treatment, but DOX did not reduce the viability of control cells (Figure 5B). DOX also decreased the GFP-expressing COS1 cells as correlated with induced expression of KLF2, KLF6, or RHOB (Figure 5C and 5D). Cell cycle analysis revealed that KLF2 and RHOB significantly induced an increase in the populations of sub-G1 and G2/M cells, respectively (Figure 5E).
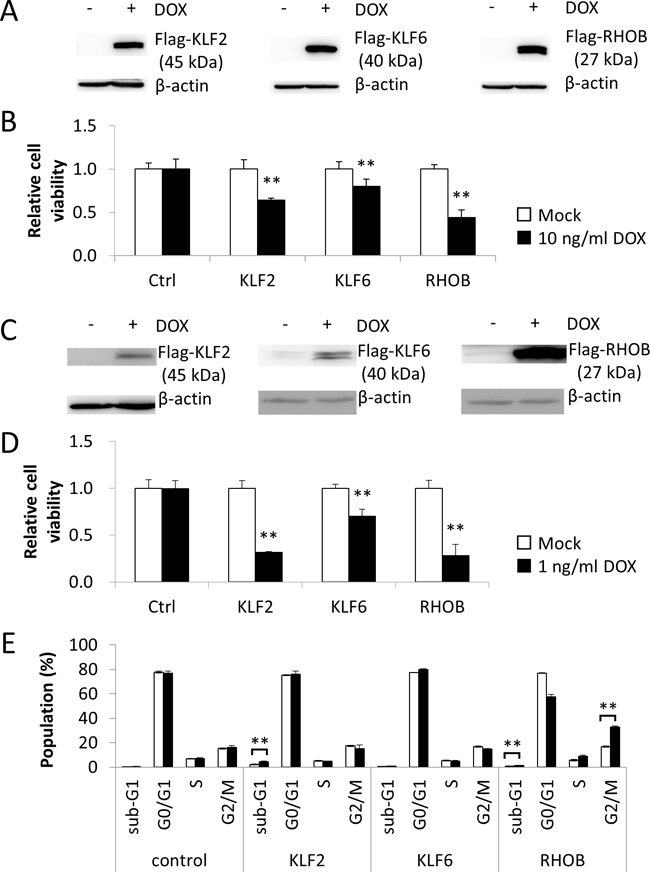
Figure 5: KLF2, KLF6, and RHOB promoted viability reduction. (A) Hela cells were transfected with plasmids containing DOX -inducible Flag-KLF2, Flag-KLF6 and Flag-RHOB genes and transfectants were selected with 3 ng/mL Puromycin for 2 weeks. Transfectants were treated with 10 g/mL DOX for 24 h and cell lysates (50 μg) were subjected to western blot analysis with anti-Flag antibody. (B) Transfectants were treated with 10 g/mL DOX for 48 h and cells expressing GFP were quantified with a LUNA-FL™ Dual Fluorescence Cell Counter. All values represent the mean of at least three independent experiments. * and ** indicate P < 0.05 and P < 0.01, respectively. (C) and (D) The same analysis of (A) and (B) were carried out using COS-1 cells. * and ** indicate P < 0.05 and P < 0.01, respectively. (E) The populations of sub-G1, G0/G1, S and G2M were calculated form the results of flow cytometer. All values represent the mean of at least three independent experiments.
Effect of combination treatment with Lovastatin and Cisplatin on viability reduction
We investigated in vitro combination treatment of cell lines with Cisplatin and Lovastatin. The combination ratio of Cisplatin vs Lovastatin was determined by using a concentration that had relative cell viability, between 0.2 and 0.8 (Table 3). The relative cell viabilities of 2 μM Cisplatin and 10 μM Lovastatin for Hela cells were 2.4 and 0.6, respectively. Therefore, the combination ratio of Cisplatin and Lovastatin used was 2 μM: 10 μM (=1:5). Other combination ratios were determined using the same method. According to the protocol of CalcuSyn software, each single and combination treatments were performed, and the combination index (CI) of ED50, ED75, and ED90 for Hela, HCP4, PC3 and PCDP5 were calculated (Table 3). These results indicated that Lovastatin had an agonistic effect on the viability reduction of Cisplatin in Cisplatin-sensitive Hela and PC3 cells and an antagonistic effect in Cisplatin-resistant HCP4 and PCDP5 cells.
