
INTRODUCTION
Over the past decades, the prognosis of osteosarcoma (OS) has been significantly improved [1–6]. Significant advances in OS early diagnosis and treatments, including surgery, radiotherapy, and chemotherapy, have been achieved [1, 5, 7–9]. Yet, the overall survival has reached a platform [10–13]. Further, the prognosis of patients with recurrent and metastatic OS is poor, but the incidence of OS is rising at a rate of 1.4% per year [11–13]. OS has become one leading cause of cancer-related mortalities among children and teenagers [10–13]. Therefore, it is needed to identify reliable oncotarget proteins for early diagnosis and treatment of OS [5, 14, 15].
The cellular sphingolipid signaling is involved in tumorigenesis and cancer progression [16–19]. Production of pro-apoptotic sphingolipids, i.e. ceramide, will potently inhibit cancer cells. Reversely, anti-apoptotic sphingolipid sphingosine-1-phosphate (S1P) will promote cancer cell survival and growth [20–22]. The balance of these sphingolipids is tightly controlled by sphingosine kinase (SphK) [20]. Over-activation or upregulation of SphK would lead to increased conversion of ceramide to S1P, causing aberrant cell growth [20, 23]. At least two SphKs have been identified, including the well-studied SphK1 and the less-known SphK2 [20]. Recent studies have proposed that SphK2 is over-expressed in many cancer cells [24–26]. Its expression and potential functions in human OS were tested in this study.
RESULTS
Over-expression of SphK2 in human OS tissues and OS cells
First, we tested the expression of SphK2 in human OS tissues. A total of ten OS tissues (“Tum”) and their surrounding normal bone tissues (“Nor”) were examined. Quantitatively real-time PCR (“qRT-PCR”) assay results showed that SphK2 mRNA expression in the OS tissues was significantly higher than that in the bone tissues (Figure 1A). SphK2 mRNA in tumor tissues was about 4-5 times higher than the normal tissues (Figure 1A). Meanwhile, SphK2 protein was also upregulated in OS tissues (the quantified results in Figure 1B), and its expression was relative low in normal tissues (Figure 1B).
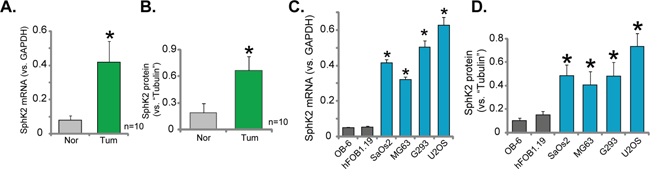
Figure 1: Over-expression of SphK2 in human OS tissues and OS cells. mRNA (A and C) and protein (B and D) expression of SphK2 in OS tissues (“Tum”) and the surrounding normal bone tissues (“Nor”), as well as in human OS cells (MG63, SaOs2, G293 and U2OS lines) and human osteoblastic cells (OB-6 and hFOB1.19 lines), were shown. Relative SphK2 protein expression (vs. Tubulin, B and D) was quantified. *p<0.05 vs. “Nor” tissues (A-B). *p<0.05 vs. OB-6 cells (C-D).
SphK2 expression in human OS cells was also tested. Four well-established human OS cell lines, MG63, SaOs2, G293 and U2OS, as well as two lines of human osteoblastic cells (OB-6 and hFOB1.19) were tested. qRT-PCR assay results in Figure 1C confirmed that SphK2 mRNA level in the OS cells was significantly higher than that in the osteoblastic cells. Consequently, SphK2 protein expression was elevated in OS cells (Data were quantified, Figure 1D). Among all the tested OS cell lines, SphK2 expression was highest in U2OS cells (Figure 1C and 1D), this cell line was selected for further studies. Together, these results clearly demonstrate that SphK2 is over-expressed in human OS tissues and OS cells.
SphK2 knockdown inhibits OS cell growth
To study the potential function of SphK2 in OS cells, we utilized shRNA method to knockdown SphK2 in U2OS cells. As discussed, a total of eight (8) distinct lentiviral shRNAs, against non-overlapping sequence of human SphK2, were designed. Of which, four of them efficiently silenced SphK2 in U2OS cells, these four shRNAs were named as SphK2 shRNA (“-1/-4”). As demonstrated, SphK2 mRNA level was significantly downregulated in the stable U2OS cells expressing the SphK2 shRNAs (Figure 2A). Consequently, SphK2 protein was also silenced (Figure 2B, data was quantified). Remarkably, the growth of U2OS cells was significantly inhibited after SphK2 knockdown (Figure 2C and 2D), as the number of viable cells (at 96 hour, Figure 2C) and colonies (at Day-8, Figure 2D) were both significantly decreased after expressing SphK2 shRNAs. Notably, scramble non-sense control shRNA (“c-sh”) showed no effect on SphK2 expression (Figure 2A-2B) nor U2OS cell growth (Figure 2C-2D). Thus, SphK2 knockdown by targeted shRNAs inhibited U2OS cell growth.
