
INTRODUCTION
The organ preference in cancer metastasis has been a subject of study and numerous hypotheses for over 125 years. In 1889, the English surgeon Stephen Paget described the propensity of various types of cancer to form metastases in specific organs, which led him to coin the metaphor that the patterns of outgrowth were due to the dependence of the “seeds” (the cancer cells) on the “congenial soil” (the target organ for metastasis) [1]. Even though the analogy continues to be cited in abundance, it has provided absolutely no mechanistic insight. Later, the pathologist James Ewing formulated a more scientific explanation by suggesting that circulatory patterns (of blood and lymph vessels) between a primary tumor and specific secondary organs were sufficient to account for most of the targeted metastases [2]. His model implied a passive role for the cancer cells that are released from their primary tumors and – consistent with their large size and low deformability – get stuck in the first capillary bed they encounter. However, serial passage of a melanoma cell line through mice generated sub-lines with increasing invasive potential and demonstrated that metastasis formation depends – at least in part – on intrinsic characteristics within the transformed cells [3]. Since the mid-1980s [4-6], it has become increasingly clear that the phenomenon of cancer metastasis can be directed by gene expression programs within the tumor cells. Metastatic potential is acquired by these tumor cells through the aberrant expression or splicing of stress response genes [7, 8]. Further, it has been elucidated that beside the positive regulators of dissemination there are gene regulation programs for metastasis suppression [9-11], which need to be inactivated for cancers to disseminate.
In regard to colorectal cancer, there is a knowledge base for the molecular characteristics of metastases to various sites. Genomic signatures are conserved in colorectal liver metastases [12] and include chromosome 20p11 gains [13]. The chemokine receptor CCL-7 [14], the adhesion molecule P-Cadherin [15], the growth factor IGF2 and the intestinal stem cell-specific transcription factor ASCL2 [12] are over-expressed, whereas MMP1 and MMP2 are under-expressed [16] in liver metastasis compared with the primary tumors. 46 genes are differentially regulated between hepatic and pulmonary metastases [17]. A gene expression signature that characterizes lung foci involves the upregulation of FN1, CCL7, MMP7, IGF1, VEGFA, and SRC [18]. The CTHRC1 (Collagen Triple Helix Repeat Containing 1) gene is associated with peritoneal carcinomatosis [19]. The expression of mesenchyme forkhead 1 (FOXC2) in the primary tumor correlates with the degree of lymph node metastasis [20].
A barrier to understanding the genetic signatures of organ preference has been the inherent heterogeneity and clonal evolution of malignant tumors. Studies of breast cancer have increasingly focused on this phenomenon [21]. While a shared pathophysiology is implied by somatic mutations within three genes (TP53, PIK3CA, GATA3) that occur in more than 10% of cases, distinct subsets of genetic and epigenetic abnormalities generate four main breast cancer types [22]. Many mutational processes emerge late, but contribute extensive genetic variation. While most genetic alterations arise in just a fraction of tumor cells, there is a dominant subclonal lineage in every tumor that represents more than half of its cells. The expansion of this dominant subclone to an appreciable mass may represent the final rate-limiting step in breast cancer progression [23]. Tumor cell dissemination, primarily into bone marrow, is an early event in the disease history [24]. There are extensive biomarker differences between the primary tumor and its metastases, as well as among multiple metastases from the same patient. Estrogen and progesterone receptors tend to be down-regulated in the metastatic growths. Variable overexpression occurs in the colonies for cyclooxygenase-2 (COX2), epidermal growth factor receptor (EGFR), MET, and mesothelin [25].
Tumor-host interactions contribute to determining organ preference in metastasis. Circulating cancer cells can recognize target organs through the use of tissue markers [26], such as the interactions of addressins or chemokines with their cognate receptors [27, 28]. The binding motifs of tri-peptides or tetra-peptides on the vasculature may also provide homing signals [29]. As such, the sequence SRL encodes a brain homing motif, CGFE is a motif in lung homing peptides recognized by membrane dipeptidase, and the integrin binding RGD motif homes to sites of neovascularization.