Table 3: Combination Index
Combination ratio |
Combination index (CI) |
|||
|---|---|---|---|---|
ED50 |
ED75 |
ED90 |
||
Hela |
1 : 5 |
0.602 |
0.495 |
0.417 |
HCP4 |
10 : 1 |
1.437 |
1.812 |
2.412 |
PC3 |
1 : 5 |
0.698 |
0.697 |
0.608 |
PCDP5 |
20 : 1 |
1.084 |
1.760 |
3.057 |
DISCUSSION
Statins are well-known inhibitors of HMGCR and are used clinically for the treatment of hyperlipidemia [5]. Recently, it was reported that statins have antitumor activity in various tumor cells, including breast, cervical, colon, endometrial, lung, pancreatic, prostate, and ovarian cancer cells, in vitro and in vivo [6, 7]. Clinical studies also indicated that the antitumor activity of statins was caused by the pleiotropic effects of statins including cell cycle arrest, induction of apoptosis, reduction of metastatic potential, inhibition of angiogenesis, and differentiation of tumors [6, 7, 21].
In this study, we investigated the antitumor effect of statins in Hela and PC3 cells as well as HCP4 and PCDP5 cells, Cisplatin-resistant cell lines established from each parent cells. Lovastatin and other statins had an antitumor effect in Hela and PC3 cells as expected. Interestingly, these statins had a strong antitumor effect in Cisplatin-resistant cells compared with each parental cell. We also investigated Lovastatin sensitivity using Cisplatin-resistant, Oxaliplatin-resistant, and Mithramycin-resistant cell lines derived from parental T24 cells. Oxaliplatin is a third-generation Cisplatin [22] and Mithramycin is a G–C-specific DNA binding antibiotic that inhibits RNA synthesis [23]. Lovastatin sensitized platinum-resistant cells but had no such sensitizing effect on Mithramycin-resistant cells. Jang et al. have reported that Irinotecan-resistant human colorectal adenocarcinoma HT-29 cells are approximately 2-fold more resistant to Simvastatin, when compared with parental cells [24]. These results suggest that the sensitizing effects of statins on cell viability may be specific for platinum-resistant cells. To evaluate this specificity, it is necessary to investigate the effect of Lovastatin using cells that have acquired resistance to platinum-based therapies through a variety of mechanisms. Lovastatin increased sub-G1 population and activated caspase cascade, these suggesting that apoptosis was involved in the viability reduction of Lovastatin. In general, agents that overcome anticancer drug resistance target drug resistance–related genes, and such agents can be used to sensitize drug-resistant cancers with the anticancer drug [25]. The most important result of this study is that statins alone sensitized Cisplatin-resistant cells. Furthermore, combination therapy with Cisplatin and Lovastatin had an agonistic effect on Cisplatin-sensitive cells, but an antagonistic effect on Cisplatin-resistant cells. This is the first report indicating that statins may have the potential to overcome Cisplatin resistance as single-agent therapy.
The MVA pathway is a metabolic pathway that uses acetyl-CoA to produce sterols and isoprenoids [5]. These products are essential for tumor growth and progression [8, 9, 26]. Several reports have demonstrated that statins, a class of HMGCR inhibitors, decreased the proliferation and induced apoptosis of cancer cells [27–30]. However, the association between the MVA pathway and drug-sensitive/resistance is not known. In this study we investigated whether the MVA pathway is involved in Cisplatin resistance. Protein and mRNA expression of HMGCS1 and HMGCR was higher in Cisplatin-resistant cells than in the parental cells, but the amount of HMG-CoA was almost the same between these cell lines. These data indicate that HMG-CoA synthesized by HMGCS1 might be is rapidly converted to MVA by HMGCR in Cisplatin-resistant cells. Cells overexpressing HMGCS1 were able to acquire resistance to Lovastatin but not Cisplatin. Furthermore, administration of MVA suppressed the effects of Lovastatin on cell viability but had no effect on Cisplatin sensitivity. These results indicated that the MVA pathway might not be involved in Cisplatin resistance.