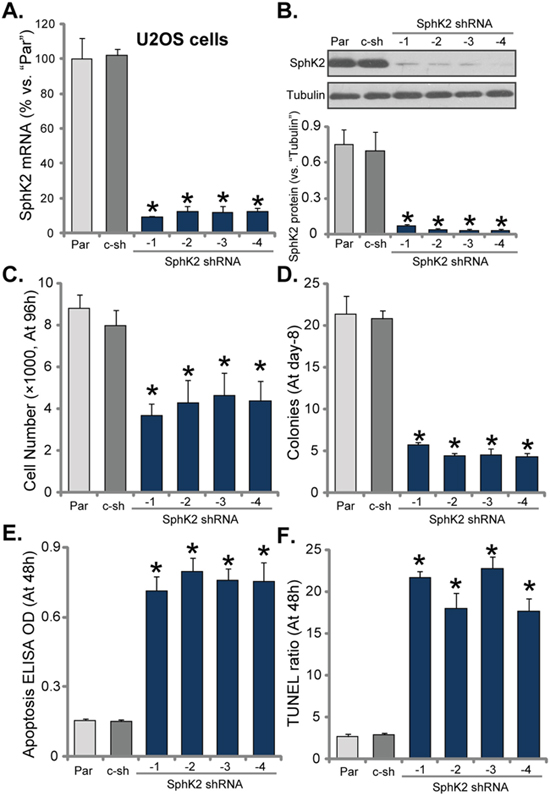
Figure 2: SphK2 knockdown inhibits OS cell growth. mRNA (A) and protein (B) expression of SphK2 in stable U2OS cells, expressing SphK2 shRNA (“-1 to -4”) or scramble non-sense control shRNA (“c-sh”), as well as in parental control U2OS cells (“Par”), were shown. Cells were also subjected to cell counting assay (C) and colony formation assay (D) to test cell growth; Cell apoptosis was examined by DNA apoptosis ELISA OD (E) and TUNEL assay (F). For each assay, exact same amount of viable cells of different genetic modification were plated initially (Same for all Figures). Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “Par” cells. Experiments in this figure were repeated three times, and similar results were obtained.
Apoptosis induction could be a main reason of growth inhibition in OS cells. Next, Histone DNA apoptosis ELISA assay and TUNEL staining assay were performed to test the potential effect of SphK2 shRNA on cell apoptosis. DNA denaturation occurs in the apoptotic cells, which binds to histone. Thus, testing histone-bound DNA could be an efficient way to examine cell apoptosis [27]. TUNEL nuclei staining assay, which tests DNA fragmentation in nuclei, is a well-established apoptosis assay [28]. As demonstrated, Histone DNA apoptosis ELISA optic density (OD, Figure 2E) and the TUNEL-nuclei percentage (Figure 2F) were both increased after expressing SphK2 shRNA in U2OS cells, suggesting that SphK2 shRNA induced apoptosis in U2OS cells. The control shRNA was again in-effective (Figure 2E and 2F). Similar results were also obtained in MG63 OS cells, where knockdown of SphK2 by targeted shRNA (“-1”) (Supplementary Figure 1A) inhibited cell growth (Supplementary Figure 1B), but induced cell apoptosis (Supplementary Figure 1C).
Exogenous over-expression of SphK2 promotes OS cell growth
To further confirm the potential function of SphK2 in promoting OS cell growth, the SphK2-expression vector (Flag-tagged, see Methods) was established. The construct was transfected to U2OS cells. Via puromycin selection, two stable U2OS cell lines expressing the construct were established, which were named as “SphK2-cDNA-L1/L2”. qRT-PCR assay results in Figure 3A demonstrated the significant upregulation of SphK2 mRNA in the stable cells with the construct. Furthermore, Western blotting assay results confirmed the expression of exogenous SphK2 (“Flag-tagged”) in the stable cells (Figure 3B). Significantly, U2OS cell growth, tested by the simple cell counting assay (Figure 3C), was facilitated after SphK2 over-expression. Similar results were also obtained in MG63 cells, where over-expression of SphK2 (Supplementary Figure 1D) facilitated cancer cell growth (Supplementary Figure 1E). The empty vector (pSuper-puro-Flag) had no significant effect on SphK2 expression (Figure 3A and 3B) and cell growth (Figure 3C). Together, these evidences conclude that exogenous over-expression of SphK2 could further promote OS cell growth.
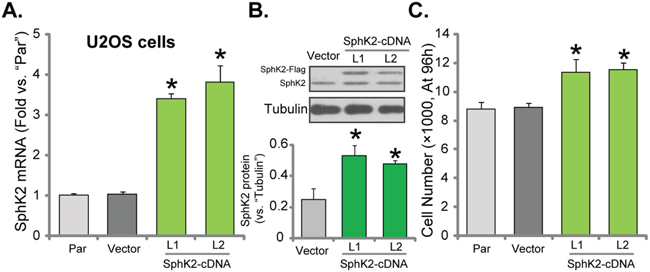
Figure 3: Exogenous over-expression of SphK2 promotes OS cell growth. mRNA (A) and protein (B) expression of SphK2 in stable U2OS cells expressing SphK2 cDNA (two lines, “SphK2-cDNA-L1/L2”) or the empty vector (pSuper-EGFP-puro-Flag), as well as in parental control cells (“Par”), were shown; Cells were also subjected to cell counting assay (C) to test cell growth. Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “Par” cells. Experiments in this figure were repeated three times, and similar results were obtained.
