
INTRODUCTION
Bladder cancer is the fourth most common cancer diagnosed in men [1]. In 2016, an estimated 76,960 new patients will be diagnosed with bladder cancer in the US [2] and about 430.000 cases worldwide [3], 16,390 will probably die from complications of this disease in the US [2] and about 160.000 worldwide [3]. Around thirty percent of the cases are already diagnosed as muscle-invasive urothelial carcinoma and most of them have locally advanced or disseminated disease at diagnosis [4]. The standard treatment for patients with muscle-invasive bladder cancer (MIBC) includes neoadjuvant cisplatin-based chemotherapy along with radical cystectomy. Although these regimens have a high response rate, they are generally non-curative, with median progression-free survival of approximately 8 months and a 5-year overall survival rate of 5–10% [1, 3]. New therapeutic approaches are urgently needed and research is focused on development of targeted therapies, which may be more effective [5] than the current protocol. Next generation sequencing of invasive urothelial carcinoma has identified the phosphatidylinositol 3-kinase/protein kinase B/mechanistic target of rapamycin (PI3K/Akt/mTOR) pathway as a potential therapeutic target [6]. mTOR pathway activation has been shown to be involved in urothelial bladder cancer tumorigenesis and to be a predictor of disease progression and cancer specific survival [7, 8]. Data of the Cancer Genome Atlas (TCGA) confirmed these findings [9]. mTOR inhibitors have been evaluated as anticancer agents, some of which are already approved for the treatment of metastatic renal cell carcinoma (temsirolimus, everolimus), mantle cell lymphoma (temsirolimus), breast cancer (everolimus) and pancreatic neuroendocrine tumors (everolimus) [10]. Disappointingly, a phase II study with everolimus, given as a single agent in bladder cancer, did not show the efficacy that might have been expected [11]. In fact, mTOR inhibition revealed heterogeneous responses, indicating anti-tumor effects in some cases, while others exhibit intrinsic or acquired resistance to the drug both in preclinical or clinical settings [10]. Mechanisms underlying resistance are various and include loss of mTOR inhibition, feedback activation of PI3K and Akt [12].
A combination with other compounds might be promising. Based on earlier studies, we postulate that modulation of the histone acetylation status by a histone deacetylase (HDAC) inhibitor might be an attractive strategy to improve an mTOR inhibitior-based regime. Notably, HDAC suppression may not only elevate the therapeutic efficacy of an mTOR inhibitor per se [13] but may overcome resistance towards this respective class of drugs [14–16]. HDAC inhibitors have been identified to restore epithelial differentiation and to abrogate growth in different cancer cells, including bladder cancer cells [17]. Several data point to the principal importance of a combined HDAC-mTOR inhibitor-based regime to optimize tumor treatment. A single molecule inhibitor targeting both HDAC activity and PI3K signaling has recently been developed, which induced greater tumor growth inhibition and pro-apoptotic activity than single-target PI3K or HDAC inhibitors in vitro and in vivo [18]. Accordingly, combining the HDAC inhibitor vorinostat with the mTOR inhibitor MLN0128 increased the expression of pro-death genes and the sensitivity to apoptotic triggers [19]. In trametinib/dabrafenib-resistant melanoma cells, addition of the HDAC inhibitor AR42 with pazopanib contributed to significantly reduced tumor growth in vitro and in vivo [20].
Since the relevance of HDAC suppression for drug-resistant bladder cancer cells has not yet been evaluated, we explored whether the HDAC inhibitor valproic acid (VPA) exerts anti-tumor properties on a panel of temsirolimus-resistant bladder cancer cell lines.
RESULTS
HDAC inhibition causes growth and proliferation blockade of both temsirolimus sensitive and resistant cells
Cell growth of RT112res was only slightly reduced when compared to RT112par cells (Figure 1A), whereas growth of UMUC-3res cells was even enhanced when compared to the respective parental control (Figure 1B). Incubation with VPA [1 mmol/ml] induced a significant growth inhibition of both RT112par and RT112res cells compared to the untreated cell sublines (Figure 1A). Growth suppression was also evoked when VPA was added to UMUC-3par or UMUC-3res cell cultures (Figure 1B).
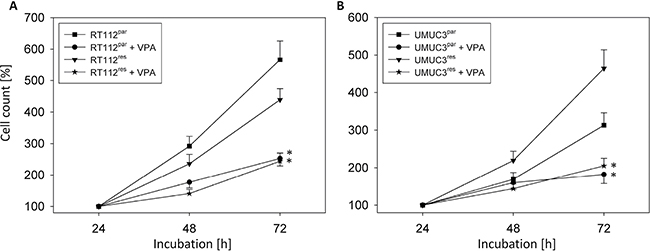
Figure 1: Growth of parental (par) and temsirolimus-resistant (res) bladder cancer cells, RT112 (A) and UMUC-3 (B). Temsirolimus-resistant cells were exposed to 1 μmol/ml temsirolimus three times a week. Cells were treated with VPA [1 mmol/ml] in the 96-well-plates for 24 h, 48 h and 72 h. Controls remained untreated. Cell number was set to 100% after 24h incubation. Bars indicate standard deviation (SD). *indicates significant difference to untreated control cells, p ≤ 0.05. n = 5.
Evaluation of tumor cell proliferation revealed distinct tumor suppressive properties of VPA exerted on RT112par and RT112res cells (Figure 2A) and on UMUC-3par and UMUC-3res cells (Figure 3A). Interestingly, stronger effects of VPA were induced on the resistant cell cultures after 24 h (RT112) and 48 h (RT112 and UMUC-3) compared to the sensitive ones. Mean percentage of RT112 proliferation blockade was calculated to 18.6% versus 60.6% (24 h values, sensitive versus resistant) and 18.0% versus 33.3% (48 h values, sensitive versus resistant; Figure 2B). Mean percentage of UMUC-3 proliferation blockade was 26.3% versus 44.8% (48 h values, sensitive versus resistant; Figure 3B). Differences in the inhibitory efficacy of VPA on UMUC-3par versus UMUC-3res were not seen after 24 h. No significant apoptotic or necrotic activity of VPA has been detected, indicating that reduced cell growth and proliferation was not caused by apoptotic events (data not shown).
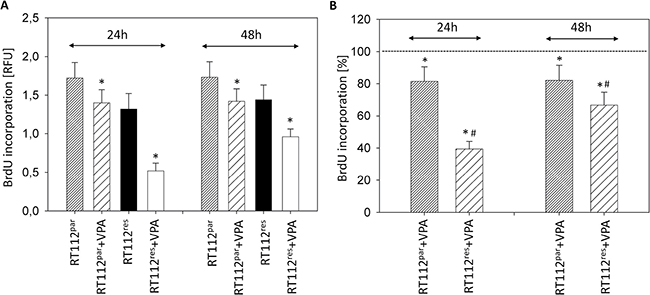
Figure 2: Proliferation of RT112par and RT112res. Temsirolimus-resistant cells were exposed to temsirolimus [1 μmol/ml] three times a week. Tumor cells were further treated with VPA [1 mmol/ml] in the BrdU assay for 24 h or 48 h. Controls remained untreated. (A) BrdU incorporation [RFU] for each sample. (B) % difference of VPA treated cells to controls without VPA. Bars indicate standard deviation (SD). *indicates significant difference to control, #indicates significant difference to parental cells, p ≤ 0.05. n = 5.