Lovastatin preferentially induced the apoptosis of Cisplatin-resistant cells compared with Cisplatin-sensitive cells. To elucidate the mechanism of apoptosis induction by Lovastatin we performed cDNA microarray analysis with two types of Cisplatin-resistant cells and found that only three genes, KLF2, KLF6, and RHOB, were commonly upregulated over 2-fold by treatment with Lovastatin. Several previous studies demonstrated that statins increased KLF2 and RHOB [31–35] and that these genes are associated with apoptosis or the cell cycle [13, 20, 36]. KLF2 and KLF6 belong to the family of Kruppel-like zinc finger transcription factors, which in humans contains at least 26 members including Sp1-like (Sp1-9) and KLF-like factors (KLF1-17) [37, 38] that regulate remarkably diverse processes including cell growth, signal transduction, and differentiation 10 11. RHOB has been reported to be downregulated in various tumors including gastric, lung, and ovarian cancer, and its overexpression inhibits proliferation, migration, and invasion [39–41]. We found that Lovastatin induced accumulation of these mRNAs by transcriptional regulation. The protein expression of RHOB in Cisplatin-resistant HCP4 cells was lower than that in Hela cells. Of note, RHOB protein in HCP4 cells was rapidly and strongly induced by Lovastatin. These results suggested that Lovastatin significantly induced KLF2, KLF6 and RHOB expression in Cisplatin-resistant cells by transcriptional regulation. It is necessary to clarify the mechanism about transcriptional regulation by Lovastatin in Cisplatin-resistant cells including signal transduction.
Even if individual treatment with an anticancer agent reduces the viability of cancer cells, this viability reduction might be antagonized when the agents are used in combination. Recently, we reported that the aurora kinase B inhibitor, AZD1152-hQPA, had an antagonistic effect on the viability reduction of Cisplatin [42]. Both AZD1152-hQPA [43] and Cisplatin [44] induced G2/M arrest. We speculate that the use of combined drugs that promote G2/M arrest may be involved in viability reduction antagonism. In this study, we did not observe the induction of G2/M arrest by Lovastatin. However, Lovastatin increased RHOB expression and increased the RHOB-induced G2/M arrest. Jang et al. have reported that Simvastatin and Irinotecan have a synergistic effect on Irinotecan-resistant HT-29 cells [24]. Like Cisplatin, Irinotecan has been reported to induce G2/M arrest [44, 45]. However, Simvastatin has contrasting effects on Cisplatin-resistant and Irinotecan-resistant cells. The underlying mechanism of acquired resistance for each drug may explain these differences. In this study, the MVA pathway was not involved in Cisplatin resistance, and so it is necessary to investigate whether this pathway has a role in Irinotecan resistance. There is a possibility of exacerbating cancer, if statins are used with Cisplatin without knowing that cancer cells are resistant to Cisplatin. In short, combination therapy with Cisplatin and statins should be performed with caution in patients who relapse after using Cisplatin. In the future, it is necessary to clarify the mechanism about antagonism in combination of Cisplatin and statins.
We have identified a new activity of statins that may have the potential to overcome Cisplatin resistance as single-agent therapy. This raises the possibility that statins might relieve the suffering of patients with Cisplatin-resistant cancer. Regarding the mechanism of viability reduction induced by statins, we found the tumor suppressor genes KLF2, KLF6, and RHOB were rapidly and strongly accumulated in Cisplatin-resistant cells compared with Cisplatin-sensitive cells; however, the underlying mechanism remains unknown. Elucidation of this mechanism will contribute to the discovery of new target genes and the development of molecular targeted agents.
MATERIALS AND METHODS
Cell culture
Human prostate cancer PC3 cells and Cisplatin-resistant PCDP5 cells, human cervical cancer Hela cells and Cisplatin-resistant HCP4 cells were previously described [46, 47]. DDP10, OX2 and MM4 cells were established with human bladder cancer T24 cells to be resistant to Cisplatin, Oxaliplatin and Mithramycin, respectively. DDP10 cells and OX2 cells were previously described [48, 49]. Mithramycin-resistant MM4 cells were established from T24 cells with increasing concentrations of Mithramycin step by step. All cells were cultured in RPMI 1640 medium with GlutaMAX™ supplement (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA). Cisplatin-resistant HCP4, PCDP5 and DDP10, Oxaliplatin-resistant OX2 and Mithramycin-resistant MM4 cells were usually cultured with medium containing 1 μM Cisplatin, 5 μM Oxaliplatin and 1 μM Mithramycin, respectively, which was removed from the medium one week before assay. Hela(MVA) cells were cultured with 100 μM DL-mevalonolactone (sc-211365, Santa Cruz Biotechnology) for one month or over. The COS-1 fibroblast-like cell lines derived from monkey kidney tissue (kindly gifted by Professor Keiko Funa, Sahlgrenska Cancer Center at the Sahlgrenska Academy, University of Gothenburg) was grown in Dulbecco’s modified Eagle’s medium with GlutaMAX™ supplement (Invitrogen) containing 10% fetal bovine serum (HyClone). All cell lines were incubated in 5% CO2 at 37°C.