Expression of microRNA-19a silences SphK2 and inhibits OS cell growth
Above results have shown that SphK2 is over-expressed in OS cells, which is important for cell growth. The potential mechanism of SphK2 upregulation in OS cells was tested. Here we focused the possible mechanism of microRNA. Via search the “TargetScan” database, we discovered one potential anti-SphK2 microRNA: microRNA-19a-3p (“miR-19a-3p”). As demonstrated, miR-19a-3p putatively targets the 3’ UTR (untranslated region) of SphK2 mRNA (Figure 4A). Next, miR-19a-mimic oligonucleotides were transfected to U2OS cells, and stable cells (three lines, “L1-L3”) expressing miR-19a were established. qRT-PCR assay results in Figure 4B confirmed that miR-19a-3p level was significantly increased in all three stable lines with miR-19a-mimic. Remarkably, SphK2 mRNA and protein were both significantly downregulated after expressing miR-19a (Figure 4C). Thus, forced-expression of miR-19a-3p downregulated SphK2 in U2OS cells. Additionally, the SphK2 mRNA 3’-UTR luciferase activity was also largely inhibited in miR-19a-expressing U2OS cells, indicating that SphK2 should be the direct target of miR-19a.
Further studies showed that U2OS cell growth, tested by cell counting assay, was also inhibited after stably expressing miR-19a-3p (Figure 4E). On the other hand, apoptosis level was increased in the stable cells with miR-19a-3p (Figure 4F and 4G). In the miR-19a-expressing cells, Histone DNA ELISA OD (Figure 4F) and TUNEL percentage (Figure 4G) were both increased. Expression of miR-control (“miR-C”), expectably, didn’t change SphK2 expression (Figure 4C) nor cell growth (Figure 4E). Similar results were also observed in the MG-63 cells. Introduction of miR-19a-mimic significantly increased miR-19a-3p expression (Supplementary Figure 2A), which downregulated SphK2 protein and mRNA (Supplementary Figure 2B), causing growth inhibition (Supplementary Figure 2C) and cell apoptosis ((Supplementary Figure 2D). Thus, expression of miR-19a-3p downregulates SphK2 and inhibits OS cell growth.
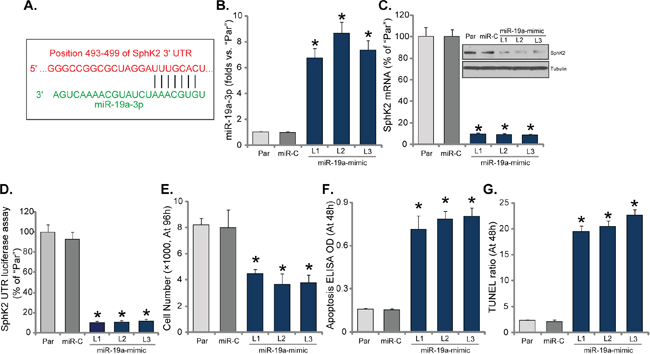
Figure 4: Expression of microRNA-19a silences SphK2 and inhibits OS cell growth. miR-19a-3p putatively targets the 3’ UTR of SphK2 mRNA (A). Expression of miR-19a-3p (B) and SphK2 (protein and mRNA, C), as well as the relative SphK2 mRNA 3’-UTR luciferase activity (D) in the stable USO2 cells with miR-19a-mimic (three lines, “L1-L3”) or miR-control (“miR-C”), and in the parental control cells (“Par”) were shown; Cells were also subjected to the cell counting assay (E) and the listed apoptosis assays (F and G). Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “Par” cells. Experiments in this figure were repeated three times, and similar results were obtained.
Downregulation of miR-19a-3p in human OS tissues and cells
Expression of miR-19a-3p in the above-mentioned human OS tissues (See Figure 1) was also tested. As demonstrated in Figure 5A, miR-19a-3p level was significantly lower in the OS tissues, as compared to the surrounding normal tissues. Further, miR-19a-3p level in all four lines of human OS cells (MG63, SaOs2, G293 and U2OS) was also lower than that in the osteoblastic cells (OB-6 and hFOB1.19 lines) (Figure 5B). These results confirmed miR-19a-3p downregulation in human OS tissues and cells.
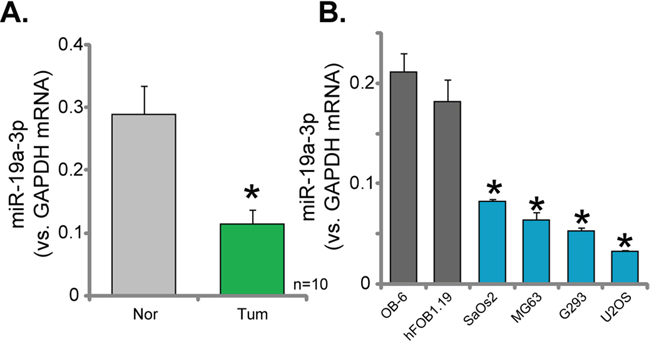
Figure 5: Downregulation of miR-19a-3p in human OS tissues and cells. RelativemiR-19a-3p expression (vs. GAPDH mRNA) in OS tissues (“Tum”) and surrounding normal bone tissues (“Nor”) (A), as well as in human OS cells (MG63, SaOs2, G293 and U2OS lines) and human osteoblastic cells (OB-6 and hFOB1.19 lines) (B), were shown. Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “Nor” tissues (A). *p<0.05 vs. OB-6 cells (B).