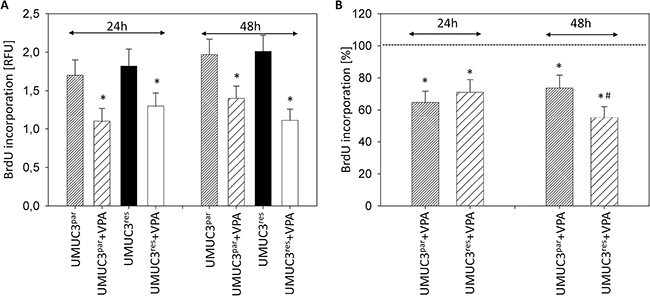
Figure 3: Proliferation of UMUC-3par and UMUC-3res. Temsirolimus-resistant cells were exposed to 1 μmol/ml temsirolimus three times a week. Tumor cells were further treated with VPA [1 mmol/ml] in the BrdU assay for 24 h or 48 h. Controls remained untreated. (A) BrdU incorporation [RFU] for each sample. (B) % difference of VPA treated cells to controls without VPA. Bars indicate standard deviation (SD). *indicates significant difference to control, #indicates significant difference to parental cells, p ≤ 0.05. n = 5.
HDAC inhibition results in G0/G1 cell cycle arrest
The number of temsirolimus-resistant RT112 and UMUC-3 cells in G2/M increased, accompanied by a decrease in the number of S-phase cells (each compared to the respective drug sensitive control, Figures 4, 5). In addition, more RT112res cells were recorded in G0/G1 (versus RT112par), whereas no differences were seen in the number of UMUC-3par versus UMUC-3res cells in this matter (Figure 4, Figure 5). VPA induced distinct accumulation of RT112par in G0/G1 (144.2 +/− 17.2%) with a simultaneous decrease of S-phase cells (44.7 +/− 8.1%; Figure 4). The number of RT112par in G2/M was not altered by VPA. With respect to RT112res cells, VPA elevated the amount of G0/G1 phase cells (121.4 +/− 14.4%) and reduced both the amount of S-phase (48.0 +/− 5.2%) and G2/M-phase cells (60.7 +/− 10.0%; Figure 4).
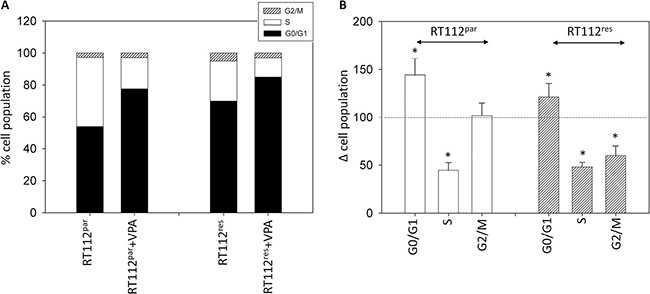
Figure 4: Cell distribution in the different cell cycle phases. (A) Percentage of parental and resistant RT112 in G01/1, S and G2/M phase is indicated. Bladder cancer cells were pre-treated with VPA [1 mmol/ml] for 3 days. Controls remained untreated. One representative of three separate experiments is shown. (B) % difference of RT112par and RT112res exposed to VPA [1 mmol/ml] compared with the corresponding untreated controls. Control phases were set to 100%. Bars indicate standard deviation (SD). *indicates significant difference to control, p ≤ 0.05. n = 5.
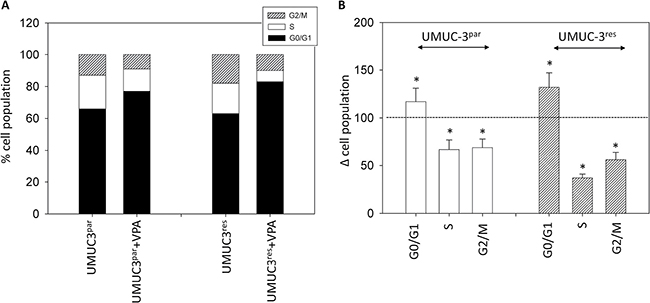
Figure 5: Cell distribution in the different cell cycle phases. (A) Percentage of parental and resistant UMUC-3 in G01/1, S and G2/M phase is indicated. Bladder cancer cells were pre-treated with VPA [1 mmol/ml] for 3 days. Controls remained untreated. One representative of three separate experiments is shown. (B) % difference of UMUC-3par and UMUC-3res exposed to VPA [1 mmol/ml] compared with the corresponding untreated controls. Control phases were set to 100%. Bars indicate standard deviation (SD). *indicates significant difference to control, p ≤ 0.05. n = 5.
Both, UMUC-3par and UMUC-3res cells were similarly modified by VPA, evidenced by a decrease of G2/M cells (parental cells: 69.3 +/− 9.7%; resistant cells: 56.8 +/ 8.4%), reduction of cells in the S-phase (parental cells: 67.5 +/− 10.2%; resistant cells: 37.0 +/ 4.1%), associated with an increase of both UMUC-3par and UMUC-3res in G0/G1 ((parental cells: 117.7 +/− 14.3%; resistant cells: 132.8 +/ 15.6%; Figure 5).
VPA causes distinct modulations of cell cycle regulating proteins
Functional alterations in growth, proliferation and cell cycle progression induced by VPA were associated with distinct modulation of cell cycle regulating protein expression and activity (Figure 6). Concerning the protein expression pattern in RT112res versus RT112par cells, the following proteins were found to be up-regulated in RT112res (Figure 6, left): Skp1 p19, cdk1, cyclin A, B and D1, pAkt, pmTOR and pRaptor. Diminished expression level in RT112res versus RT112par cells was related to p27, cdk2, cdk4, pRictor and pp70S6k. The acetylation status of histone H3 (aH3) did not change during resistance development (Figure 6, left).
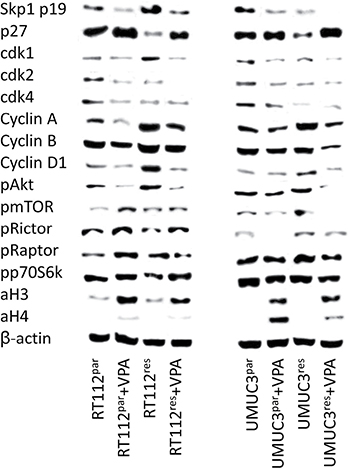
Figure 6: Protein expression profile of cell cycle regulating and targeted proteins in parental and temsirolimus-resistant RT112 (left) and UMUC-3 (right) cells after 3 days exposure to VPA [1 mmol/ml] and untreated controls. ß-actin served as internal control. One representative of three separate experiments is shown.