Antibodies and treatment agents
Anti-HMGCS1 (sc-33829), anti-HMGCR (sc-27578), anti-RHOB (sc-180), and anti-PARP-1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti–cleaved caspase 3 (#9661) and anti–cleaved caspase 9 (#9501) antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-Flag M2 (F3165) and anti–β-actin (A2228) antibodies were from Sigma-Aldrich (St. Louis, MO). Simvastatin (196-17801), Pravastatin sodium salt (162-19821), Lovastatin (125-04581), Compactin (033-17031), Fluvastatin sodium (069-05571), Atorvastatin calcium trihydrate (012-23901), and Pitavastatin calcium (163-24861) were purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan).
Plasmid construction
To obtain cDNAs of HMGCS1, KLF2, KLF6, and RHOB, a cDNA library (Human Universal QUICK-Clone™ cDNA II, Clontech Laboratories, Inc., Palo Alto, USA) was PCR amplified using specific primer pairs (Supplementary Table 3). The PCR product of HMGCS1 cDNA containing Flag sequence at N-terminal was ligated into the MCS of pEB GFP-T2A-Puro expression plasmid (Supplementary Figure 1). Flag-tagged KLF2, KLF6 and RHOB cDNAs were ligated into the MCS of pEB Tet-On GFP-T2A-Puromycin expression plasmid (Supplementary Figure 3). To prepare the KLF2, KLF6, and RHOB promoter resions, genomic DNA was amplified using specific primer pairs (Supplementary Table 4). The PCR products of promoter resions were cloned and ligated into the MCS of Lenti Luc2P GFP-T2A-Puromycin reporter plasmid (Supplementary Figure 4).
Transfection of expression plasmids
Hela or COS1 cells were transfected with expression plasmids (Supplementary Figure 1 and 3) capable of replication in mammalian cells through EBNA1 and OriP gene expression (Wako Pure Chemical Industries, Tokyo, Japan). Transfection was performed with X-tremeGENE 9 DNA transfection reagent (Roche Life Sciences, Indianapolis, IN) according to the manufacturer’s protocol [23]. After transfection and culture in medium containing 3 ng/mL Puromycin (Invitrogen, Carlsbad, CA) for 2 weeks, more than 90% of the cells expressed GFP. Ectopic Flag-HMGCS1 was expressed by the CMV promoter, and Flag-KLF2, Flag-KLF6, and Flag-RHOB were expressed by the Tet-On system (Supplementary Figure 3).
Infection of lentivirus
A Lenti Luc2P GFP-T2A-Puromycin reporter plasmid (Supplementary Figure 4) was transfected to HEK293TN cells (System Biosciences, CA, USA) with pPACKH1 Lentivector Packaging Kit (#LV500A-1; System Biosciences) according to the manufacturer’s protocol. After 12 hours, the culture medium was changed, and cells were further cultured for 24 hours. Culture medium containing lentivirus was collected and centrifuged for 5 min at 8,000 rpm. The supernatant was transferred to Hela or HCP4 cells. After 48 hours, the medium was changed to fresh medium containing 3 ng/mL Puromycin, and the cells were cultured for 2 weeks. Before the assay, we confirmed that more than 90% of the cells expressed GFP.
Cell viability assays
Hela cells (1 × 103) or HCP4 cells (2 × 103) were seeded into 96-well plates for 24 h and then treated with Cisplatin or statins at the maximum concentration indicated and 2-fold serial dilutions. For combination treatment with Lovastatin and Cisplatin, the indicated fixed concentration ratios were used. After 72 h, the surviving cells were stained with cell proliferation assay kit (TetraColor ONE; Seikagaku Corporation, Tokyo, Japan) for 2–3 h at 37°C and absorbance was measured at 450 nm, according to the manufacturer’s protocol. To measure the half maximal inhibitory concentration (IC50) in each experiment, CalcuSyn software version 2.0 (Biosoft, Cambridge, UK) was used. For combination treatment of Cisplatin and Lovastatin, the combination index (CI) calculated by CalcuSyn software was employed as previously reported [42]. For analysis of transfectants, cells expressing GFP were counted using a LUNA-FL™ Dual Fluorescence Cell Counter (Logos Biosystems, Gyunggi-do, South Korea) according to the manufacturer’s protocol.