Despite variation from tumor heterogeneity, clonal evolution and host factors, consistent genetic adjustments are required within the tumor cells to complete the process of metastasis. Although research over the past fifteen years has identified some genes, the expression of which is associated with cancer dissemination to specific sites [30-32, 17, 18], gene regulation maps that reflect organ specificity in cancer spread are still lacking. Here we expand on earlier microarray analyses [33, 34] and investigate metastasis-specificity in gene expression signatures from various primary tumors, their metastases, and host tissue in target sites to identify genetically encoded programs that distinguish the disseminated growths.
RESULTS
Gene expression differences delineate metastases from their primary cancers and from their target tissues
To assess overall relatedness across GEO data sets (Supplement 1), we compared samples by principal component analysis. The existence of tight clusters suggests shared features among the metastases; their frequent grouping away from primary tumors and from host tissues implies uniqueness of the metastatic gene expression (top graphs in Figures 1A and 1B). Breast cancer (primary tumor and metastases) displays three distinct clusters, comprising metastases that group far from the primary tumors and only select target sites that group with their primary tumors. Colon cancer has metastases that spread broadly away from the primary tumors (Figure 1A). While metastases to liver and lymph nodes mostly group at a distance to the host tissue (with the exception of breast cancer metastases to the liver and vulva metastases to the lymph nodes), they form distinct clusters (Figure 1B), implying the existence of common genetically encoded programs that uniquely characterize the metastases. We set out to identify the features that set metastases apart from their sources and from their targets.
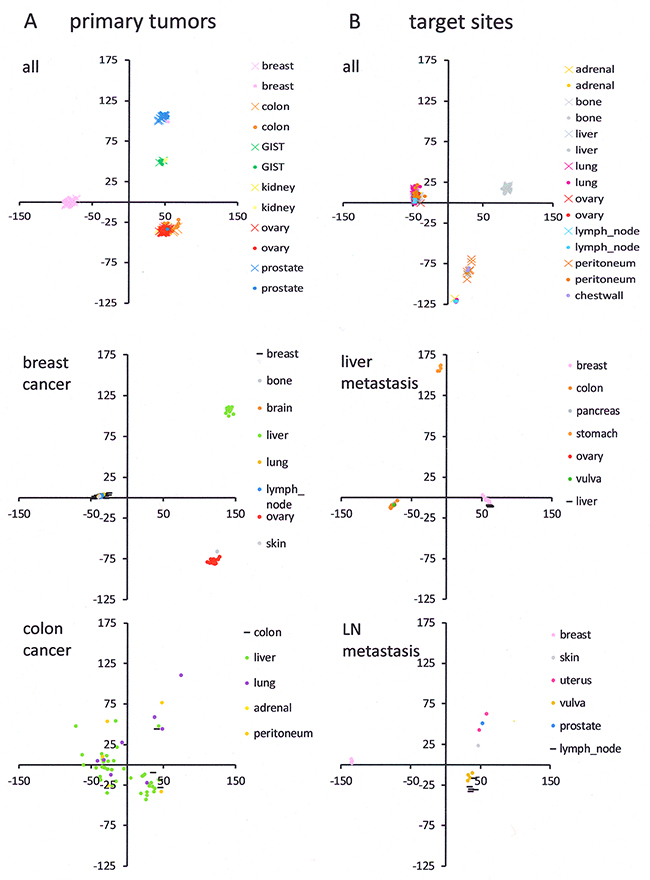
Figure 1: Principal component analysis. (A) Comparison of primary tumors to their metastases in various organs. (B) Comparison of healthy host tissue in metastatic target sites to the metastases in those sites, having originated from various primary tumors. The top graph (A, B) shows the PCA results of the entire data set (the metastases are represented by color-filled circles, the reference values for primary tumors in A and for host tissue in B are depicted as color-matched x, while each gene expression profile is one data point there is overlap). The graphs for the individual sites of cancer origin or individual metastatic target sites (middle and bottom) are subset analyses (the PCA was done separately for each subset shown), selectively for a specific primary tumor and its colonies (A) or a specific healthy host tissue and metastases that target it (B). The primary tumor sites (A) or the host sites (B) are displayed as horizontal black lines. The round symbols, indicating the principal component coordinates for the metastases, are color coded by target site (A) or source tumor (B). Note that all graphs (x-axis PC1 and y-axis PC2) have the same scale to facilitate the comparison of cluster tightness. With the exception of colon cancer, the PC1 contribution was 50-65% and the PC2 contribution was 15-22%.