Silence of SphK2 inhibits U2OS tumor growth in nude mice
At last, the potential effect of SphK2 on OS cell growth in vivo was tested. Parental U2OS cells, as well as cells stably expressing SphK2-shRNA or miR-19a-3p, were injected s.c. to the nude mice. Xenografted U2OS tumors were established within 2-3 weeks. Weekly tumor growth was recorded. Estimated tumor growth curve results in Figure 6A demonstrated that U2OS tumors expressing SphK2-shRNA or miR-19a-3p grew significantly slower than the control tumors (from parental cells). The volumes of tumor expressing SphK2-shRNA or miR-19a-3p were much lower than the control tumors (Figure 6A). At the end of experiment (week-7), tumors of each group were isolated and weighted. Results in Figure 6B confirmed that SphK2-shRNA- or miR-19a-3p-expressing tumors were dramatically lighter than the control tumors. Notably, mice body weights were not significantly different between the groups (Figure 6C). When analyzing tumor tissue lysates, we showed that SphK2 expression was indeed downregulated in tumors expressing SphK2-shRNA or miR-19a-3p (Figure 6D, two sets). These results suggest that knockdown of SphK2, by targeted-shRNA or miR-19a-3p, inhibited U2OS tumor growth in nude mice.
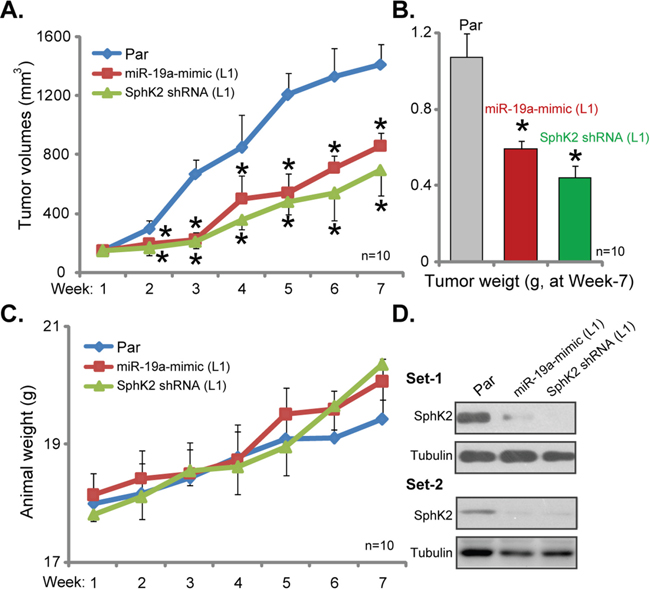
Figure 6: Silence of SphK2 inhibits U2OS tumor growth in nude mice. Parental U2OS cells (“Par”), as well as cells stably expressing SphK2-shRNA (“L1”) or miR-19a-3p (“L1”), were injected s.c. to the nude mice; Estimated tumor volume (A) and mice body weight (deducting tumor weight, C) were recorded weekly for total six weeks. At the end of experiment (week-7), tumors of the each group were isolated and weighted (B), expression of listed proteins in tumor lysates was shown (Two sets, D). *p<0.05 vs. “Par” tumors.
DISCUSSIONS
The oncogenic/pro-cancerous function of SphK1 has been well-established [23, 29, 30]. On the other hand, the potential function of SphK2 in human cancers has not been extensively studied until recently [24, 26, 31, 32]. Although early studies have proposed a possible pro-apoptotic/anti-cancer function by of SphK2 [33], emerging recent evidences have confirmed that SphK2, like SphK1, is also oncogenic [24, 26, 31, 32]. It is known that SphK2 could also enzymatically produce S1P from sphingosine, although its efficiency is less potent than SphK1 [26, 34]. Meanwhile, SphK2 activation can induce G1/S transition and promote cell proliferation [26, 34]. SphK2 is also shown to be important for oncogenic AKT activation and ERK2 expression. On the other hand, SphK2 knockdown resulted in AKT inactivation and ERK2 downregulation, causing proliferation and migration suppression [34]. SphK2 is important for MYC expression to promote acute lymphoblastic leukemia progression [35]. Over-expression of SphK2 is associated with multi-drug resistance [26, 36]. ABC294640, a selective SphK2 inhibitor, has anti-proliferative and antitumor activity in a variety of cellular and animal models [31, 37, 38]. The SphK2 inhibitor is now under phase II clinical trials [31, 32, 38–42]. Thus, targeting SphK2 should have therapeutic potential for treating cancer [24, 26, 31, 35, 43].