Flow cytometry
The flow cytometric analysis has been described previously [50]. Hela and HCP4 cells (2.5 × 105 cells/well) were seeded in 6-well plates and incubated overnight before treatment with 0, 1, and 10 μM Lovastatin. After 24 h, 48 h, and 72 h, cells were harvested, washed twice with ice-cold PBS, resuspended in 70% ethanol, and stored at −20°C until use. Cells transfected with Flag-KLF2, Flag-KLF6, and Flag-RHOB expression plasmids were treated with 1 ng/mL DOX for 48 h, washed twice with ice-cold PBS, and stained with the Muse Cell Cycle Kit (Millipore). The cells were analyzed using a flow cytometer (EC800 Analyzer, Sony Co., Ltd., Tokyo, Japan).
Western blot analysis
Preparation of whole cell lysates and western blot analysis were performed as described previously [50]. Briefly, 50 μg of whole cell lysate was separated by 8–15% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The bound antibody was visualized using an enhanced chemiluminescence kit (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) and the signal intensity was quantitated using Multi Gauge software version 3.0 (Fujifilm, Tokyo, Japan).
RNA extraction, reverse transcription, and quantitative real-time PCR analysis
Total RNA of Hela and HCP4 cells under the indicated conditions was extracted by RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol and used for real-time PCR or mRNA microarray analysis as described previously [50]. Briefly, quantitative real-time RT-PCR analysis was carried out with the appropriate primer sets (Supplementary Table 5) using the 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The comparative cycle time (ΔΔCT) method was used to quantify gene expression. Values were normalized to those for human β-actin. All samples were run in duplicate in each experiment.
Microarray analysis
Total RNA of HCP4 cells with or without Lovastatin treatment was extracted by miRNeasy Mini Kit (Qiagen) and 1 μg or 0.25 μg of total RNAs were used for 3D-Gene mRNA. Microarray analysis was performed using the 3D-Gene mRNA microarray platforms (TORAY, Kamakura, Japan). Briefly, for 3D-Gene mRNA microarray analysis, total RNA was transcriptionally amplified once using Arcturus® Paradise® PLUS 2 Round Kit–Amino Allyl (Life Technologies Corporation, Carlsbad, CA USA) according to the manufacturer’s protocol. Obtained amino-allyl labeled antisense RNA (10 μg of aRNA) was labeled with Cy5-dye (GE Healthcare, Buckinghamshire, England) according to the manufacturer’s protocol. The Cy5-labeled aRNA pools and hybridization buffer, and hybridized for 16 h. The hybridization was performed using the supplier’s protocols (www.3d-gene.com). Detected signals for each gene were normalized by global normalization method (the median of the detected signal intensity was adjusted to 25.
Metabolic assay
Hela cells (3.47 × 103) or HCP4 cells (6.15 × 103) were plunged into 2 mL of 5% Mannitol/Milli-Q water containing internal standards (Solution ID: 304-1002, Human Metabolome Technologies, Inc., Tsuruoka, Japan) at 0°C to inactivate enzymes. Sample preparation and metabolome measurements by CE-MS analysis were carried out through a facility service at Human Metabolome Technologies Inc., Tsuruoka, Japan [51].
Reporter assays
Hela or HCP4 cells were infected with lentivirus including KLF2-, KLF6-, RHOB-promoter-Luc2P constructs as described above. Each cell type (1 × 105/well) was seeded in a 12-well plate. After 24 hours, cells were treated with the indicated concentration of statins and cultured further for 24 hours. Luciferase activity was detected using a Picagene kit (Toyoinki, Tokyo, Japan) and measured with a luminometer (Luminescencer JNII RAB-2300; ATTO, Tokyo, Japan). Results were normalized to the protein concentration determined by the Bradford method and are representative of at least three independent experiments.
Statistical assays
Results were compared by Student’s t-test and data were expressed as mean ± S.D. Statistical significance was defined as P < 0.05.