VPA similarly acted on RT112res versus RT112par cells with respect to Skp1 p19, cdk1, cdk2, cyclin A, B and D1 and pAkt (all: protein suppression) and p27, pRictor and aH3 (all: protein elevation, Figure 6, left). Acetylation of histone H4 (aH4) was not detectable in both RT112res and RT112par cells; however, a very slight band appeared when cells were exposed to VPA. Differences between both cell sublines following VPA application have also been recorded. Cdk4 was diminished by VPA in RT112par cells exclusively; pRaptor was enhanced in RT112par but reduced in RT112res cells. Phosphorylation of p70S6k (pp70S6k) became elevated in RT112res cells but remained unchanged in RT112par.
In accordance with the RT112 data, UMUC-3res have been characterized by an up-regulation of cyclin A, cyclin D1, pmTOR and pRaptor, and a down-regulation of p27, cdk4 and pp70S6k, each compared to UMUC-3par cells (Figure 6, right). In contrast to the RT112 data, Skp1 p19 (distinctly), cdk1, cdk2 (both moderately) were suppressed in UMUC-3res, pRictor increased and cyclin B and pAkt remained unchanged. Acetylation of histones H3 and H4 (aH3, aH4) was not detectable in UMUC-3res and UMUC-3par cells.
In presence of VPA, Skp1 p19, cdk1, cdk2, cyclin A, cyclin B, pAkt, pmTOR and pRictor were all diminished in both UMUC-3res and UMUC-3par cells (Figure 6, right). Independent of the UMUC-3 cell subline, VPA additionally induced an up-regulation of p27 and strong expression of aH3 and aH4 (UMUC-3par > UMUC-3res). Different effects of VPA on resistant versus sensitive tumor cells have also been observed. cdk4 increased in UMUC-3par but decreased in UMUC-3res, whereas cyclin D1 and pRaptor were not modified in UMUC-3par but reduced in UMUC-3res.
Decrease of cdk2 and cyclin A is involved in VPA-induced growth inhibition
Common to all tumor cells analysed, cdk2 and cyclin A were distinctly suppressed by VPA. Therefore we evaluated the particular role of the cdk2-cyclin A axis in tumor growth control by blocking their function using siRNA. Knock-down of cdk2 and cyclin A resulted in significant cell growth inhibition in all tumor sublines, compared to the untreated cells and the mock control (Figure 7C–7F). Down-regulation of cyclin A had a stronger effect than blocking cdk2. Knock-down efficacy of cdk2 and cyclin A protein expression was verified by Western blot analysis (Figure 7A, 7B).
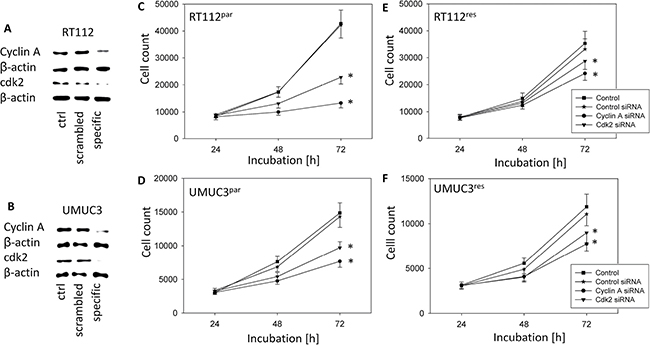
Figure 7: Functional blocking with siRNA targeting cdk2 and cyclin A of RT112 (upper panel) and UMUC-3 (lower panel). All Stars Negative Control siRNA served as transfection control (mock). Controls remained untreated. (A) and (B) Protein expression profile of cell cycle regulating proteins of RT112 and UMUC-3 cells after functional blocking with siRNA targeting cdk2 and cyclin A. ß-actin served as internal control. One representative of three separate experiments is shown. (C–F) Tumor cell growth of blocked bladder cancer cells, (C) RT112par, (D) UMUC-3par, (E) RT112res and (F) UMUC-3res. Bars indicate standard deviation (SD). *indicates significant difference to control, p ≤ 0.05. n = 5.
DISCUSSION
To our knowledge, this is the first manuscript dealing with temsirolimus-driven resistance mechanism in bladder cancer and the potential of VPA in combating resistance. Common to both resistant cell lines, RT112res and UMUC-3res, cyclin A, cyclin D1, pmTOR and pRaptor were up-regulated during chronic drug treatment with temsirolimus. Concerning the mTOR molecule, two functionally distinct sub-structures exist: mTOR complex 1 (mTORC1), which (among others) contains the Rapamycin-sensitive adapter protein of mTOR (Raptor), and mTOR complex 2 (mTORC2), which includes the Rapamycin-insensitive companion of mTOR (Rictor). The mechanistic details of mTORC1-mTORC2 crosstalk are not completely understood. Evidence has been provided confirming Raptor (mTORC1) as the main driving force for mitosis induction and progression and, inversely, resistance induction caused by chronic mTOR blockade has been associated with increased Raptor activation [14]. Since cyclin D1 is under the control of mTORC1 [21, 22], it might not be surprising to see an up-regulation of cyclin D1 in the context of pRaptor increase. A positive correlation between cyclin A and mTOR expression has also been shown [15, 23], although it is not yet clear whether cyclin A might be regulated by mTORC1, mTORC2 or by both.
In contrast to the behavior of cyclin A, cyclin D1, pmTOR and pRaptor, p27 was massively reduced in the temsirolimus-resistant tumor cell lines. Clinical studies on bladder cancer patients point to a negative correlation between p27 expression and recurrence-free survival, as well as between p27 expression and overall survival in this matter [24–26]. Based on a bladder cancer cell model, expression of p27 seems to be directly mediated through an mTOR-dependent mechanism [27], presumably via mTORC1 [28]. Therefore we assume that long-term administration of temsirolimus to bladder cancer cells may result in a feedback mechanism characterized by re-activation of Raptor, associated with cyclin A and cyclin D1 elevation and loss of p27. This scenario may accelerate mitotic cycling and tumor progression. However, apart from similarities among the cell lines, cell line specific alterations have also been noted. E.g. RT112res revealed enhanced Skp1 p19 and cdk1 and reduced pRictor, whereas the opposite was true for UMUC-3res cells. Bearing in mind that cell growth of RT112 versus UMUC-3 cells was differentially influenced by chronic temsirolimus treatment (RT112res < RT112par and UMUC-3res > UMUC-3par), a different molecule expression pattern might reflect different drug sensitivity. However, this assumption is speculative and requires further evaluation.
HDAC suppression by VPA contributed to a significant reduction of bladder cancer cell growth and proliferation, not only of the parental but, most importantly, of those sublines with acquired resistance towards temsirolimus. Due to this, we conclude that HDAC inhibition might be an innovative strategy to overcome mTOR-driven resistance processes. The principal significance of histone modifications on bladder cancer progression has already been well documented. HDAC inhibition mediates apoptosis [29], delays cell cycle progression [30] and blocks adhesive events of bladder cancer cells [31]. Novel data revealed an increased transcription of DNA repair genes by VPA [32]. There is also evidence that targeting HDAC might reverse resistance towards a considerable panel of chemotherapeutic drugs such as cisplatin [33], methotrexate [34], paclitaxel [35], gemcitabine [36], temozolomide [37], gefitinib [38] and epirubicin [39]. Since blocking of HDAC also counteracts resistance to tyrosine kinase inhibitors [40], epigenetic repression might be hypothesized as being an effective strategy to optimize current anti-tumor protocols [41–43].