The preclinical evidences of this study suggested that SphK2 is possibly a rational therapeutic target for human OS. First, SphK2 expression is upregulated in multiple human OS tissues, as well as in OS cell lines. Its level was however quite low in normal bone tissues and in human osteoblastic cells. Second, stable knockdown of SphK2 by targeted-shRNAs largely inhibited U2OS cell growth, and induced cell apoptosis. On the other hand, exogenous over-expression of SphK2 could further promote U2OS cell growth. Third, forced-expression of miR-19a-3p, the potential anti-SphK2 miRNA, silenced SphK2 and inhibited U2OS cell growth. Fourth, SphK2 silence, by targeted-shRNA or miR-19a-3p, dramatically inhibited U2OS tumor growth in nude mice. Fifth, our preliminary studies have demonstrated that ABC294640, the SphK2 specific inhibitor [31, 38, 42], inhibited OS cell growth in vitro and in vivo. Thus, SphK2 could be a key oncotarget protein for human OS.
miRNA binds to the 3′ UTR of the targeted mRNA, causing translational inhibition and/or mRNA degradation [44–46]. It has been aware that dysregulation of miRNA participates in a number of cancerous behaviors, including cell survival, growth and cell cycle progression as well as apoptosis and migration, metabolism [47–50]. Dysregulation of miRNA is also the characteristic marker of human OS [51–53]. One novel finding of this study is that miR-19a-3p could be the specific SphK2-targeting miRNA. miR-19a-3p putatively targets the 3’UTR of SphK2 mRNA. Significantly, forced-expression of miR-19a-3p downregulated SphK2 and efficiently inhibited U2OS cell growth. More importantly, miR-19a-3p level was decreased in the OS tissues/cells, corresponding to SphK2 upregulation. It is therefore possible that miR-19a-3p downregulation is the cause of SphK2 upregulation in human OS cells, although this hypothesis warrants further investigations.
It should be noted that the same miRNA (miR-19a-3p in our study) might have other target proteins and exert different and sometimes contradictory functions in cancer progression [54]. Furthermore, expression a single miRNA, miR-19a for example, could exhibit reverse activities under different contexts [55–57]. Therefore, it will be interesting to further understand the functional complexity of miR-19a, and to verify it as a potential anti-OS miRNA.
MATERIALS AND METHODS
Reagents
Puromycin was purchased from Sigma Aldrich (St. Louis, MO). All the antibodies were purchased from Cell Signaling (Beverly, MA).
Human tissue specimens
The human OS tissues along with the surrounding normal bone tissues were obtained at the time of surgery, and were separated carefully under microscope. Tissues were washed with PBS plus antibiotics, and were minced into small pieces, which were then mechanically dissociated and lysed by the tissue lysis buffer (Biyuntian, Nantong China). Tissue lysates were stored in liquid nitrogen for further analysis. A total of ten OS patients were enrolled, which were provided with written-informed consent. The protocol was according to the principles expressed in the Declaration of Helsinki, and was approved by the Ethic Board of Nantong University.
Cell culture
The human OS cell lines, including MG63, SaOs2, G293 and U2OS, as well as two lines of human osteoblastic cells (OB-6 and hFOB1.19) [49, 58] were obtained from the Cell Bank of Shanghai Institute of Biological Science (Shanghai, China). All the cell lines were cultured in DMEM/F12 supplemented with 8-10% FBS, and were maintained at 37°C in the presence of 5% CO2.
Clonogenic assay of cell growth
Cells were plated at 500 cells/well onto the six-well plate and were incubated for 8 days, which were then fixed with methanol-acetic acid solution and stained with crystal violet. The number of colonies was manually counted under the light microscopy.
Fragmented DNA detection by ELISA
Nucleosomal DNA fragmentation, the characteristic marker of cell apoptosis, was tested via measuring Histone-bound DNA using a specific two-site ELISA kit (Roche, Shanghai, China), including an anti-histone primary antibody and a secondary anti-DNA antibody. ELISA OD at 450 nm was recorded as the quantitative measurement of cell apoptosis [59].
TUNEL assay of apoptosis
U2OS cells with distinct genetic treatment were subjected to the TUNEL dye assay. Positive TUNEL staining is a well-known indicator of cell apoptosis. At least 200 cells of five random views of same condition were analyzed. TUNEL ratio (vs total cell nuclei, Hoechst staining) was recorded.
Western blotting assay
Equivalent amount of lysate proteins (30 μg per lane) were separated by 8-10% of SDS-PAGE gels, and were transferred to PVDF membranes (Millipore, Shanghai, China). The blots were then blocked, and were probed with the indicated primary and secondary antibodies. The protein signals were visualized under the ECL detection kit. β-Tubulin (“Tubulin”) was always tested as the loading control. Quantification of the signal was performed through the Image J software [60], analyzing total gray of each band.
SphK2 shRNA
A total of eight distinct lentiviral shRNAs, against non-overlapping sequence of human SphK2, were designed, synthesized and verified by Genepharm Company (Shanghai, China). The lentiviral shRNA (10 μL/mL, per well) was added to cultured U2OS/MG-63 cells for 24 hours. Stable cells were selected by puromycin (2.5 μg/mL, Sigma) for another 6-8 days. SphK2 expression in the stable cells was tested by Western blotting assay. Control cells were infected with lentiviral scramble control shRNA (Santa Cruz Biotech).
SphK2 over-expression
The full length human SphK2 cDNA, provided by Genepharm Company, was inserted into the pSuper-puro-EGFP-Flag vector (Addgene, Shanghai, China) to establish SphK2 expression construct. The latter was transfected to U2OS/MG-63 cells via Lipofectamine 2000 regent [61]. Stable cells were selected by puromycin (2.5 μg/mL, Sigma) for another 6-8 days. Both endogenous and exogenous (Flag-tagged) SphK2 expression in the stable cells was tested by Western blotting assay.
