
INTRODUCTION
Lung cancer is the leading cause of cancer-related death worldwide [1]. A significant proportion of non-small cell lung cancers (NSCLCs) carry epidermal growth factor receptor (EGFR) gene mutations [2–6], which have been identified in approximately 10–30% of NSCLCs [7, 8]. Somatic mutation of the EGFR tyrosine kinase domain promotes the active conformation of this receptor, hence inducing its constitutive activation [9–11]. Activated EGFR transduces downstream signals, including the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathway, and phosphatidylinositol-3-kinase/protein kinase B (AKT) [12–14]. The most common or “classic” mutations are in-frame deletions around the LREA motif in exon 19, which account for approximately 45% of EGFR mutations, and the exon 21 L858R point mutation, accounting for approximately 40% of EGFR mutations. Other less common EGFR mutations include G719X (3% of EGFR mutations), L861Q (2% of EGFR mutations) [12], and exon 20 insertion mutations (4–10% of EGFR mutations) [15–17].
EGFR tyrosine kinase inhibitors (EGFR-TKIs) have been developed to inhibit EGFR-mediated signaling. These molecules bind reversibly or irreversibly to the ATP binding pocket of EGFR, thus inhibiting activation. The exon 19 deletions, the L858R, the G719X, and the L861Q mutations result in sensitivity to the first-generation EGFR-TKIs, gefitinib and erlotinib. The response rates to gefitinib or erlotinib among NSCLC patients harboring the classic EGFR mutations are around 60–80% [7, 18]. Acquired resistance can occur after treatment with first- or second-generation EGFR-TKIs and the EGFR T790M mutation accounts for about 60% of this resistance [19, 20]. EGFR T790M is thought to induce resistance to these EGFR-TKIs by decreasing the affinity of EGFR-TKIs and increasing the affinity of ATP to tyrosine kinase domain ATP binding pocket of EGFR [20, 21]. Third-generation EGFR-TKIs irreversibly bind to the EGFR ATP binding pocket via a covalent interaction with the C797 residue, thereby blocking the increased affinity for ATP conferred by the EGFR T790M mutation. Some of third-generation EGFR-TKIs such as osimertinib [22]and nazartinib, which was formerly called EGF816, have demonstrated clinically significant efficacy and safety in NSCLC patients harboring EGFR T790M mutations, although the development of other third-generation candidates (rociletinib, olmutinib and naquotinib) has been halted [23, 24]. Osimertinib treatment produced a high objective response rate of approximately 60% for tumors with T790M mutations that showed resistance to first-generation EGFR-TKIs [22]. Nazartinib is undergoing clinical evaluation [25].
Unlike classic EGFR mutations, there is a paucity of data regarding the EGFR-TKI sensitivity of patients with lung cancers expressing less common EGFR mutations. For tumors expressing some of these mutations such as G719X, L861Q, and S768I, afatinib was effective [26]. However, the efficacy of third-generation EGFR-TKIs in patients with these mutations, in the presence or absence of the T790M mutation, is unclear.
On the other hand, most EGFR exon 20 insertion mutations confer resistance to first- and second-generation EGFR-TKIs [15, 27, 28]. One exception is EGFR A763_Y764insFQEA, which we previously reported as a first-generation EGFR-TKI-sensitizing mutation [29]. Our previous study reported the potential efficacy of osimertinib against tumors with exon 20 insertions associated with first- and second-generation EGFR-TKI resistance [30]. We created an in vitro model to determine the therapeutic windows for EGFR-TKIs, where the ratios of the 50% inhibitory concentrations (IC50) in Ba/F3 cells transduced with either mutated or wild type EGFRs were calculated [30]. We identified a therapeutic window of osimertinib for several EGFR exon 20 insertion mutations. The efficacy of nazartinib against cells expressing EGFR exon 20 insertion mutations has also been reported previously [31]. These authors revealed that nazartinib potently inhibited major subtypes of exon 20 insertion mutations, with EC50 values of 7, 11, and 190 nmol/L against D770_V771dupSVD, V769_D770insASV, and H773_V774insNPH, respectively. Furthermore, they showed an antitumor effect of nazartinib in a mouse patient-derived xenograft model expressing H773_V774insNPH. Therefore, osimertinib and nazartinib are expected to be potentially effective for tumors with exon 20 insertion mutations.
There are no direct comparison data relating to the efficacies of EGFR-TKIs for tumors expressing different EGFR mutations, including the less common mutations. The present study aimed to characterize the efficacy of third-generation EGFR-TKIs and to compare them with that of other EGFR-TKIs using in vitro modeling. This analysis of the efficacy of EGFR-TKIs provides preclinical information to inform the proper selection of EGFR-TKIs for the treatment of patients with cancers harboring EGFR mutations.
RESULTS
Therapeutic windows for osimertinib and nazartinib in Ba/F3 cells expressing classic EGFR mutations, in the presence or absence of T790M
We generated stable EGFR-expressing Ba/F3 cell lines in order to directly compare the sensitivities of multiple EGFR mutations to EGFR-TKIs using MTS assays (Figure 1A). The viabilities of Ba/F3 cells harboring exon 19del or L858R were dramatically reduced by afatinib. Erlotinib also effectively inhibited the proliferation of these cells, although it was less potent than afatinib. The potency of nazartinib was comparable to that of erlotinib, while osimertinib was more potent than either erlotinib or nazartinib. Although low concentrations of erlotinib did not inhibit the proliferation of Ba/F3 cells harboring EGFR T790M mutations (EGFR exon 19del + T790M or L858R + T790M), afatinib, osimertinib, and nazartinib effectively reduced the viability of these cells.