In good accordance with our results, combined HDAC-mTOR blockade delayed the time to resistance towards the mTOR inhibitor ridaforolimus in a clinical renal cancer study [44] and in vitro studies showed a distinct impact of HDAC suppression on growth and proliferation of tumor cells with acquired resistance to the mTOR inhibitor everolimus [14, 15]. The effect of VPA on bladder cancer progression was associated with an accumulation in the G0/G1-phase and concomitant decrease of S-phase cells. Reports point to a G0/G1-phase arrest, paralleled by a reduction of S-phase cells, as the main mechanism of VPA on several tumor entities such as oral squamous cell carcinoma [45], ovarian cancer [46], endometrial cancer [47], renal cell [15] and prostate carcinoma cells [48]. Interestingly, G2/M phase cells were also diminished in both UMUC-3par and UMUC-3res cultures, whereas VPA lowered the number of RT112res but not of RT112par G2/M phase cells. The different response of the RT112 cell sublines to VPA (in terms of G2/M counts) can be interpreted in two ways. Either this phenomenon reflects a different mode of action of VPA on the resistant versus sensitive cells, or an unspecific effect is seen here. In support of the first assumption, the BrdU incorporation assay demonstrated stronger effects of VPA on the resistant RT112 subline, particularly after 24 h incubation. However, only very few RT112par cells have been counted in G2/M, making a further reduction under VPA unlikely (favoring the second assumption). Indeed, experiments on renal cell cancer cells showed a variable influence of VPA on G2/M cell populations which depended on the basal G2/M count [49]. Nevertheless, both assumptions are hypothetical and not underlined by fundamental data.
As a common mechanism, delay in cell cycle progression of RT112 and UMUC-3 cells caused by VPA was associated with up-regulation of p27 and acetylation of H3 and H4. Immunohistochemical evaluation of bladder cancer specimens revealed that p27 may serve as a prognostic biomarker as well as a promising therapeutic target [50, 51]. In fact, loss of p27 expression correlated with overall and recurrence-free survival [24, 52] and was associated with stage, grade, DNA ploidy and lymph-node involvement [53]. Recently, acetylation of histone H3 or H4 was demonstrated to be directly linked to the p27 promoter [54, 55]. Since aH3 and aH4 expression levels correlated with the p27 level in our experiments, we presume that epigenetic regulation of gene transcription by histone acetylation is (at least partially) responsible for increasing p27 and p27 driven cell growth control. Remarkably, the influence of VPA on p27 was strongest in the drug-resistant bladder cancer cell lines. p27 has been associated with G0/G1 S-phase transition and BrdU incorporation rate [56, 57], which might explain why BrdU uptake was diminished to a lesser extent by VPA in the temsirolimus sensitive cell lines compared to the resistant ones (RT112res−24 + 48 h; UMUC-3res-24 h) and why the S-phase was diminished to a greater extent in UMUC-3res compared to UMUC-3par.
As a further common mechanism, VPA acted on the cyclin cdk axis by suppressing cdk 1 and 2 along with cyclin A and B. This is important as these molecules are crucially involved in cell growth regulation. Patient data have demonstrated a positive correlation between cyclin A-cdk2 level and metastatic progression of bladder cancer [58–60]. siRNA knockdown studies on UMUC-3 and RT112 cell lines revealed that VPA induced loss of cdk2 and cyclin A might be one prominent mechanism causing VPA to slow down mitotic activity. Former experiments on bladder cancer cell culture models showed that the cyclin B-cdk1 axis is also closely involved in tumor growth and proliferation, and that down-regulation of cyclin B/cdk1 causes a distinct delay in cell cycle progression [30, 61].
The mechanism of VPA on cdk-cyclin expression is of high clinical relevance. Aberrant activation of the cdk-cyclin family with subsequent proliferation through the deregulation of cell cycle control has been recognized as one of the key hallmarks of cancer [62]. Recent publications highlight the role of cdk members as potent targets for cancer therapeutics, with the hope of achieving cdk-cyclin inhibition in preventing the emergence of resistance to multiple targeted therapies across various cancer types [63]. In this regard, VPA may exert the resistance-preventing properties as desired by Whittacker and coworkers [63]. Importantly, VPA is well established in the treatment of epilepsy and relatively safe, with a low toxicity and convenient pharmacokinetic properties, which may recommend VPA as a useful adjuvant in the treatment of bladder cancer once resistance has been developed. Although reports are not available dealing with this issue, VPA increased the sensitivity of bladder cancer to mitomycin C, cisplatin and doxorubicin in vitro and in vivo [64], which is in accordance with our statement.
Nevertheless, there are also some discrepant data which need closer discussion. VPA reduced Akt-mTOR signaling in both drug-resistant and drug-sensitive UMUC-3 sublines. In contrast, mTOR and Raptor were activated by VPA in the drug-sensitive RT112par cells, and pRictor was in fact enhanced in both RT112par and RT112res. Data provided in the literature are also inconsistent. Suppression of the Akt/mTOR signal pathway in prostate cancer cell lines by VPA has been documented by Xia et al. [65], whereas others observed elevated Akt activation by VPA in the same culture system [13]. Based on in vivo rat models, VPA exposure decreased pmTOR in one experimental approach [66] but increased pmTOR in the other approach [67]. Two scenarios should be considered when interpreting the results. Activation of Akt-mTOR might point to resistance development towards VPA. In fact, we recently not only provided evidence of a diminished Akt content in VPA sensitive tumor-bearing animals, but also a massive accumulation of Akt in VPA non-responders [68]. Moreover, histone acetylation may activate Akt-mTOR signaling. Cross-communication has recently been observed in a prostate cancer cell line in a manner that enhanced histone H3 and H4 acetylation triggered elevated Akt-mTOR activity, particularly seen with pRictor [69]. The clinical relevance of this finding is not yet clear. VPA counteracted temsirolimus-driven resistance processes in two different bladder cancer cell lines. This property qualifies VPA as a highly valuable compound which may minimize the rapid onset of resistance induction caused by chronic suppression of mTOR. Since VPA may also (re)activate Akt-mTOR under certain circumstances, the question arises about the optimum treatment option. We have recently demonstrated that simultaneous targeting of both HDAC and mTOR delays the time to resistance towards the mTOR inhibitor [16]. With respect to these data, combined use of an HDAC and mTOR inhibitor might be superior to a regimen based on mTOR inhibition followed by HDAC blockade, once resistance towards the mTOR inhibitor has been developed. However, this assumption requires further evaluation. Preclinical evaluation of dual mTOR-HDAC inhibition in non-Hodgkin lymphoma cells showed that the mechanisms of effectiveness of both drugs were largely retained [70]. Patients with renal cell carcinoma experienced prolonged disease stabilization under co-administration of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in a phase I study [44]. In a similar protocol, simultaneous application of the mTOR inhibitor sirolimus and vorinostat led to stable disease in hepatocellular carcinoma patients [71].