Metastases are characterized by gene expression changes in four functional groups
We set up a meta-analysis type study to comprehensively evaluate relevant gene expression profiles of solid tumor metastases retrieved from GEO. The initial differential expression analysis compared metastases from various primary sites to the same target organ (all source sites combined for each target organ). Up-regulated genes identify those with higher expression levels in the metastasis samples as compared to the healthy host tissues, down-regulated genes are lower in the metastases than in the host tissue (Supplement 2). We then performed pathway enrichment analysis to identify biological processes significantly affected by the up- and down-regulated genes. Regardless of the metastatic target site, common gene expression differences from the host tissues predominantly reflect functional entities that regulate metabolism, vascularization/tissue remodeling, motility/extracellular matrix interactions, as well as pathways for inorganic ion transport/homeostasis (Figure 2). These four functional groups comprise about one fourth (25.5%) of the top changes in gene ontology categories as compared to the host organ (Supplement 2). In addition, site-specific alterations have consistent characteristics (manuscript in preparation). In sum, the gene expression profile of metastasis is characterized by a core program component plus a specific program component that adjusts to the target site.
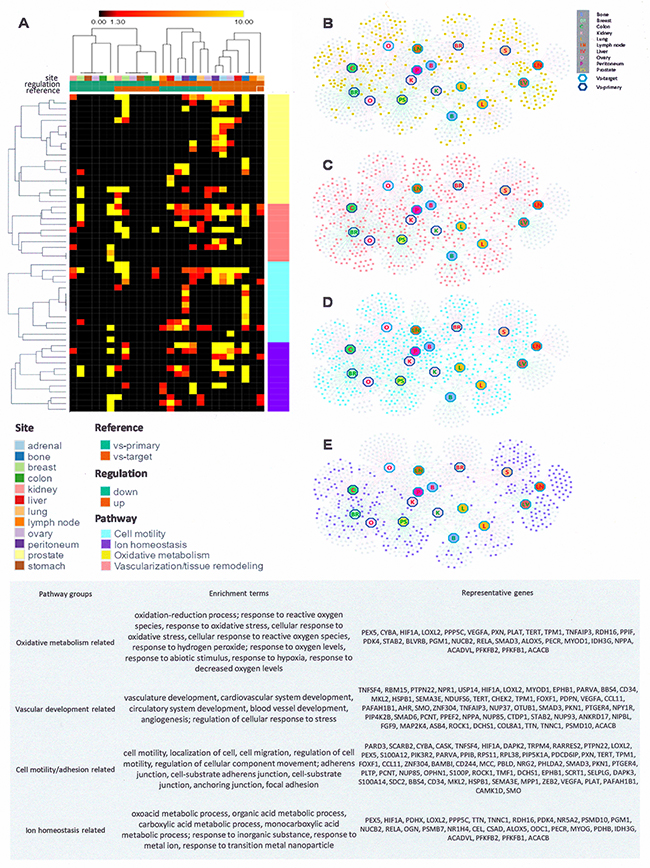
Figure 2: The gene expression core program of metastasis. (A) Heatmap of enrichment terms. Differentially expressed genes in metastases (as compared to their originating primary tumors or to their target host sites) are augmented in four major pathways (colored sectioning on the right). Each row represents an enriched pathway group and each column represents a specific analysis. The columns are arranged as indicated in the bars on the top; the map has been ordered with hierarchical clustering in Spearman correlation distance. (B-E) Networks of differentially expressed genes that are involved the four major pathways of metastasis. Each square represents a gene, and each hexagon represents metastases that up-regulate (red label) or down-regulate (green label) those genes compared with either primary cells (dark blue border) or target cells (light turquoise border). (B) Genes involved in pathways of the oxidative metabolism are highlighted in yellow. (C) Genes involved in vascularization related pathways are highlighted in red. (D) Genes involved in pathways of cell mobility are highlighted in turquoise. (E) Genes involved in ion homeostasis related pathways are highlighted in purple. (Bottom panel) Table of enrichment terms (GO terms and KEGG pathways) in the four major pathway groups. Representative genes are those that are differentially expressed in at least three distinct metastasis comparisons. The functional enrichment analysis of the gene lists was performed using the ToppGene Suite.