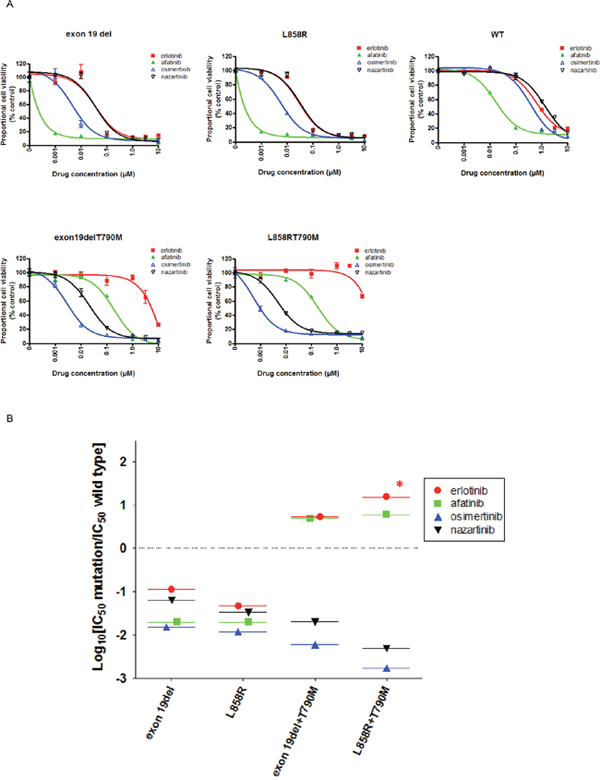
Figure 1: The sensitivity of Ba/F3 cells expressing wild type (WT) or mutant EGFR to EGFR-TKIs. (A) MTS assays were conducted in Ba/F3 cells expressing the indicated EGFR genotypes. Data points represent the mean ± standard deviation. (B) Selectivity index (SI) values were calculated for cells expressing the indicated EGFR mutations versus wild type.*; SI index >1.
In the clinic, skin rashes and diarrhea are common or life-threatening side effects of EGFR-TKIs. These side effects are most likely caused by the inhibition of wild type EGFR expressed in epithelial cells within the lungs, gastrointestinal tract, and skin. Hence, the clinical utility of EGFR-TKIs is hampered by their lack of selectivity for mutated versus wild type EGFR; this results in a narrow therapeutic window, with a high level of toxicity at the clinical doses required to effectively inhibit mutated EGFR. We therefore evaluated the effects of EGFR-TKIs on wild type EGFR using the MTS assay. Wild type EGFR-expressing Ba/F3 cells were efficiently inhibited by afatinib, while low concentrations of nazartinib did not inhibit their viability. The IC50 value for afatinib was 30 nM, as compared to 1031 nM for nazartinib, indicating that afatinib had more potent effects on wild type EGFR. The IC50 values for each EGFR-TKI examined in this study are summarized in Table 1.
Table 1: IC50 values (nM) of Ba/F3 cells expressing EGFR mutations
Mutations |
erlotinib |
afatinib |
osimertinib |
nazartinib |
|---|---|---|---|---|
exon 19del |
73 |
0.6 |
7.9 |
66 |
L858R |
30 |
0.6 |
6.2 |
35 |
G719S |
101 |
1.5 |
158 |
91.2 |
L861Q |
410 |
3.6 |
35.8 |
116 |
A763_Y764insFQEA |
152 |
<0.1 |
24 |
78 |
Y764_V765insHH |
2948 |
140 |
143 |
387 |
A767_V769dupASV |
3073 |
77 |
134 |
257 |
D770_N771insNPG |
2342 |
36 |
28 |
84 |
exon19del+T790M |
3429 |
146 |
3.1 |
52 |
L858R+T790M |
>10000 |
179 |
0.9 |
5.1 |
G719S+T790M |
>10000 |
34 |
97 |
77 |
L861Q+T790M |
>10000 |
163 |
73 |
101 |
exon19del+T790M+C797S |
5153 |
390 |
655 |
2146 |
L858R+T790M+C797S |
8360 |
826 |
1024 |
3834 |
WT |
638 |
30 |
516 |
1031 |
We used in vitro modeling to calculate the selectivity index (SI): the ratio of IC50 values in Ba/F3 cells transduced with mutant or wild type EGFRs (Figure 1B). In this model, a low SI value indicates a highly mutant EGFR-selective EGFR-TKI. For Ba/F3 cells harboring exon 19del or L858R, osimertinib showed the lowest SI values of the EGFR-TKIs included in this study; this indicated a wide therapeutic window for osimertinib in patients with these mutations. Nazartinib showed slightly higher SI values than osimertinib in cells expressing classic EGFR mutations. For Ba/F3 cells harboring EGFR T790M, osimertinib and nazartinib showed striking mutation specificity. Their SI values of ≤ -2 indicated IC50 values for cells expressing the T790M mutation that were > 100-fold lower than those for cells expressing wild type EGFR. On the other hand, erlotinib and afatinib did not demonstrate mutation specificity for EGFR T790M mutations. These SI values were > 0, indicating IC50 values for cells expressing EGFR T790M were higher than those for cells expressing wild type EGFR.
Effect of EGFR-TKIs on EGFR downstream signals and apoptosis in cells Ba/F3 expressing classic EGFR mutations in the presence or absence of the T790M mutation
To examine EGFR signaling pathways, we performed immunoblotting (Supplementary Figure 1A). In Ba/F3 cells harboring exon 19del or L858R, afatinib dramatically inhibited the phosphorylation of EGFR, AKT, and ERK. Erlotinib, osimertinib, and nazartinib inhibited the phosphorylation of EGFR, AKT, and ERK to a similar extent. In Ba/F3 cells harboring the EGFR T790M mutation (exon 19del + T790M or L858R + T790M), erlotinib did not inhibit the phosphorylation of EGFR, AKT, and ERK. However, afatinib, osimertinib, and nazartinib did effectively inhibit the phosphorylation of these molecules. These data indicated that the sensitivity of Ba/F3 cells to EGFR-TKIs was reflected by the inhibition of the EGFR and its downstream signaling pathways.