Abbreviations
aRNA, antisense RNA; AZD1152-hQPA, AZD1152-hydroxyquinazoline pyrazol anilide; CMV, cytomegalovirus; COS1, fibroblast-like cell lines derived from monkey kidney tissue; DDP10, Cisplatin-resistant T24 cells; DOX, doxycycline; EBNA1, Epstein-Barr nuclear antigen 1; GFP, green fluorescence protein; HCP4, Cisplatin-resistant Hela cells; Hela, human cervical cancer cells; Hela(MVA), Hela cells cultured in the presence of mevalonic acid; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; HMGCS1, 3-hydroxy-3-methylglutaryl-coenzyme A synthase 1; IC50, inhibitory concentration of 50%; KLF2, Krüppel-like factor 2; KLF6, Krüppel-like factor 6; KLFs, Krüppel-like factors; Luc2P, destabilized and optimized firefly luciferase with the -Pro-Glu-Ser-Thr; MCS, multicloning sites; MM4, Mithramycin-resistant T24 cells; MVA, mevalonic acid; OriP, origin of viral replication; OX2, Oxaliplatin-resistant T24 cells, PARP-1, poly (adenosine diphosphate ribose) polymerase 1; PC3, human prostate cancer cells; PCDP5, Cisplatin-resistant PC3 cells; RHOB, ras homolog family member B; RT-PCR, reverse transcription-polymerase chain reaction; rtTA, teteracycline (Tc)-dependent and -inducible transcriptional activator; shRNA, short hairpin ribonucleic acid; T24, human bladder cancer cells; T2A, Thosea asigna virus 2A; TRE, tetracycline response element
CONFLICTS OF INTEREST
The authors have no conflict of interest.
GRANT SUPPORT
This work was supported in part by Center for Clinical and Translational Research of Kyushu University and Grant-in-Aid for Scientific Research(C) Grant Number JP17K08479.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
2. Heintz AP, Odicino F, Maisonneuve P, Quinn MA, Benedet JL, Creasman WT, Ngan HY, Pecorelli S, Beller U. Carcinoma of the ovary. FigO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006; 95:S161–192.
3. Quinn MA, Benedet JL, Odicino F, Maisonneuve P, Beller U, Creasman WT, Heintz AP, Ngan HY, Pecorelli S. Carcinoma of the cervix uteri. FigO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006; 95:S43–103.
4. Creasman WT, Odicino F, Maisonneuve P, Quinn MA, Beller U, Benedet JL, Heintz AP, Ngan HY, Pecorelli S. Carcinoma of the corpus uteri. FigO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006; 95:S105–143.
5. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990; 343:425–430.
6. Altwairgi AK. Statins are potential anticancerous agents (review). Oncol Rep. 2015; 33:1019–1039.
7. Osmak M. Statins and cancer: current and future prospects. Cancer Lett. 2012; 324:1–12.
8. Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016. 16:718–731.
9. Yeganeh B, Wiechec E, Ande SR, Sharma P, Moghadam AR, Post M, Freed DH, Hashemi M, Shojaei S, Zeki AA, Ghavami S. Targeting the mevalonate cascade as a new therapeutic approach in heart disease, cancer and pulmonary disease. Pharmacol Ther. 2014; 143:87–110.
10. Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001; 188:143–160.
11. Bieker JJ. Kruppel-like factors: three fingers in many pies. J Biol Chem. 2001; 276:34355–34358.
12. Wu J, Lingrel JB. KLF2 inhibits Jurkat T leukemia cell growth via upregulation of cyclin-dependent kinase inhibitor p21WAF1/CIP1. Oncogene. 2004; 23:8088–8096.
13. Wang F, Zhu Y, Huang Y, McAvoy S, Johnson WB, Cheung TH, Chung TK, Lo KW, Yim SF, Yu MM, Ngan HY, Wong YF, Smith DI. Transcriptional repression of WEE1 by Kruppel-like factor 2 is involved in DNA damage-induced apoptosis. Oncogene. 2005; 24:3875–3885.
14. Zhang W, Levi L, Banerjee P, Jain M, Noy N. Kruppel-like factor 2 suppresses mammary carcinoma growth by regulating retinoic acid signaling. Oncotarget. 2015; 6:35830–35842. https://doi.org/10.18632/oncotarget.5767.
15. Ito G, Uchiyama M, Kondo M, Mori S, Usami N, Maeda O, Kawabe T, Hasegawa Y, Shimokata K, Sekido Y. Kruppel-like factor 6 is frequently down-regulated and induces apoptosis in non-small cell lung cancer cells. Cancer Res. 2004; 64:3838–3843.