In addition to impairing the tumor cell growth of the resistant cells, there is apparent evidence that the combined inhibition of HDAC and mTOR might also have an impact on the metastatic spread. This aspect is interesting, as advanced cancers and therapy resistance are accompanied by the occurrence of metastases. In non-small-cell lung cancer (NSCLC) combined HDAC and mTOR inhibition resulted in a synergistic decrease of migration and invasion in vitro and diminished metastasis rates in vivo [72]. Notably, the effect of HDAC blockade on the metastatic properties has also been demonstrated on osteosarcoma cells [73]. Whether treatment with an HDAC inhibitor may also modulate the metastatic potential of temsirolimus-resistant bladder cancer cells is as yet speculative. The results of ongoing studies in our group will shed light on this aspect. From our present data, we postulate that VPA might reverse bladder cancer cell therapy resistance to temsirolimus by at least partially blocking the cdk2/cyclin A axis. Further investigations should evaluate the effect of concomitant HDAC and mTOR inhibition in bladder cancer cells.
MATERIALS AND METHODS
Cell cultures and treatment
RT112 and UMUC-3 (ATCC/LGC Promochem GmbH, Wesel, Germany) bladder carcinoma cells were grown and cultured in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 20 mmol HEPES buffer, 1% glutamax and 1% penicillin/streptomycin (all: Gibco/Invitrogen; Karlsruhe, Germany) in a humidified, 5% CO2 incubator. RT112 is an invasive (pathological stage T2) moderately differentiated (grade 2/3) model of human bladder cancer, UMUC-3 a high grade 3 invasive bladder cancer. In all experiments, treated to non-treated tumor cell cultures were compared. Resistance towards temsirolimus was induced by treating tumor cells with stepwise ascending concentrations from 1 nmol/ml up to 1 μmol/ml. The tumor cells were further exposed to 1 μmol/ml temsirolimus three times a week for over one year. Tumor cells, resistant to temsirolimus, were designated UMUC3res and RT112res. The parental control cells are named UMUC3par and RT112par. Valproic acid (VPA) (G. L. Pharma GmbH, Lannach, Austria) was applied at a final concentration of 1 mmol/ml to the cells for 1-3 days. Control cell cultures remained untreated. To examine toxic effects of amygdalin, cell viability was determined by trypan blue (Gibco/Invitrogen).
Measurement of tumor cell growth, proliferation and apoptosis
Cell growth was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay (Roche Diagnostics, Penzberg, Germany). Bladder cancer cells (50 μl, 1 × 105 cells/ml) were seeded onto 96-well tissue culture plates. After 24, 48 and 72 h, 10 μl MTT (0.5 mg/ml) were added for an additional 4 h. Thereafter, cells were lysed in a buffer containing 10% SDS in 0.01 M HCl. The plates were incubated overnight at 37°C, 5% CO2. Absorbance at 550 nm was determined for each well using a microplate enzyme-linked immunosorbent assay (ELISA) reader. After subtracting background absorbance, results were expressed as mean cell number.
Cell proliferation was measured using a BrdU cell proliferation enzyme-linked immunosorbent assay (ELISA) kit (Calbiochem/Merck Biosciences, Darmstadt, Germany). Tumor cells (50 μl, 1 × 105 cells/ml), seeded onto 96-well plates, were incubated with 20 μl BrdU-labeling solution per well for 8 h, fixed and detected using anti-BrdU mAb according to the manufacturer’s instructions. Absorbance was measured at 450 nm using a microplate ELISA reader.
To evaluate whether tumor cell growth was impaired or reduced due to apoptosis, the expression of Annexin V/propidium iodide (PI) was evaluated using the Annexin V-FITC Apoptosis Detection kit (BD Pharmingen, Heidelberg, Germany). Tumor cells were washed twice with PBS, and then incubated with 5 μl of Annexin V-FITC and 5 μl of PI in the dark for 15 min at RT. Cells were analyzed by flow cytometry using FACScalibur (BD Biosciences, Heidelberg, Germany). The percentage of apoptotic (early and late), necrotic and vital cells in each quadrant was calculated using CellQuest software (BD Biosciences).
Percentage of cells in different cell cycle phases
Cell cycle analysis was carried out on subconfluent cell cultures. Tumor cell populations were stained with PI, using a Cycle TEST PLUS DNA Reagent Kit (Becton Dickinson, Heidelberg, Germany) and then subjected to flow cytometry using FACScan (Becton Dickinson). 10,000 events were collected from each sample. Data acquisition was carried out using CellQuest software and cell cycle distribution was calculated using the ModFit software (Becton Dickinson). The number of gated cells in G1, G2/M or S-phase was expressed as %.
Expression of cell cycle regulating proteins
Cell cycle regulating proteins were investigated by Western blot. Tumor cell lysates were applied to a 7%–15% polyacrylamide gel (depending on protein size) and electrophoresed for 90 min at 100 V. The protein was then transferred to nitrocellulose membranes (1 h, 100 V). After blocking with non-fat dry milk for 1h, the membranes were incubated overnight with monoclonal antibodies directed against the following cell cycle proteins, all from BD Biosciences, Heidelberg, Germany: Cdk1 (IgG1, clone 1, dilution 1:2,500), cdk2 (IgG2a, clone 55, dilution 1:2,500), cdk4 (IgG1, clone 97, dilution 1:250), cyclin A (IgG1, clone 25, dilution 1:250), cyclin B (IgG1, clone 18, dilution 1:1,000), cyclin D1 (IgG1, clone G124-326, dilution 1:250), Skp1 p19 (IgG1, clone 52/p19, dilution 1:5,000), p27 (IgG1, clone 57, dilution 1:500). HRP-conjugated goat anti-mouse IgG (Upstate Biotechnology, Lake Placid, NY, USA; dilution 1:5,000) served as the secondary antibody. The membranes were briefly incubated with ECL detection reagent (ECLTM, Amersham/GE Healthcare, Munich, Germany) to visualize the proteins and then analyzed by the Fusion FX7 system (Peqlab, Erlangen, Germany). ß-actin (1:1,000; Sigma, Taufenkirchen, Germany) served as internal control.
Expression of targeted proteins
To evaluate target specificity of temsirolimus and VPA, mTOR signaling and histone acetylation were evaluated. The following monoclonal antibodies were employed to determine mTOR signaling: anti-phospho Akt (pAkt; clone 104A282, both: mouse IgG1, dilution 1:500, BD Biosciences), anti-phospho mTOR (pmTOR; IgG, Ser2448, clone D9C2, dilution 1:1,000), anti-phospho rictor (pRictor; IgG, Thr1135, D30A3, dilution 1:1,000), anti-phospho raptor (IgG, Ser792; both: MerckMillipore, dilution 1:1,000) and anti-phospho p70S6k (pp70S6k; clone 108D2, dilution 1:1,000, New England Biolabs). To investigate histone acetylation, cell lysates were marked with anti-acetylated histone H3 (rabbit IgG, clone Y28, dilution 1:500, Epitomics, USA) and anti-acetylated histone H4 (Lys8, rabbit IgG, dilution 1:500, Upstate Biotechnology, USA). HRP-conjugated goat anti-mouse or goat anti-rabbit IgG (both: dilution 1:5,000, Upstate Biotechnology, Lake Placid, NY, USA) were used as secondary antibodies. The membranes were briefly incubated with ECL detection reagent (ECL™, Amersham/GE Healthcare, Munich, Germany) to visualize the proteins and then analyzed by the Fusion FX7 system (Peqlab, Erlangen, Germany). β-actin (dilution 1:1,000, Sigma, Taufenkirchen, Germany) served as the internal control.