Next, to biologically confirm the therapeutic windows for third-generation EGFR-TKIs in cells expressing the T790M mutation, we analyzed apoptosis in Ba/F3 cells expressing wild type EGFR or EGFR exon 19del + T790M. We stained these cells with annexin V-APC and propidium iodide after 48-h exposures to the indicated EGFR-TKIs (0.1 μM). The proportions of annexin V-positive and/or propidium iodide-positive cells were examined by flow cytometry (Supplementary Figure 1B). As expected, afatinib induced apoptosis in Ba/F3 cells harboring either wild type EGFR or EGFR exon 19del + T790M, indicating a narrow therapeutic window for afatinib in the presence of EGFR exon 19del + T790M. The proportion of annexin V-positive wild type EGFR-expressing cells was 91.0%, as compared with 32.8% of EGFR exon 19del + T790M-expressing cells. In contrast, osimertinib and nazartinib produced only minor effects on apoptosis in Ba/F3 cells harboring wild type EGFR, and were more effective in cells harboring EGFR exon 19del + T790M. The proportions of annexin V-positive wild type EGFR-expressing cells were 27.9% and 25.5%, respectively, in the presence of osimertinib and nazartinib, while the equivalent values for cells expressing EGFR exon 19del + T790M were 72.5% and 62.8%, respectively. These data indicated a wide therapeutic window for osimertinib and nazartinib in the treatment of tumors expressing the EGFR T790M mutation.
Investigation of the sensitivity/resistance profiles of EGFR-TKIs in lung cancer cell lines
To explore the relevance of the sensitivity/resistance profiles derived from stably transduced Ba/F3 cells, we also performed MTS assays in human lung cancer-derived cell lines in the presence and absence of EGFR-TKIs (Figure 2A). The lung cancer cell lines included in this study were PC-9 (EGFR exon 19del), PC-9ER (EGFR exon 19del + T790M), and H1975 (EGFR L858R + T790M). For PC-9 cells, afatinib produced the most dramatic inhibitory effect, with an IC50 value of 1.3 nM. Although less potent than afatinib, erlotinib also effectively inhibited the proliferation of PC-9 cells, with an IC50 value of 28 nM. The potencies of osimertinib and nazartinib were comparable to that of erlotinib in this cell line, with IC50 values of 23 nM and 36 nM, respectively. Low concentrations of erlotinib did not inhibit the proliferation of cell lines harboring the EGFR T790M mutation (PC-9ER and H1975). However, afatinib and the third-generation EGFR-TKIs, osimertinib and nazartinib, effectively inhibited the proliferation of these lung cancer cells. The calculated IC50 values of afatinib, osimertinib, and nazartinib for PC-9ER cells were 677 nM, 166 nM, and 276 nM, respectively. The calculated IC50 values of afatinib, osimertinib, and nazartinib for H1975 cells were 80 nM, 4.6 nM, and 52 nM, respectively. These IC50 values are summarized in Supplementary Table 1. These data indicated that the sensitivity/resistance profile data produced in stably transfected Ba/F3 cells were comparable to those generated using human lung cancer cell lines.
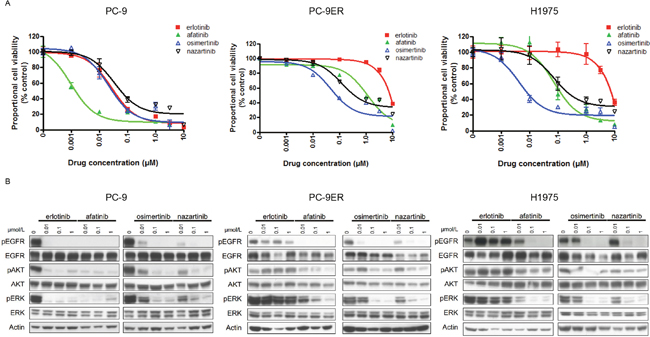
Figure 2: The sensitivities of lung cancer cell lines to EGFR-TKIs. (A) MTS assays of PC-9, PC9-ER, and H1975 cells exposed to the indicated EGFR-TKIs. Data points represent the mean ± standard deviation. (B) PC-9, PC9-ER, and H1975 cells were treated with the indicated concentrations of EGFR-TKIs for 4 h prior to immunoblotting for the phosphorylated (p) and non-phosphorylated forms of EGFR, AKT, and ERK. Actin was used as a loading control.
Furthermore, immunoblotting was performed to determine whether the sensitivity of lung cancer cells to EGFR-TKIs was mediated through inhibition of EGFR signaling (Figure 2B). Consistent with the results of the MTS assay, afatinib caused the most potent inhibition of the phosphorylation of EGFR and its downstream proteins, AKT and ERK, in PC-9 cells. Although they were less potent than afatinib, erlotinib, osimertinib, and nazartinib also effectively inhibited the phosphorylation of EGFR, AKT, and ERK in PC-9 cells. Our study of lung cancer cells harboring the EGFR T790M mutation showed that all of the tested EGFR-TKIs, except for erlotinib, effectively inhibited the phosphorylation of the EGFR, AKT, and ERK. In summary, these data indicated that the sensitivity/resistance profiles observed in Ba/F3 cells were also present in human lung cancer cells.