Differential expression analysis was also performed comparing primary tumors of specific tissue origin to their respective metastases (all target sites combined for each primary tumor site). Up-regulated genes identify those with higher expression levels in the metastases as compared to the primary cancer of origin, down-regulated genes are lower in the metastases (Supplement 3). We performed pathway enrichment analysis to identify biological pathways significantly affected among up- and down-regulated genes for all metastasis sites from each source site. General changes in the metastases, compared to their primary tumors, occur predominantly in pathways that regulate metabolism, vascularization/tissue remodeling, extracellular matrix interactions/motility, and inorganic ion homeostasis (see Figure 2; Supplement 3). Further, there are changes that are unique for specific metastases-versus-primary tumor comparisons. The results were confirmed in an analysis of the largest GEO data set on breast cancer (Supplement 4).
Metastases are distinguished from their target sites by common gene expression signatures
Select genes are up- or down- regulated compared to host tissue in all or most of the target sites analyzed (Table 1). The deregulation of numerous zinc finger proteins and other transcription factors may reflect the control by designated genetic programs. At all 7 metastatic target sites evaluated, two genes (BSN, YAF2) are induced and one gene (CKAP5) is suppressed in the metastases. Multiple genes are induced or suppressed in 5 or 6 target sites. Those sets of genes seem to constitute essential mediators of metastasis, possibly conveying the ability to survive and expand in a non-cognate microenvironment. It is implied that they are critical for determining whether a cancer forms clinically relevant metastases.
Table 1: Up- or down-regulated common genes in metastases compared to target
UP |
DOWN |
||||||
|---|---|---|---|---|---|---|---|
gene symbol |
target tissue |
gene symbol |
target tissue |
gene symbol |
target tissue |
gene symbol |
target tissue |
BSN |
li:pe:ov:ln:bo:ad:lu |
UTP11L |
pe:li:ov:bo:lu |
CKAP5 |
li:pe:ov:ln:bo:ad:lu |
FSTL4 |
li:ln:ov:bo:lu |
YAF2 |
li:pe:ov:ln:bo:ad:lu |
NUSAP1 |
li:pe:ln:ov:ad |
NAB1 |
li:pe:ov:bo:ad:lu |
CDC37L1 |