Blocking studies
Since cdk2 and cyclin A revealed distinct down-regulation by VPA they might be responsible for growth inhibition induced by VPA. To determine whether cdk2 and cyclin A have an impact on growth of the used tumor cell lines, both proteins were blocked. Therefore, tumor cells (3 × 105/6-well) were transfected with small interfering RNA (siRNA) directed against cdk2 (gene ID: 1017, target sequence: AGGTGGTGGCGCTTAAGAAAA) or cyclin A (gene ID: 890, target sequence: GCCAGCTGTCAGGATAATAAA) with a siRNA/transfection reagent (HiPerFect Transfection Reagent; Qiagen, Hilden, Germany) ratio of 1:6. Untreated cells and cells treated with 5 nmol control siRNA (All stars negative control siRNA; Qiagen, Hilden, Germany) served as controls. Knock-down was verified by western blot analysis. Tumor cell growth was analyzed by the MTT assay as indicated above.
Statistics
All experiments were performed 3-6 times. Statistical significance was determined by the Wilcoxon–Mann-Whitney-U-test. Differences were considered statistically significant at a p-value less than 0.05.
Author contributions
All authors of this research paper have directly participated in conception and design, acquisition of data or analysis and interpretation of data. EJ, and RB are responsible for conception and design of the study. RN, JR, SM, JM contributed substantially to the acquisition of data. FR, IT, EJ and RB analyzed and interpreted the data. AH and RB revised the article for important content and finally approved the manuscript submitted.
CONFLICTS OF INTEREST
None.
FUNDING
This work was supported by the “Prof. Dr. Karl und Gerhard Schiller-Stiftung” and “Friedrich-Ebert-Stiftung”.
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9–29.
2. American Cancer Society. Cancer Facts & Figures. Atlanta, GA: American Cancer Society; 2016.
3. Morales-Barrera R, Suárez C, de Castro AM, Racca F, Valverde C, Maldonado X, Bastaros JM, Morote J, Carles J. Targeting fibroblast growth factor receptors and immune checkpoint inhibitors for the treatment of advanced bladder cancer: New direction and New Hope. Cancer Treat Rev. 2016; 50:208–216.
4. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359–86.
5. Smolensky D, Rathore K, Cekanova M. Molecular targets in urothelial cancer: detection, treatment, and animal models of bladder cancer. Drug Des Devel Ther. 2016; 10:3305–3322.
6. Iyer G, Al-Ahmadie H, Schultz N, Hanrahan AJ, Ostrovnaya I, Balar AV, Kim PH, Lin O, Weinhold N, Sander C, Zabor EC, Janakiraman M, Garcia-Grossman IR, et al. Prevalence and co-occurrence of actionable genomic alterations in high-grade bladder cancer. J Clin Oncol. 2013; 31:3133–40.
7. Park SJ, Lee TJ, Chang IH. Role of the mTOR Pathway in the Progression and Recurrence of Bladder Cancer: An Immunohistochemical Tissue Microarray Study. Korean J Urol. 2011; 52:466–73.
8. Sun CH, Chang YH, Pan CC. Activation of the PI3K/Akt/mTOR pathway correlates with tumour progression and reduced survival in patients with urothelial carcinoma of the urinary bladder. Histopathology. 2011; 58:1054–63.
9. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507:315–22.
10. Pinto-Leite R, Arantes-Rodrigues R, Sousa N, Oliveira PA, Santos L. mTOR inhibitors in urinary bladder cancer. Tumour Biol. 2016; 37:11541–11551.
11. Seront E, Rottey S, Sautois B, Kerger J, D'Hondt LA, Verschaeve V, Canon JL, Dopchie C, Vandenbulcke JM, Whenham N, Goeminne JC, Clausse M, Verhoeven D, et al. Phase II study of everolimus in patients with locally advanced or metastatic transitional cell carcinoma of the urothelial tract: clinical activity, molecular response, and biomarkers. Ann Oncol. 2012; 23:2663–70.
12. Siska PJ, Beckermann KE, Rathmell WK, Haake SM. Strategies to overcome therapeutic resistance in renal cell carcinoma. Urol Oncol. 2017; 35:102–110.
13. Wedel S, Hudak L, Seibel JM, Makarević J, Juengel E, Tsaur I, Wiesner C, Haferkamp A, Blaheta RA. Impact of combined HDAC and mTOR inhibition on adhesion, migration and invasion of prostate cancer cells. Clin Exp Metastasis. 2011; 28:479–91.
14. Juengel E, Maxeiner S, Rutz J, Justin S, Roos F, Khoder W, Tsaur I, Nelson K, Bechstein WO, Haferkamp A, Blaheta RA. Sulforaphane inhibits proliferation and invasive activity of everolimus-resistant kidney cancer cells in vitro. Oncotarget. 2016; 7:85208–85219. https://doi.org/10.18632/oncotarget.13421.
15. Juengel E, Nowaz S, Makarevi J, Natsheh I, Werner I, Nelson K, Reiter M, Tsaur I, Mani J, Harder S, Bartsch G, Haferkamp A, Blaheta RA. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol Cancer. 2014; 13:152.
16. Juengel E, Dauselt A, Makarević J, Wiesner C, Tsaur I, Bartsch G, Haferkamp A, Blaheta RA. Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal cell carcinoma cells. Cancer Lett. 2012; 324:83–90.
17. Tang HM, Kuay KT, Koh PF, Asad M, Tan TZ, Chung VY, Lee SC, Thiery JP, Huang RJ. An epithelial marker promoter induction screen identifies histone deacetylase inhibitors to restore epithelial differentiation and abolishes anchorage independence growth in cancers. Cell Death Discov. 2016; 2:16041.
18. Qian C, Lai CJ, Bao R, Wang DG, Wang J, Xu GX, Atoyan R, Qu H, Yin L, Samson M, Zifcak B, Ma AW, DellaRocca S, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res. 2012; 18:4104–13.
19. Beagle BR, Nguyen DM, Mallya S, Tang SS, Lu M, Zeng Z, Konopleva M, Vo TT, Fruman DA. mTOR kinase inhibitors synergize with histone deacetylase inhibitors to kill B-cell acute lymphoblastic leukemia cells. Oncotarget. 2015; 6:2088–100. https://doi.org/10.18632/oncotarget.2992.
20. Booth L, Roberts JL, Sander C, Lee J, Kirkwood JM, Poklepovic A, Dent P. The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget. 2017; 8:16367–16386. https://doi.org/10.18632/oncotarget.14829.