Therapeutic windows of nazartinib and osimertinib in Ba/F3 cells expressing EGFR exon 20 insertion mutations
We investigated the effects of EGFR-TKIs on the viability of Ba/F3 cells transduced with four representative EGFR exon 20 insertion mutations (A763_Y764insFQEA, Y764_V765insHH, A767_V769dupASV, and D770_N771insNPG) using MTS assays (Figure 3A). All of the tested EGFR-TKIs inhibited the growth of Ba/F3 cells harboring EGFR A763_Y764insFQEA. A similar response pattern was observed in a human lung cancer-derived cell line (BID007) harboring EGFR A763_Y764insFQEA (Supplementary Figure 2). Afatinib, osimertinib, and nazartinib showed comparable efficacies against cells expressing other first- and second-generation EGFR-TKI-resistant EGFR exon 20 insertion mutations, Y764_V765insHH, A767_V769dupASV, and D770_N771insNPG. The calculated IC50 values are summarized in Table 1. We applied the aforementioned in vitro model to estimate the therapeutic windows for EGFR-TKIs in cells expressing EGFR exon 20 insertion mutations (Figure 3B). In Ba/F3 cells harboring EGFR A763_Y764insFQEA, all EGFR-TKIs showed comparable SI values of around -1. In contrast, erlotinib and afatinib had SI values > 0 in cells expressing EGFR exon 20 insertion mutations associated with resistance to first- and second-generation EGFR-TKIs. In these cell lines, osimertinib and nazartinib had SI values of around -1, indicating IC50 values of about 10-fold lower than those observed in cells expressing wild type EGFR. These data indicated that both osimertinib and nazartinib could have therapeutic windows and act effectively against cells expressing these exon 20 insertion mutations.
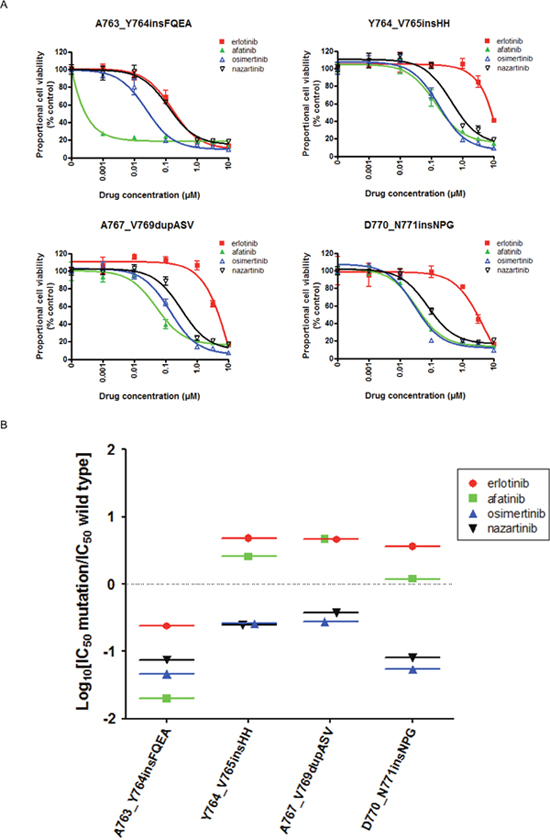
Figure 3: The EGFR-TKI sensitivity of Ba/F3 cells harboring the indicated EGFR genotypes. (A) MTS assays were conducted in Ba/F3 cells harboring the indicated EGFR exon 20 insertion mutations. Data points represent the mean ± standard deviation. (B) The selectivity index (SI) values are shown for the indicated EGFR-TKIs in Ba/F3 cells expressing the indicated EGFR exon 20 insertion mutations.
Effects of EGFR-TKIs on EGFR downstream signals and apoptosis in cells expressing EGFR exon 20 insertion mutations
We performed immunoblotting to investigate whether the sensitivity of Ba/F3 cells harboring EGFR exon 20 insertion mutations was associated with inhibition of the EGFR signaling pathway (Supplementary Figure 3A). As expected, afatinib dramatically inhibited the phosphorylation of EGFR, AKT, and ERK in cells expressing EGFR A763_Y764insFQEA. Osimertinib and nazartinib produced similar inhibitions of EGFR, AKT, and ERK phosphorylation. These inhibition patterns were also observed in BID007 cells (Supplementary Figure 3B). In cells expressing other EGFR exon 20 insertion mutations associated with resistance to first- and second-generation EGFR-TKIs (Y764_V765insHH, A767_V769dupASV, and D770_N771insNPG), afatinib, osimertinib, and nazartinib produced similar inhibition patterns.
We performed assays to determine the level of apoptosis in Ba/F3 cells harboring Y764_V765insHH and A767_V769dupASV, following exposure to 0.1 μM afatinib, osimertinib, or nazartinib for 48 h. The proportions of annexin V- and/or propidium iodide-positive cells were determined by flow cytometry (Supplementary Figure 4). In cells expressing Y764_V765insHH, the proportions of annexin V-positive cells following exposure to afatinib, osimertinib, or nazartinib were 64.2%, 72.9%, or 51.3%, respectively. The corresponding proportions in cells expressing A767_V769dupASV were 88.5%, 73.3%, or 48.9%, respectively. These data indicated that afatinib, osimertinib, and nazartinib induced apoptosis in cells expressing different EGFR exon 20 insertion mutations.