Quantitative RT-PCR
Total cellular RNA was extracted through the Trizol reagents (Invitrogen, Shanghai, China), and cDNA was synthesized from 0.5 μg mRNA using High Capacity cDNA Reverse Transcription Kit according to the manufacturer's instruction. Quantitative real-time PCR (“qRT-PCR”) assay by Power SYBR Green RT-PCR Reagents Kit was performed via the ABI7500 system. The primers for SphK2 mRNA were 5′-TTCTATTGGTCAATCCCTTTGG-3′ and 5′-AGCCCGTTCAGCACCTCA-3′. The primers GAPDH mRNA were described early [62]. The 2-ΔΔCt was calculated to yield SphK2 mRNA fold expression (relative to GAPDH). miR-19a-3p expression was tested via the TaqMan microRNA assay (Applied Biosystems, Shanghai, China), from 5 ng of total RNA [63].
miR-19a transfection
U2OS/MG-63 cells were transfected with 20 nM of miR-19a mimic oligonucleotides (Ambion, Shanghai, China) by Lipofectamine 2000 (Invitrogen). After two days, cells were split and were transfected with miR-19a mimic again. This process was repeated for eight rounds for a total of 16 days when necessary, until stable cell lines were established. Expression of miR-19a-3p in the stable OS cells was tested by the qRT-PCR assay. The Ambion Pre-miRNA Precursor Negative Control (“miR-C”) was introduced to OS cells as the control cells.
SphK2 mRNA 3’-UTR luciferase activity assay
The reporter vector with the 3’-UTR of SphK2 carrying a putative miR-19a-3p binding site (Position 493-499) was designed, constructed, sequence-verified by Genepharm (Shanghai, China). The complementary oligonucleotides for the selected region were hybridized to form double-stranded DNA and inserted into pmIR-ReporterTM firefly luciferase vector (Genepharm, Shanghai, China). The construct was transfected together with the miR-19a mimic to U2OS cells. Cells were subjected to the luciferase assay using the commercial available kit (Promega, Shanghai, China). The luciferase of β-galactosidase was utilized as an internal control.
U2OS xenograft assay
All experimental protocols involving the nude mice were approved by Nantong University’s Ethics Board. Female nude mice (6-8 weeks age, 17.5-18.8 g weight) were subcutaneously (s.c.) inoculated with 5 × 106 U2OS cells (in 0.2 mL DMEM/10% FBS), with/out SphK2-shRNA or miR-19a-3p, into the right flanks. When U2OS xenograft volumes reached about 150 mm3, the recordings were started. The xenografted tumor volumes along with mice body weights were recorded every week for a total of six weeks [64]. At the end of experiment (week-7), tumors of each group were isolated and weighted. Tumor tissues were subjected to Western blotting assay of indicated signaling proteins.
Statistics
Data were expressed as the mean ± SD (Standard Deviation). Comparisons between groups were performed via one-way ANOVA (SPSS 18.0) followed by Bonferroni post hoc test. p values < 0.05 were considered statistically significant.
CONCLUSIONS
In summary, SphK2 over-expression promotes OS cell growth. SphK2 could be a novel and key oncotarget protein that is critical for OS cell progression.
Author contributions
All authors carried out the experiments, participated in the design of the study and performed the statistical analysis, participated in its design and coordination and helped to draft the manuscript.
CONFLICTS OF INTEREST
The listed authors have no conflicts of interest.
FUNDING
This work was supported by a grant from the National Natural Science Foundation of China (No. 81300955), three grants from the Scientific Research Program of Nantong (No. MS22015047, MS12016009, wq2015011), and also a grant from the Ke-Jiao-Qiang-Wei-Qing-Nian-Yi-Xue-Zhong-Dian-Ren-Cai Financial Assistance of Jiangsu Province (No. QNRC2016412).
REFERENCES
1. Bishop MW, Janeway KA, Gorlick R. Future directions in the treatment of osteosarcoma. Curr Opin Pediatr. 2016; 28:26-33.
2. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol. 2015; 33:3029-3035.
3. Ferrari S, Serra M. An update on chemotherapy for osteosarcoma. Expert Opin Pharmacother. 2015; 16:2727-2736.
4. Denduluri SK, Wang Z, Yan Z, Wang J, Wei Q, Mohammed MK, Haydon RC, Luu HH, He TC. Molecular pathogenesis and therapeutic strategies of human osteosarcoma. J Biomed Res. 2015; 30:5-18.
5. Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer. 2014; 14:722-735.
6. Yang J, Zhang W. New molecular insights into osteosarcoma targeted therapy. Curr Opin Oncol. 2013; 25:398-406.
7. He JP, Hao Y, Wang XL, Yang XJ, Shao JF, Guo FJ, Feng JX. Review of the molecular pathogenesis of osteosarcoma. Asian Pac J Cancer Prev. 2014; 15:5967-5976.
8. Chou AJ, Gorlick R. Chemotherapy resistance in osteosarcoma: current challenges and future directions. Expert Rev Anticancer Ther. 2006; 6:1075-1085.
9. Carrle D, Bielack SS. Current strategies of chemotherapy in osteosarcoma. Int Orthop. 2006; 30:445-451.
10. Luetke A, Meyers PA, Lewis I, Juergens H. Osteosarcoma treatment - where do we stand? A state of the art review. Cancer Treat Rev. 2014; 40:523-532.
11. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7-30.
12. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7-30.
13. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5-29.
14. Zhu YR, Zhou XZ, Zhu LQ, Yao C, Fang JF, Zhou F, Deng XW, Zhang YQ. The anti-cancer activity of the mTORC1/2 dual inhibitor XL388 in preclinical osteosarcoma models. Oncotarget. 2016; 7:49527-49538. https://doi.org/10.18632/oncotarget.10389.
15. Zhen YF, Li ST, Zhu YR, Wang XD, Zhou XZ, Zhu LQ. Identification of DNA-PKcs as a primary resistance factor of salinomycin in osteosarcoma cells. Oncotarget. 2016; 7:79417-79427. https://doi.org/10.18632/oncotarget.12712.
16. Soriano JM, Gonzalez L, Catala AI. Mechanism of action of sphingolipids and their metabolites in the toxicity of fumonisin B1. Prog Lipid Res. 2005; 44:345-356.
17. Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004; 4:604-616.
18. Mullen TD, Obeid LM. Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med Chem. 2012; 12:340-363.
19. Goldkorn T, Chung S, Filosto S. Lung cancer and lung injury: the dual role of ceramide. Handb Exp Pharmacol. 2013; 216:93-113.
20. Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim Biophys Acta. 2002; 1585:193-201.
21. Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012; 22:50-60.
22. Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013; 12:688-702.
23. Vadas M, Xia P, McCaughan G, Gamble J. The role of sphingosine kinase 1 in cancer: oncogene or non-oncogene addiction? Biochim Biophys Acta. 2008; 1781:442-447.
24. Xiao M, Liu Y, Zou F. Sensitization of human colon cancer cells to sodium butyrate-induced apoptosis by modulation of sphingosine kinase 2 and protein kinase D. Exp Cell Res. 2012; 318:43-52.
25. Weigert A, Schiffmann S, Sekar D, Ley S, Menrad H, Werno C, Grosch S, Geisslinger G, Brune B. Sphingosine kinase 2 deficient tumor xenografts show impaired growth and fail to polarize macrophages towards an anti-inflammatory phenotype. Int J Cancer. 2009; 125:2114-2121.
26. Liu W, Ning J, Li C, Hu J, Meng Q, Lu H, Cai L. Overexpression of Sphk2 is associated with gefitinib resistance in non-small cell lung cancer. Tumour Biol. 2016; 37:6331-6336.
27. Frankfurt OS, Krishan A. Enzyme-linked immunosorbent assay (ELISA) for the specific detection of apoptotic cells and its application to rapid drug screening. J Immunol Methods. 2001; 253:133-144.
28. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992; 119:493-501.
29. Stevenson CE, Takabe K, Nagahashi M, Milstien S, Spiegel S. Targeting sphingosine-1-phosphate in hematologic malignancies. Anticancer Agents Med Chem. 2011; 11:794-798.
30. Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr Drug Targets. 2008; 9:662-673.
31. Yang J, Yang C, Zhang S, Mei Z, Shi M, Sun S, Shi L, Wang Z, Wang Y, Li Z, Xie C. ABC294640, a sphingosine kinase 2 inhibitor, enhances the antitumor effects of TRAIL in non-small cell lung cancer. Cancer Biol Ther. 2015; 16:1194-1204.
32. Gestaut MM, Antoon JW, Burow ME, Beckman BS. Inhibition of sphingosine kinase-2 ablates androgen resistant prostate cancer proliferation and survival. Pharmacol Rep. 2014; 66:174-178.
33. Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H, Payne SG, Bektas M, Ishii I, Chun J, Milstien S, Spiegel S. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem. 2003; 278:40330-40336.
34. Gao P, Smith CD. Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol Cancer Res. 2011; 9:1509-1519.
35. Wallington-Beddoe CT, Powell JA, Tong D, Pitson SM, Bradstock KF, Bendall LJ. Sphingosine kinase 2 promotes acute lymphoblastic leukemia by enhancing MYC expression. Cancer Res. 2014; 74:2803-2815.
36. Shi WN, Cui SX, Song ZY, Wang SQ, Sun SY, Yu XF, Li Y, Zhang YH, Gao ZH, Qu XJ. Overexpression of SphK2 contributes to ATRA resistance in colon cancer through rapid degradation of cytoplasmic RXRalpha by K48/K63-linked polyubiquitination. Oncotarget. 2017; 8:39605-39617. https://doi.org/ 10.18632/oncotarget.17174.
37. Xun C, Chen MB, Qi L, Tie-Ning Z, Peng X, Ning L, Zhi-Xiao C, Li-Wei W. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J Exp Clin Cancer Res. 2015; 34:94.
38. French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 2010; 333:129-139.
39. Gao P, Peterson YK, Smith RA, Smith CD. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS One. 2012; 7:e44543.
40. Chumanevich AA, Poudyal D, Cui X, Davis T, Wood PA, Smith CD, Hofseth LJ. Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis. 2010; 31:1787-1793.
41. Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S, Smith CD, Burow ME, Beckman BS. Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther. 2011; 11:678-689.
42. Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LV, Ashe HB, Wiese TE, Smith CD, Burow ME, Beckman BS. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010; 151:5124-5135.