21. Wysocki PJ. mTOR in renal cell cancer: modulator of tumor biology and therapeutic target. Expert Rev Mol Diagn. 2009; 9:231–41.
22. Koo J, Wang X, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY. GSK3 is required for rapalogs to induce degradation of some oncogenic proteins and to suppress cancer cell growth. Oncotarget. 2015; 6:8974–87. https://doi.org/10.18632/oncotarget.3291.
23. Li A, Fan S, Xu Y, Meng J, Shen X, Mao J, Zhang L, Zhang X, Moeckel G, Wu D, Wu G, Liang C. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J Cell Mol Med. 2017; 21:1619–1635.
24. Sarsik B, Doganavsargil B, Simsir A, Yazici A, Pehlivanoglu B, Cal C, Sen S. p21 and p27 Immunoexpression in Upper Urinary Tract Urothelial Carcinomas. Pathol Oncol Res. 2016; 22:839–45.
25. Kim K, Sung CO, Park BH, Ku JY, Go H, Ro JY, Kim J, Cho YM. Immunoprofile-based subgrouping of urothelial bladder carcinomas for survival prediction. Hum Pathol. 2015; 46:1464–70.
26. Lotan Y, Bagrodia A, Passoni N, Rachakonda V, Kapur P, Arriaga Y, Bolenz C, Margulis V, Raj GV, Sagalowsky AI, Shariat SF. Prospective evaluation of a molecular marker panel for prediction of recurrence and cancer-specific survival after radical cystectomy. Eur Urol. 2013; 64:465–71.
27. Kopsiaftis S, Sullivan KL, Garg I, Taylor JA, Claffey KP. AMPKα2 Regulates Bladder Cancer Growth through SKP2-Mediated Degradation of p27. Mol Cancer Res. 2016; 14:1182–1194.
28. Davaadelger B, Duan L, Perez RE, Gitelis S, Maki CG. Crosstalk between the IGF-1R/AKT/mTORC1 pathway and the tumor suppressors p53 and p27 determines cisplatin sensitivity and limits the effectiveness of an IGF-1R pathway inhibitor. Oncotarget. 2016; 7:27511–26. https://doi.org/10.18632/oncotarget.8484.
29. Cao QF, Qian SB, Wang N, Zhang L, Wang WM, Shen HB. TRPM2 mediates histone deacetylase inhibition-induced apoptosis in bladder cancer cells. Cancer Biother Radiopharm. 2015; 30:87–93.
30. Vallo S, Xi W, Hudak L, Juengel E, Tsaur I, Wiesner C, Haferkamp A, Blaheta RA. HDAC inhibition delays cell cycle progression of human bladder cancer cells in vitro. Anticancer Drugs. 2011; 22:1002–9.
31. Juengel E, Meyer dos Santos S, Schneider T, Makarevic J, Hudak L, Bartsch G, Haferkamp A, Wiesner C, Blaheta RA. HDAC inhibition suppresses bladder cancer cell adhesion to collagen under flow conditions. Exp Biol Med (Maywood). 2013; 238:1297–304.
32. Xu XS, Wang L, Abrams J, Wang G. Histone deacetylases (HDACs) in XPC gene silencing and bladder cancer. J Hematol Oncol. 2011; 4:17.
33. Qi Z, Liu M, Liu Y, Zhang M, Yang G. Tetramethoxychalcone, a chalcone derivative, suppresses proliferation, blocks cell cycle progression, and induces apoptosis of human ovarian cancer cells. PLoS One. 2014; 9:e106206.
34. Chen SY, Zheng XW, Cai JX, Zhang WP, You HS, Xing JF, Dong YL. Histone deacetylase inhibitor reverses multidrug resistance by attenuating the nucleophosmin level through PI3K/Akt pathway in breast cancer. Oncol. 2016; 49:294–304.
35. Wang L, Li H, Ren Y, Zou S, Fang W, Jiang X, Jia L, Li M, Liu X, Yuan X, Chen G, Yang J, Wu C. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. 2016; 7:e2063.
36. Lee HS, Park SB, Kim SA, Kwon SK, Cha H, Lee DY, Ro S, Cho JM, Song SY. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci Rep. 2017; 7:41615.
37. Li ZY, Li QZ, Chen L, Chen BD, Wang B, Zhang XJ, Li WP. Histone Deacetylase Inhibitor RGFP109 Overcomes Temozolomide Resistance by Blocking NF-κB-Dependent Transcription in Glioblastoma Cell Lines. Neurochem Res. 2016; 41:3192–3205.
38. Wang J, Yang T, Xu G, Liu H, Ren C, Xie W, Wang M. Cyclin-Dependent Kinase 2 Promotes Tumor Proliferation and Induces Radio Resistance in Glioblastoma. Transl Oncol. 2016; 9:548–556.
39. Braunstein M, Liao L, Lyttle N, Lobo N, Taylor KJ, Krzyzanowski PM, Kalatskaya I, Yao CQ, Stein LD, Boutros PC, Twelves CJ, Marcellus R, Bartlett JM, Spears M. Downregulation of histone H2A and H2B pathways is associated with anthracycline sensitivity in breast cancer. Breast Cancer Res. 2016; 18:16.
40. Tanimoto A, Takeuchi S, Arai S, Fukuda K, Yamada T, Roca X, Ong ST, Yano S. Histone Deacetylase 3 Inhibition Overcomes BIM Deletion Polymorphism-Mediated Osimertinib Resistance in EGFR-Mutant Lung Cancer. Clin Cancer Res. 2017; 23:3139–3149.
41. Damaskos C, Valsami S, Kontos M, Spartalis E, Kalampokas T, Kalampokas E, Athanasiou A, Moris D, Daskalopoulou A, Davakis S, Tsourouflis G, Kontzoglou K, Perrea D, et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017; 37:35–46.
42. Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee MJ, Trepel JB. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013; 31:2128–35.
43. Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M, Minton SE. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011; 104:1828–35.
44. Zibelman M, Wong YN, Devarajan K, Malizzia L, Corrigan A, Olszanski AJ, Denlinger CS, Roethke SK, Tetzlaff CH, Plimack ER. Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Invest New Drugs. 2015; 33:1040–7.
45. Sang Z, Sun Y, Ruan H, Cheng Y, Ding X, Yu Y. Anticancer effects of valproic acid on oral squamous cell carcinoma via SUMOylation in vivo and in vitro. Exp Ther Med. 2016; 12:3979–3987.
46. Takai N, Kawamata N, Gui D, Said JW, Miyakawa I, Koeffler HP. Human ovarian carcinoma cells: histone deacetylase inhibitors exhibit antiproliferative activity and potently induce apoptosis. Cancer. 2004; 101:2760–70.
47. Takai N, Desmond JC, Kumagai T, Gui D, Said JW, Whittaker S, Miyakawa I, Koeffler HP. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin Cancer Res. 2004; 10:1141–9.
48. Tsaur I, Hudak L, Makarević J, Juengel E, Mani J, Borgmann H, Gust KM, Schilling D, Bartsch G, Nelson K, Haferkamp A, Blaheta RA. Intensified antineoplastic effect by combining an HDAC-inhibitor, an mTOR-inhibitor and low dosed interferon alpha in prostate cancer cells. J Cell Mol Med. 2015; 19:1795–804.