Therapeutic windows of osimertinib and nazartinib in Ba/F3 cells expressing EGFR G719S, L861Q, and/or T790M
We used MTS assays to investigate the effects of EGFR-TKIs on the viability of Ba/F3 cells transduced with the less common EGFR mutations, G719S and L861Q, in the presence or absence of T790M (Figure 4A). The resultant IC50 values are summarized in Table 1. The viability of Ba/F3 cells harboring G719S and L861Q mutations was dramatically reduced by afatinib. For these mutations, the SI values of all EGFR-TKIs were < 0, and afatinib showed the lowest SI values in cells expressing G719S (Figure 4B). In the presence of T790M, the sensitivities to erlotinib and afatinib reduced. For G719S+T790M or L861Q+T790M, the IC50 values of osimertinib and nazartinib were around 100 nM, 10- to 100-fold higher than those observed in cells expressing classic+T790M mutations (Table 1). Moreover, the SI values of osimertinib and nazartinib for these mutations were around -1 (Figure 4B), higher than those observed in cells expressing classic mutations. These data indicate the possibility that third-generation EGFR-TKIs would have lower efficacies in tumors expressing the less common EGFR mutations, as compared with those expressing the classic EGFR mutations, regardless of T790M mutation.
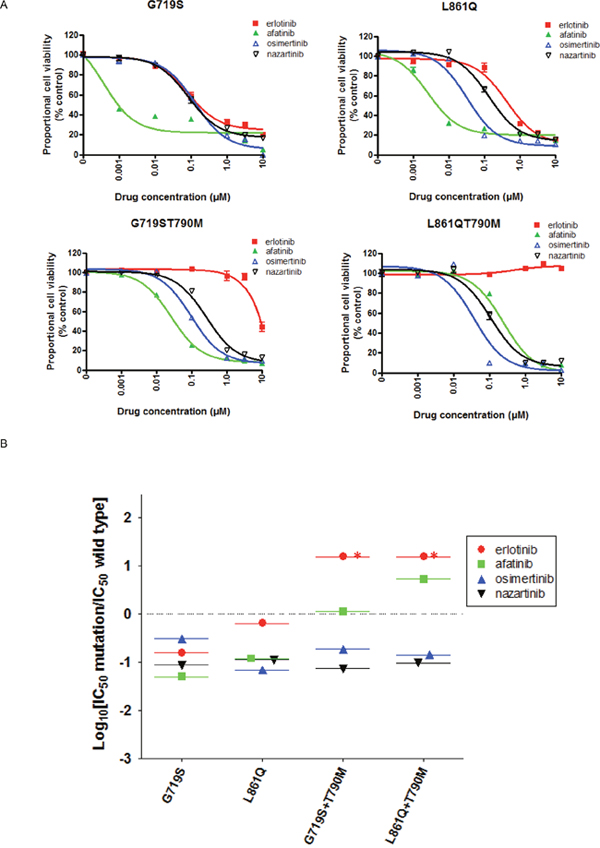
Figure 4: The EGFR-TKI sensitivity of Ba/F3 cells harboring the indicated EGFR genotypes. (A) MTS assays were conducted in Ba/F3 cells harboring the indicated EGFR uncommon mutations, G719S and L861Q in the presence or absence of T790M. Data points represent the mean ± standard deviation. (B) The selectivity index (SI) values are shown for the indicated EGFR-TKIs in Ba/F3 cells expressing the indicated EGFR uncommon mutations. *; SI index >1.
Effects of EGFR-TKIs on EGFR downstream signals and apoptosis in cells expressing EGFR G719S, L861Q, and/or T790M
We performed immunoblotting to investigate the effect of EGFR-TKIs on the EGFR signaling pathway (Supplementary Figure 5A). Afatinib dramatically inhibited the phosphorylation of EGFR, AKT, and ERK, particularly in cells expressing EGFR G719S+T790M. Osimertinib and nazartinib produced similar inhibitions of EGFR, AKT, and ERK phosphorylation in cells expressing G719S+T790M and L861Q+T790M. The level of apoptosis in Ba/F3 cells harboring these mutations were consistent with the IC50 values for these EGFR-TKIs (Supplementary Figure 5B).
The efficacy of osimertinib in NSCLC patients harboring G719X in the presence or absence of T790M
To seek the clinical relevance of the obtained data about the G719X mutation, we reviewed clinical data. In the AURA trial, a phase I/II clinical trial (NCT01802632), a total of seven patients with the G719X mutation received at least one dose of osimertinib. EGFR T790M was detected in three of these patients. Of the 7 patients, 1 (14%) had a confirmed partial response, 3 (43%) had stable disease, and 3 (43%) had progressive disease. In addition, we identified an interesting case in our institution. A 63-year-old woman was diagnosed with stage IV adenocarcinoma of the lung via biopsy using bronchoscopy. This tumor expressed the EGFR G719S mutation. The timeline of her anticancer treatment is shown in Figure 5A. Treatment with erlotinib maintained stable pulmonary lesions, but did not control her brain metastases; she therefore underwent γ-knife and brain surgery. After 10 months, the left upper lobe tumor had expanded and she received afatinib as a second-line therapy. After 11 months, the left upper lobe lesion enlargement led to a second computed tomography-guided biopsy. Molecular analysis found EGFR G719S and T790M mutations. The patient was then treated with osimertinib (80 mg/day). However, after 3 months, she met the definition of progressive disease (+ 127%) because the left upper lobe tumor had expanded (Figure 5B) and left adrenal gland involvement was detected (not shown). These findings may reflect the limited efficacy of third-generation EGFR-TKIs for tumors expressing G719X, in the presence or absence of T790M.