16. Sangodkar J, Shi J, DiFeo A, Schwartz R, Bromberg R, Choudhri A, McClinch K, Hatami R, Scheer E, Kremer-Tal S, Martignetti JA, Hui A, Leung WK, et al. Functional role of the KLF6 tumour suppressor gene in gastric cancer. Eur J Cancer. 2009; 45:666–676.
17. DiFeo A, Narla G, Martignetti JA. Emerging roles of Kruppel-like factor 6 and Kruppel-like factor 6 splice variant 1 in ovarian cancer progression and treatment. Mt Sinai J Med. 2009; 76:557–566.
18. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005; 21:247–269.
19. Prendergast GC. Actin' up: RhoB in cancer and apoptosis. Nat Rev Cancer. 2001; 1:162–168.
20. Huang M, Prendergast GC. RhoB in cancer suppression. Histol Histopathol. 2006; 21:213–218.
21. Bockorny B, Dasanu CA. HMG-CoA reductase inhibitors as adjuvant treatment for hematologic malignancies: what is the current evidence? Ann Hematol. 2015; 94:1–12.
22. Raymond E, Faivre S, Woynarowski JM, Chaney SG. Oxaliplatin: mechanism of action and antineoplastic activity. Semin Oncol. 1998; 25:4–12.
23. Yarbro JW, Kennedy BJ, Barnum CP. Mithramycin inhibition of ribonucleic acid synthesis. Cancer Res. 1966; 26:36–39.
24. Jang HJ, Hong EM, Jang J, Choi JE, Park SW, Byun HW, Koh DH, Choi MH, Kae SH, Lee J. Synergistic Effects of Simvastatin and Irinotecan against Colon Cancer Cells with or without Irinotecan Resistance. Gastroenterol Res Pract. 2016; 2016:7891374.
25. Kigawa J. New strategy for overcoming resistance to chemotherapy of ovarian cancer. Yonago Acta Med. 2013; 56:43–50.
26. Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene. 2012; 31:4967–4978.
27. Wong WW, Dimitroulakos J, Minden MD, Penn LZ. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002; 16:508–519.
28. Kato S, Smalley S, Sadarangani A, Chen-Lin K, Oliva B, Branes J, Carvajal J, Gejman R, Owen GI, Cuello M. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. J Cell Mol Med. 2010; 14:1180–1193.
29. Bjarnadottir O, Kimbung S, Johansson I, Veerla S, Jonsson M, Bendahl PO, Grabau D, Hedenfalk I, Borgquist S. Global Transcriptional Changes Following Statin Treatment in Breast Cancer. Clin Cancer Res. 2015; 21:3402–3411.
30. Stine JE, Guo H, Sheng X, Han X, Schointuch MN, Gilliam TP, Gehrig PA, Zhou C, Bae-Jump VL. The HMG-CoA reductase inhibitor, simvastatin, exhibits anti-metastatic and anti-tumorigenic effects in ovarian cancer. Oncotarget. 2016; 7:946–960. https://doi.org/10.18632/oncotarget.5834.
31. Von Zee CL, Richards MP, Bu P, Perlman JI, Stubbs EB Jr. Increased RhoA and RhoB protein accumulation in cultured human trabecular meshwork cells by lovastatin. Invest Ophthalmol Vis Sci. 2009; 50:2816–2823.
32. Chan KC, Wu CH, Huang CN, Lan KP, Chang WC, Wang CJ. Simvastatin inhibits glucose-stimulated vascular smooth muscle cell migration involving increased expression of RhoB and a block of Ras/Akt signal. Cardiovasc Ther. 2012; 30:75–84.
33. Zhao J, Natarajan SK, Chronos N, Singh JP. Cerivastatin represses atherogenic gene expression through the induction of KLF2 via isoprenoid metabolic pathways. Cell Mol Biol Lett. 2015; 20:825–839.
34. Miao Q, Zeng X, Ma G, Li N, Liu Y, Luo T, Lian J, Chen S. Simvastatin suppresses the proangiogenic microenvironment of human hepatic stellate cells via the Kruppel-like factor 2 pathway. Rev Esp Enferm Dig. 2015; 107:63–71.