43. Raje MR, Knott K, Kharel Y, Bissel P, Lynch KR, Santos WL. Design, synthesis and biological activity of sphingosine kinase 2 selective inhibitors. Bioorg Med Chem. 2012; 20:183-194.
44. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012; 13:271-282.
45. Paez-Colasante X, Figueroa-Romero C, Sakowski SA, Goutman SA, Feldman EL. Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nat Rev Neurol. 2015; 11:266-279.
46. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015; 16:421-433.
47. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6:857-866.
48. Li P, Fan JB, Gao Y, Zhang M, Zhang L, Yang N, Zhao X. miR-135b-5p inhibits LPS-induced TNFalpha production via silencing AMPK phosphatase Ppm1e. Oncotarget. 2016; 7:77978-77986. https://doi.org/10.18632/oncotarget.12866.
49. Fan JB, Ruan JW, Liu W, Zhu LQ, Zhu XH, Yi H, Cui SY, Zhao JN, Cui ZM. miR-135b expression downregulates Ppm1e to activate AMPK signaling and protect osteoblastic cells from dexamethasone. Oncotarget. 2016; 7:70613-70622. https://doi.org/10.18632/oncotarget.12138.
50. Lulli V, Buccarelli M, Martini M, Signore M, Biffoni M, Giannetti S, Morgante L, Marziali G, Ilari R, Pagliuca A, Larocca LM, De Maria R, Pallini R, et al. miR-135b suppresses tumorigenesis in glioblastoma stem-like cells impairing proliferation, migration and self-renewal. Oncotarget. 2015; 6:37241-37256. https://doi.org/10.18632/oncotarget.5925.
51. Sampson VB, Yoo S, Kumar A, Vetter NS, Kolb EA. MicroRNAs and potential targets in osteosarcoma: review. Front Pediatr. 2015; 3:69.
52. Wang W, Zhou X, Wei M. MicroRNA-144 suppresses osteosarcoma growth and metastasis by targeting ROCK1 and ROCK2. Oncotarget. 2015; 6:10297-10308. https://doi.org/10.18632/oncotarget.3305.
53. Salah Z, Arafeh R, Maximov V, Galasso M, Khawaled S, Abou-Sharieha S, Volinia S, Jones KB, Croce CM, Aqeilan RI. miR-27a and miR-27a* contribute to metastatic properties of osteosarcoma cells. Oncotarget. 2015; 6:4920-4935. https://doi.org/10.18632/oncotarget.3025.
54. Yang J, Zhang Z, Chen C, Liu Y, Si Q, Chuang TH, Li N, Gomez-Cabrero A, Reisfeld RA, Xiang R, Luo Y. MicroRNA-19a-3p inhibits breast cancer progression and metastasis by inducing macrophage polarization through downregulated expression of Fra-1 proto-oncogene. Oncogene. 2014; 33:3014-3023.
55. Sochor M, Basova P, Pesta M, Dusilkova N, Bartos J, Burda P, Pospisil V, Stopka T. Oncogenic microRNAs: miR-155, miR-19a, miR-181b, and miR-24 enable monitoring of early breast cancer in serum. BMC Cancer. 2014; 14:448.
56. Wu Q, Yang Z, An Y, Hu H, Yin J, Zhang P, Nie Y, Wu K, Shi Y, Fan D. MiR-19a/b modulate the metastasis of gastric cancer cells by targeting the tumour suppressor MXD1. Cell Death Dis. 2014; 5:e1144.
57. Chen Q, Xia HW, Ge XJ, Zhang YC, Tang QL, Bi F. Serum miR-19a predicts resistance to FOLFOX chemotherapy in advanced colorectal cancer cases. Asian Pac J Cancer Prev. 2013; 14:7421-7426.
58. Ding H, Wang T, Xu D, Cha B, Liu J, Li Y. Dexamethasone-induced apoptosis of osteocytic and osteoblastic cells is mediated by TAK1 activation. Biochem Biophys Res Commun. 2015; 460:157-163.
59. Cai S, Li Y, Bai JY, Zhang ZQ, Wang Y, Qiao YB, Zhou XZ, Yang B, Tian Y, Cao C. Galphai3 nuclear translocation causes irradiation resistance in human glioma cells. Oncotarget. 2017; 8:35061-35068. https://doi.org/10.18632/oncotarget.17043.
60. Gong YQ, Huang W, Li KR, Liu YY, Cao GF, Cao C, Jiang Q. SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget. 2016; 7:60123-60132. https://doi.org/ 10.18632/oncotarget.11164.
61. Wang B, Wang XB, Chen LY, Huang L, Dong RZ. Belinostat-induced apoptosis and growth inhibition in pancreatic cancer cells involve activation of TAK1-AMPK signaling axis. Biochem Biophys Res Commun. 2013; 437:1-6.
62. Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci U S A. 2002; 99:4221-4226.
63. Matsumura T, Sugimachi K, Iinuma H, Takahashi Y, Kurashige J, Sawada G, Ueda M, Uchi R, Ueo H, Takano Y, Shinden Y, Eguchi H, Yamamoto H, et al. Exosomal microRNA in serum is a novel biomarker of recurrence in human colorectal cancer. Br J Cancer. 2015; 113:275-281.
64. Zhu YR, Min H, Fang JF, Zhou F, Deng XW, Zhang YQ. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against osteosarcoma. Cancer Biol Ther. 2015; 16:602-609.