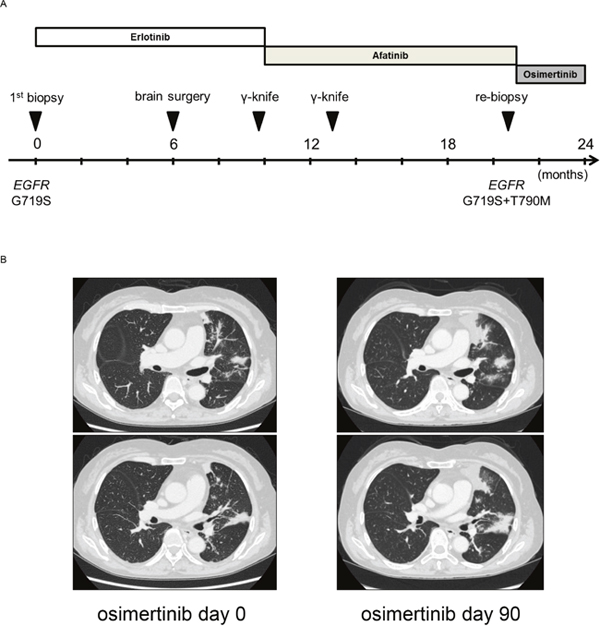
Figure 5: (A) Clinical course after diagnosis of NSCLC. (B) Clinical response to osimertinib in a patient with NSCLC harboring EGFR G719S+T790M. Chest computed tomography images of the patient were captured before and after osimertinib treatment.
EGFR-TKI resistance in cells expressing the EGFR C797S mutation
To date, several mechanisms underlying acquired resistance to third-generation EGFR-TKIs have been reported in preclinical models and clinical specimens; these include the tertiary EGFR C797S mutation [32–35] that prevents the covalent binding of EGFR-TKIs, EGFR L718Q mutation [36, 37], MET amplification [31, 32], HER2 amplification [32], mutated EGFR allele amplification [38], or wild type EGFR allele amplification [39]. Cys797 is the amino acid that is covalently modified by third-generation EGFR-TKIs and its substitution by Ser797 abolishes this interaction. We investigated the effects of EGFR-TKIs on the viability of Ba/F3 cells harboring delL747_P753insS + T790M + C797S (exon 19del + T790M + C797S) or L858R + T790M + C797S using MTS assays (Supplementary Figure 6A). None of the tested EGFR-TKIs inhibited the proliferation of these cells at low concentrations. The IC50 values of EGFR-TKIs for EGFR C797S-positive cell lines are summarized in Table 1. Furthermore, the SI values of the tested EGFR-TKIs were > 0 (Supplementary Figure 6B). In summary, these data confirmed that Ba/F3 cells expressing the EGFR C797S mutation were resistant to these EGFR-TKIs.
DISCUSSION
The present study compared the effects of erlotinib, afatinib, osimertinib, and nazartinib on cells expressing a range of EGFR mutations; these included classic mutations (exon 19 deletions and L858R), the T790M mutation, less common mutations (G719S, L861Q and exon 20 insertion mutations), and the C797S mutation. The therapeutic window for these EGFR-TKIs is an important determinant of their potential toxicity in humans. To quantify this, we used our previously reported in vitro model to determine the ratio of their IC50 values in Ba/F3 cells transduced with mutant or wild type EGFRs [30]. In general, the SI values obtained in this study were tightly matched to previously published data [30], indicating the robustness of this model.
In addition, the findings of our studies in stably transduced Ba/F3 cell lines and in human lung cancer-derived cell lines were consistent. All the tested EGFR-TKIs demonstrated wide therapeutic windows in cells expressing the classic EGFR mutations, exon 19 deletions and L858R. Interestingly, osimertinib had a wider therapeutic window than nazartinib. On the contrary, in cells expressing the EGFR T790M mutation, osimertinib and nazartinib showed markedly wider therapeutic windows than either erlotinib or afatinib. These data indicate that osimertinib and nazartinib could provide effective and safe EGFR-TKIs for patients with EGFR T790M-positive tumors. This is consistent with human clinical trials, in which osimertinib and nazartinib have shown promising safety and efficacy [22].
Unlike classic EGFR mutations, there is a paucity of data regarding the EGFR-TKI sensitivity of patients with lung cancers expressing less common EGFR mutations. Preclinical data have indicated that EGFR exon 18 mutations (exon 18 deletions, E709K, and G719A) and others (S768I and L861Q) exhibited higher sensitivity to afatinib than to erlotinib or osimertinib [40, 41]. In the present study, afatinib had a wider therapeutic window for G719S than for L861Q. A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6 found that the response rate to afatinib was higher for G719X (77.8%) than for L861Q (56.3%) [26]. Furthermore, we identified limited efficacy of third-generation EGFR-TKIs for these mutations, irrespective of the T790M mutation. Interestingly, osimertinib was not effective for the patient with NSCLC harboring the G719S+T790M mutations. In the AURA trial, 7 patients with G719X-positive tumors received at least one dose of osimertinib. The objective response rate was 14% and the disease control rate was 57% [22]. In summary, the efficacy of third-generation EGFR-TKIs is likely to be lower in patients with the EGFR G719X mutation than in those with EGFR classic mutations, both in the presence and absence of the T790M mutation. Osimertinib and nazartinib showed similar potencies and greater mutation specificity than afatinib in cells expressing exon 20 insertion mutations, which are associated with resistance to first- and second-generation EGFR-TKIs. Dose-adjusted clinical trials will be required to examine the efficacy of osimertinib or nazartinib for patients with NSCLCs harboring these types of exon 20 insertion mutations.
Structurally, the EGFR T790M mutation induces resistance to first- and second-generation EGFR-TKIs by altering the gatekeeper residue in the ATP-site and increasing the affinity for ATP [21]. The EGFR C797S mutation impairs the covalent binding between the cysteine residue at position 797 of EGFR and third-generation EGFR-TKIs [33]. In general, the data obtained in this study was reasonable from the structural view point.