35. Tuomisto TT, Lumivuori H, Kansanen E, Hakkinen SK, Turunen MP, van Thienen JV, Horrevoets AJ, Levonen AL, Yla-Herttuala S. Simvastatin has an anti-inflammatory effect on macrophages via upregulation of an atheroprotective transcription factor, Kruppel-like factor 2. Cardiovasc Res. 2008; 78:175–184.
36. Andreoli V, Gehrau RC, Bocco JL. Biology of Kruppel-like factor 6 transcriptional regulator in cell life and death. IUBMB Life. 2010; 62:896–905.
37. Suske G, Bruford E, Philipsen S. Mammalian SP/KLF transcription factors: bring in the family. Genomics. 2005; 85:551–556.
38. van Vliet J, Crofts LA, Quinlan KG, Czolij R, Perkins AC, Crossley M. Human KLF17 is a new member of the Sp/KLF family of transcription factors. Genomics. 2006; 87:474–482.
39. Zhou J, Zhu Y, Zhang G, Liu N, Sun L, Liu M, Qiu M, Luo D, Tang Q, Liao Z, Zheng Y, Bi F. A distinct role of RhoB in gastric cancer suppression. Int J Cancer. 2011; 128:1057–1068.
40. Mazieres J, Antonia T, Daste G, Muro-Cacho C, Berchery D, Tillement V, Pradines A, Sebti S, Favre G. Loss of RhoB expression in human lung cancer progression. Clin Cancer Res. 2004; 10:2742–2750.
41. Liu Y, Song N, Ren K, Meng S, Xie Y, Long Q, Chen X, Zhao X. Expression loss and revivification of RhoB gene in ovary carcinoma carcinogenesis and development. PLoS One. 2013; 8:e78417.
42. Sakai S, Izumi H, Yoshiura Y, Nakayama Y, Yamaguchi T, Harada Y, Koi C, Kurata H, Morimoto Y. In vitro evaluation of a combination treatment involving anticancer agents and an aurora kinase B inhibitor. Oncol Lett. 2016; 12:4263–4269.
43. Ghanizadeh-Vesali S, Zekri A, Zaker F, Zaghal A, Yousefi M, Alimoghaddam K, Ghavamzadeh A, Ghaffari SH. Significance of AZD1152 as a potential treatment against Aurora B overexpression in acute promyelocytic leukemia. Ann Hematol. 2016; 95:1031–1042.
44. Sorenson CM, Barry MA, Eastman A. Analysis of events associated with cell cycle arrest at G2 phase and cell death induced by cisplatin. J Natl Cancer Inst. 1990; 82:749–755.
45. Kaku Y, Tsuchiya A, Kanno T, Nishizaki T. Irinotecan induces cell cycle arrest, but not apoptosis or necrosis, in Caco-2 and CW2 colorectal cancer cell lines. Pharmacology. 2015; 95:154–159.
46. Miyamoto N, Izumi H, Noguchi T, Nakajima Y, Ohmiya Y, Shiota M, Kidani A, Tawara A, Kohno K. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. J Biol Chem. 2008; 283:18218–18226.
47. Akiyama M, Izumi H, Wang KY, Yamaguchi T, Kuma A, Kitamura N, Harada Y, Oya R, Yamaguchi K, Iwai Y, Kohno K. Hypersensitivity to aurora kinase inhibitors in cells resistant against platinum-containing anticancer agents. Anticancer Agents Med Chem. 2014; 14:1042–1050.
48. Kotoh S, Naito S, Yokomizo A, Kumazawa J, Asakuno K, Kohno K, Kuwano M. Increased expression of DNA topoisomerase I gene and collateral sensitivity to camptothecin in human cisplatin-resistant bladder cancer cells. Cancer Res. 1994; 54:3248–3252.
49. Kashiwagi E, Izumi H, Yasuniwa Y, Baba R, Doi Y, Kidani A, Arao T, Nishio K, Naito S, Kohno K. Enhanced expression of nuclear factor I/B in oxaliplatin-resistant human cancer cell lines. Cancer Sci. 2011; 102:382–386.
50. Yamaguchi T, Kurita T, Nishio K, Tsukada J, Hachisuga T, Morimoto Y, Iwai Y, Izumi H. Expression of BAF57 in ovarian cancer cells and drug sensitivity. Cancer Sci. 2015; 106:359–366.
51. Soga T, Heiger DN. Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Anal Chem. 2000; 72:1236–1241.