49. Jones J, Juengel E, Mickuckyte A, Hudak L, Wedel S, Jonas D, Blaheta RA. The histone deacetylase inhibitor valproic acid alters growth properties of renal cell carcinoma in vitro and in vivo. J Cell Mol Med. 2009; 13:2376–85.
50. Chen M, Ye Y, Zou B, Guo S, Zhou F, Lu K, Liu J, Xu Z, Han H, Liu Z, Li Y, Yao K, Liu C, Qin Z. C14orf166 is a high-risk biomarker for bladder cancer and promotes bladder cancer cell proliferation. J Transl Med. 2016; 14:55.
51. Zhang Q, Zhuang J, Deng Y, Zhao X, Tang B, Yao D, Zhao W, Chang C, Lu Q, Chen W, Zhang S, Ji C, Cao L, Guo H. GOLPH3 is a potential therapeutic target and a prognostic indicator of poor survival in bladder cancer treated by cystectomy. Oncotarget. 2015; 6:32177–92. https://doi.org/10.18632/oncotarget.4867.
52. Fahmy M, Mansure JJ, Brimo F, Yafi FA, Segal R, Althunayan A, Hicks J, Meeker A, Netto G, Kassouf W. Relevance of the mammalian target of rapamycin pathway in the prognosis of patients with high-risk non-muscle invasive bladder cancer. Hum Pathol. 2013; 44:1766–72.
53. Kapur P, Lotan Y, King E, Kabbani W, Mitra AP, Mosbah A, Abol-Enein H, Ghoneim M, Youssef RF. Primary adenocarcinoma of the urinary bladder: value of cell cycle biomarkers. Am J Clin Pathol. 2011; 135:822–30.
54. Huang Y, Wu R, Su ZY, Guo Y, Zheng X, Yang CS, Kong AN. A naturally occurring mixture of tocotrienols inhibits the growth of human prostate tumor, associated with epigenetic modifications of cyclin-dependent kinase inhibitors p21 and p27. J Nutr Biochem. 2017; 40:155–163.
55. Byun SW, Chang YJ, Chung IS, Moss SF, Kim SS. Helicobacter pylori decreases p27 expression through the delta opioid receptor-mediated inhibition of histone acetylation within the p27 promoter. Cancer Lett. 2012; 326:96–104.
56. Uehara N, Yoshizawa K, Tsubura A. Vorinostat enhances protein stability of p27 and p21 through negative regulation of Skp2 and Cks1 in human breast cancer cells. Oncol Rep. 2012; 28:105–10.
57. Lin F, Lin P, Zhao D, Chen Y, Xiao L, Qin W, Li D, Chen H, Zhao B, Zou H, Zheng X, Yu X. Sox2 targets cyclinE, p27 and survivin to regulate androgen-independent human prostate cancer cell proliferation and apoptosis. Cell Prolif. 2012; 45:207–16.
58. Munari E, Chaux A, Maldonado L, Compérat E, Varinot J, Bivalacqua TJ, Hoque MO, Netto GJ. Cyclin A1 expression predicts progression in pT1 urothelial carcinoma of bladder: a tissue microarray study of 149 patients treated by transurethral resection. Histopathology. 2015; 66:262–9.
59. Liang PI, Li WM, Wang YH, Wu TF, Wu WR, Liao AC, Shen KH, Wei YC, Hsing CH, Shiue YL, Huang HY, Hsu HP, Chen LT, et al. HuR cytoplasmic expression is associated with increased cyclin A expression and poor outcome with upper urinary tract urothelial carcinoma. BMC Cancer. 2012; 12:611.
60. Akli S, Zhang XQ, Bondaruk J, Tucker SL, Czerniak PB, Benedict WF, Keyomarsi K. Low molecular weight cyclin E is associated with p27-resistant, high-grade, high-stage and invasive bladder cancer. Cell Cycle. 2012; 11:1468–76.
61. Ou TT, Wang CJ, Lee YS, Wu CH, Lee HJ. Gallic acid induces G2/M phase cell cycle arrest via regulating 14-3-3β release from Cdc25C and Chk2 activation in human bladder transitional carcinoma cells. Mol Nutr Food Res. 2010; 54:1781–90.
62. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74.
63. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017; 173:83–105.
64. Wang D, Jing Y, Ouyang S, Liu B, Zhu T, Niu H, Tian Y. Inhibitory effect of valproic acid on bladder cancer in combination with chemotherapeutic agents in vitro and in vivo. Oncol Lett. 2013; 6:1492–1498.
65. Xia Q, Zheng Y, Jiang W, Huang Z, Wang M, Rodriguez R, Jin X. Valproic acid induces autophagy by suppressing the Akt/mTOR pathway in human prostate cancer cells. Oncol Lett. 2016; 12:1826–1832.
66. Nicolini C, Ahn Y, Michalski B, Rho JM, Fahnestock M. Decreased mTOR signaling pathway in human idiopathic autism and in rats exposed to valproic acid. Acta Neuropathol Commun. 2015; 3:3.
67. Qin L, Dai X, Yin Y. Valproic acid exposure sequentially activates Wnt and mTOR pathways in rats. Mol Cell Neurosci. 2016; 75:27–35.
68. Juengel E, Makarević J, Tsaur I, Bartsch G, Nelson K, Haferkamp A, Blaheta RA. Resistance after chronic application of the HDAC-inhibitor valproic acid is associated with elevated Akt activation in renal cell carcinoma in vivo. PLoS One. 2013; 8:e53100.
69. Makarević J, Tawanaie N, Juengel E, Reiter M, Mani J, Tsaur I, Bartsch G, Haferkamp A, Blaheta RA. Cross-communication between histone H3 and H4 acetylation and Akt-mTOR signalling in prostate cancer cells. J Cell Mol Med. 2014; 18:1460–6.
70. Bianchi G, Ghobrial IM. Team work matters: dual inhibition puts non-hodgkin lymphoma under siege. Clin Cancer Res. 2014; 20:5863–5.
71. Park H, Garrido-Laguna I, Naing A, Fu S, Falchook GS, Piha-Paul SA, Wheler JJ, Hong DS, Tsimberidou AM, Subbiah V, Zinner RG, Kaseb AO, Patel S, et al. Phase I dose-escalation study of the mTOR inhibitor sirolimus and the HDAC inhibitor vorinostat in patients with advanced malignancy. Oncotarget. 2016; 7:67521–67531. https://doi.org/10.18632/oncotarget.11750.
72. Piao J, Chen L, Quan T, Li L, Quan C, Piao Y, Jin T, Lin Z. Superior efficacy of co-treatment with the dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A against NSCLC. Oncotarget. 2016; 7:60169–60180. https://doi.org/10.18632/oncotarget.11109.
73. Mu X, Brynien D, Weiss KR. The HDAC inhibitor Vorinostat diminishes the in vitro metastatic behavior of Osteosarcoma cells. Biomed Res Int. 2015; 2015:290368.