The present study was conducted in vitro. To develop improved strategies for using EGFR-TKIs, further in vivo and human clinical trials will be required because EGFR-TKIs may have different pharmacokinetic and pharmacodynamic properties that will influence their in vivo concentrations.
In summary, the present study provides fundamental osimertinib and nazartinib sensitivity/resistance data in cells expressing a range of EGFR mutations, including those that occur relatively infrequently. The findings highlight the diverse mutation-related sensitivity to EGFR-TKIs. These data may help in the selection of EGFR-TKIs for the treatment of NSCLC patients.
MATERIALS AND METHODS
Cell lines
Four human NSCLC cell lines were used: PC-9 [EGFR exon 19 deletion (delE746-A750)]; PC-9ER [EGFR exon 19 deletion (delE746-A750) + T790M]; BID007 [EGFR exon 20 insertion (A763_Y764insFQEA)]; and H1975 [EGFR L858R + T790M]. PC9 and BID007 cells were a kind gift from Dr. Susumu Kobayashi (Beth Israel Deaconess Medical Center, Boston, MA, USA). H1975 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). PC-9ER cells became resistant to erlotinib after chronic exposure to this molecule and acquisition of the EGFR T790M mutation. Cell authentication for H1975 was performed in June 2015.
Reagents
Erlotinib and afatinib were purchased from LC Laboratories (Woburn, MA, USA). Osimertinib was purchased from Selleck Chemicals (Houston, TX, USA). Nazartinib (#B5889) was purchased from Apexbio. Antibodies recognizing total EGFR (#2232), total AKT (#9272), phospho-AKT (S473; D9E; #4060), total p44/42 MAPK (#9102S), and phospho-p44/42 MAPK (T202/204; #9101S) were purchased from Cell Signaling Technology (Beverly, MA, USA). The phospho-EGFR (Y1068) antibody (44788G) was purchased from Invitrogen/Life Technologies (Carlsbad, CA, USA), and the actin antibody was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Ba/F3 stable cell lines
Ba/F3 cells stably expressing either wild type or mutated EGFR were created as previously described [29]. Ba/F3 cells harboring EGFR mutations were cultured in RPMI-1640 growth medium, supplemented with 10% fetal bovine serum, at 37°C in a humidified 5% CO2 incubator. Ba/F3 cells expressing wild type EGFR were cultured in RPMI-1640 growth medium, supplemented with 10% fetal bovine serum, at 37°C in a humidified 5% CO2 incubator with EGF (10 ng/mL). The EGFR mutations examined in this study included: delL747_P753insS (exon 19del); L858R; delL747_P753insS + T790M (exon 19del + T790M); L858R + T790M; A763_Y764insFQEA; Y764_V765insHH; A767_V769dupASV; D770_N771insNPG; G719S; L861Q; G719S + T790M; L861Q + T790M; delL747_P753insS + T790M + C797S (exon 19del + T790M + C797S); and L858R + T790M + C797S.
Cell proliferation assay
The MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay was performed as previously described [29]. PC-9, PC-9ER, H1975, and BID007 cells were seeded in 96-well plates. Twenty-four hours after seeding, the appropriate medium with or without EGFR-TKI was added to each well. Control cells were treated with the same concentration of the vehicle, dimethyl sulfoxide (DMSO). Seventy-two hours after treatment, absorbance was measured. For Ba/F3 cells, the cells were seeded with or without EGFR-TKI. Seventy-two hours after seeding, absorbance was measured. All experiments were performed at least three times.
Immunoblotting analysis
Cells were treated with EGFR-TKI at concentrations of 0.01–1 μmol/L for 4 h. Cells were lysed in Cell Lysis Buffer (Cell Signaling Technology). Equal amounts of protein were loaded per lane on sodium dodecyl sulfate-polyacrylamide gels. Separated proteins were transferred to polyvinylidene fluoride membranes. The membranes were incubated overnight with primary antibodies at 4°C and then incubated with secondary antibodies for 1 h. For the detection of proteins, the membranes were incubated with agitation in LumiGLO reagent and peroxide (Cell Signaling Technology), and then exposed to X-ray film.
Apoptosis assay
Ba/F3 cells harboring wild type EGFR, exon 19del + T790M, Y764_V765insHH, A767_V769dupASV, G719S + T790M, or L861Q + T790M were seeded in 6-well plates. The cells were treated with EGFR-TKIs (0.1 μM) for 48 h. Control cells were treated with the same concentration of the vehicle, DMSO. We analyzed the apoptotic status of cells using the Annexin V Apoptosis Detection Kit APC (eBioscience, San Diego, CA, USA), according to the manufacturer’s protocol. The proportion of apoptotic cells was evaluated by flow cytometric analysis, using the Gallios flow cytometer system (Beckman Coulter, Brea, CA, USA).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism software, version 4.0 (GraphPad Software, La Jolla, CA, USA). The IC50 values were calculated using GraphPad Prism software.
Human data
This study included NSCLC patient data. The study was reviewed and approved by the Institutional Review Board of Keio University School of Medicine (Tokyo, Japan). Tumor samples were obtained with written informed consent. EGFR mutation analyses were performed on genomic DNA extracted from tumor samples by using the PNA-LNA PCR clamp method.
ACKNOWLEDGMENTS
We thank Ms. Mikiko Shibuya for her excellent technical assistance.
CONFLICTS OF INTEREST
There are no potential conflicts of interest to disclose.
GRANT SUPPORT
This work was supported in part by Grants-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan to K.S. (15K09229), T.B. (15H04833) and H.Y. (17K09667).
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
2. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497-1500.
3. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350:2129-2139.
4. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004; 64:8919-8923.
5. Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004; 101:13306-13311.
6. Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005; 97:339-346.
7. Janne PA, Johnson BE. Effect of epidermal growth factor receptor tyrosine kinase domain mutations on the outcome of patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2006; 12:4416s-4420s.
8. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543-550.
9. Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007; 11:217-227.
10. Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011; 42:9-22.
11. Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006; 125:1137-1149.
12. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007; 7:169-181.
13. Nyati MK, Morgan MA, Feng FY, Lawrence TS. Integration of EGFR inhibitors with radiochemotherapy. Nat Rev Cancer. 2006; 6:876-885.
14. Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007; 4:1669-1679; discussion 1680.
15. Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012; 13:e23-31.
16. Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, Zakowski MF, Kris MG, Ladanyi M. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013; 12:220-229.
17. Oxnard GR, Lo PC, Nishino M, Dahlberg SE, Lindeman NI, Butaney M, Jackman DM, Johnson BE, Janne PA. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol. 2013; 8:179-184.
18. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007; 98:1817-1824.
19. Oxnard GR, Arcila ME, Sima CS, Riely GJ, Chmielecki J, Kris MG, Pao W, Ladanyi M, Miller VA. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011; 17:1616-1622.
20. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005; 352:786-792.
21. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008; 105:2070-2075.
22. Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015; 372:1689-1699.
23. Van Der Steen N, Caparello C, Rolfo C, Pauwels P, Peters GJ, Giovannetti E. New developments in the management of non-small-cell lung cancer, focus on rociletinib: what went wrong? Onco Targets Ther. 2016; 9:6065-6074.
24. Tan CS, Cho BC, Soo RA. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor -mutant non-small cell lung cancer. Lung Cancer. 2016; 93:59-68.
25. Juan O, Yousaf N, Popat S. First-line epidermal growth factor receptor (EGFR) kinase inhibitors for EGFR mutant non-small cell lung cancer: and the winner is. Clin Oncol (R Coll Radiol). 2017; 29:e1-e4.
26. Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, Yamamoto N, Yu CJ, Ou SH, Zhou C, Massey D, Zazulina V, Wu YL. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015; 16:830-838.
27. Yuza Y, Glatt KA, Jiang J, Greulich H, Minami Y, Woo MS, Shimamura T, Shapiro G, Lee JC, Ji H, Feng W, Chen TH, Yanagisawa H, et al. Allele-dependent variation in the relative cellular potency of distinct EGFR inhibitors. Cancer Biol Ther. 2007; 6:661-667.
28. Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005; 2:e313.
29. Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, Huberman MS, Cohen DW, Nakayama S, Ishioka K, Yamaguchi N, Hanna M, Oxnard GR, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013; 5:216ra177.
30. Hirano T, Yasuda H, Tani T, Hamamoto J, Oashi A, Ishioka K, Arai D, Nukaga S, Miyawaki M, Kawada I, Naoki K, Costa DB, Kobayashi SS, et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget. 2015; 6:38789-38803. https://doi.org/10.18632/oncotarget.5887.
31. Jia Y, Juarez J, Li J, Manuia M, Niederst MJ, Tompkins C, Timple N, Vaillancourt MT, Pferdekamper AC, Lockerman EL, Li C, Anderson J, Costa C, et al. EGF816 exerts anticancer effects in non-small cell lung cancer by irreversibly and selectively targeting primary and acquired activating mutations in the EGF receptor. Cancer Res. 2016; 76:1591-1602.
32. Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, Soria JC. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. 2015; 26:2073-2078.
33. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015; 21:560-562.
34. Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, Sequist LV, Engelman JA. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015; 21:3924-3933.
35. Yu HA, Tian SK, Drilon AE, Borsu L, Riely GJ, Arcila ME, Ladanyi M. Acquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol. 2015; 1:982-984.
36. Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS, Janne PA. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res. 2015; 21:3913-3923.
37. Bersanelli M, Minari R, Bordi P, Gnetti L, Bozzetti C, Squadrilli A, Lagrasta CA, Bottarelli L, Osipova G, Capelletto E, Mor M, Tiseo M. L718Q mutation as new mechanism of acquired resistance to AZD9291 in EGFR-mutated NSCLC. J Thorac Oncol. 2016; 11:e121-123.
38. Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, Fulton L, Hata AN, Lockerman EL, Kalsy A, Digumarthy S, Muzikansky A, Raponi M, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015; 5:713-722.
39. Nukaga S, Yasuda H, Tsuchihara K, Hamamoto J, Masuzawa K, Kawada I, Naoki K, Matsumoto S, Mimaki S, Ikemura S, Goto K, Betsuyaku T, Soejima K. Amplification of EGFR wild type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. 2017; 77:2078-2089.
40. Kobayashi Y, Togashi Y, Yatabe Y, Mizuuchi H, Jangchul P, Kondo C, Shimoji M, Sato K, Suda K, Tomizawa K, Takemoto T, Hida T, Nishio K, Mitsudomi T. EGFR Exon 18 mutations in lung cancer: molecular predictors of augmented sensitivity to afatinib or neratinib as compared with first- or third-generation TKIs. Clin Cancer Res. 2015; 21:5305-5313.
41. Banno E, Togashi Y, Nakamura Y, Chiba M, Kobayashi Y, Hayashi H, Terashima M, de Velasco MA, Sakai K, Fujita Y, Mitsudomi T, Nishio K. Sensitivities to various epidermal growth factor receptor-tyrosine kinase inhibitors of uncommon epidermal growth factor receptor mutations L861Q and S768I: what is the optimal epidermal growth factor receptor-tyrosine kinase inhibitor? Cancer Sci. 2016; 107:1134-1140.