pe:li:ov:ln:bo |
MYO9B |
li:pe:ov:ln:bo:ad |
RAB1B |
li:pe:ov:ad:lu |
SLC1A1 |
pe:ov:li:ln:bo:lu |
CMTM6 |
li:pe:ov:ad:lu |
SNTA1 |
pe:li:ov:ln:bo:lu |
A2M |
pe:li:ov:bo:ad |
ADH1A |
pe:li:ln:ov:bo:ad |
ADAMTS1 |
pe:li:ov:ln:ad |
ZNF551 |
ov:li:pe:ln:ad:lu |
SHBG |
pe:ov:li:ad:lu: |
CCL24 |
li:pe:ov:bo:ad:lu: |
FNBP4 |
li:pe:ov:bo:ad: |
GALNT10 |
pe:li:ov:ln:ad:lu |
PLCL2 |
pe:li:ov:ad:lu |
TGM1 |
li:pe:ov:ln:bo:lu |
FOXA1 |
ov:li:pe:ad:lu |
TAS2R4 |
pe:li:ov:bo:ad:lu |
JPH2 |
li:pe:ov:bo:ad |
CDCA3 |
li:ov:pe:bo:ad:lu |
ZFYVE21 |
pe:ov:bo:ad:lu |
SIRT4 |
pe:li:ov:ln:ad:lu |
RBM7 |
pe:ov:ln:bo:ad |
SPG21 |
li:pe:ov:ln:bo:ad |
RBBP5 |
li:pe:ov:ln:lu |
EIF2B5 |
pe:li:ov:ln:bo:ad |
TAOK2 |
li:pe:bo:ad:lu |
TSC22D4 |
pe:li:ov:bo:ad:lu |
SERPINI2 |
ln:ov:bo:ad:lu |
SON |
li:pe:ov:ln:ad:lu |
PPCDC |
li:ov:pe:ad:lu |
TRPC4AP |
pe:ov:ln:bo:ad:lu |
CD151 |
pe:li:ln:ov:ad |
FLRT1 |
li:ov:pe:ln:bo:lu |
TMOD2 |
pe:ov:bo:ad:lu |
ZNF35 |
pe:li:ov:ln:bo:ad |
ARFRP1 |
pe:li:ov:ad:lu |
SP110 |
pe:li:ln:bo:ad:lu |
XRCC1 |
pe:li:ov:ln:ad |
CX3CL1 |
li:pe:ov:ln:bo:ad |
AMELX |
li:ln:ov:bo:lu |
TREH |
li:pe:ln:ov:bo:ad |
CDS2 |
li:ov:pe:ln:bo |
VGF |
ov:pe:li:ln:ad:lu |
HIST1H3E |
li:pe:ov:bo:ad |
SMO |
pe:li:ov:bo:ad:lu |
PRRX2 |
pe:li:ov:bo:ad |
CDKL2 |
pe:ov:ln:bo:ad:lu |
GIMAP6 |
pe:ov:ln:bo:ad |
TIAM1 |
li:pe:ov:bo:ad:lu |
TRIOBP |
li:pe:ov:bo:lu |
CYP46A1 |
li:pe:ov:ln:bo:ad |
MTF2 |
pe:ov:li:bo:ad |
ZKSCAN1 |
li:ov:ln:bo:ad:lu |
DAK |
li:pe:ov:bo:ad |
CD160 |
li:ov:ln:pe:ad |
XPO6 |
li:pe:ov:ln:bo |
ARL4D |
pe:li:ov:ln:bo:ad |
PBXIP1 |
li:pe:ov:ad:lu |
CHFR |
pe:li:ov:bo:ad |
CDK2 |
pe:li:ov:ad:lu |
RIN1 |
pe:li:ov:ln:ad:lu |
RNF10 |
li:pe:ov:bo:ad |
TAF12 |
li:ov:bo:ad:lu |
TACR3 |
pe:ov:bo:ad:lu |
SALL1 |
pe:li:ov:ad:lu |
RPL10A |
li:pe:bo:ad:lu |
ZNF667 |
li:pe:ov:ln:ad |
ANKRD7 |
li:pe:ln:bo:ad |
SLC9A3 |
li:pe:bo:ad:lu |
PRDM14 |
pe:ov:li:ad:lu |
SNCAIP |
li:pe:ov:bo:lu |
BPI |
pe:ov:li:bo:ad |
SOCS3 |
pe:ov:bo:ad:lu |
RAP1A |
li:pe:ov:bo:ad |
MAS1 |
li:pe:ov:ln:bo |
DOLK |
pe:li:ov:ad:lu |
G6PD |
li:pe:bo:ad:lu |
RHOF |
pe:li:ov:ad:lu |
RFK |
li:pe:ov:ln:ad |
ARL14 |
li:ln:ov:ad:lu |
P2RX1 |
pe:li:ov:ad:lu |
PLXNB1 |
pe:li:ov:ad:lu |
GFOD1 |
li:pe:ov:bo:ad |
NEIL3 |
li:pe:ln:ad:lu |
CD84 |
pe:ln:bo:ad:lu |
VEGFC |
li:ov:pe:bo:ad |
PRKD2 |
pe:ln:bo:ad:lu |
TSGA10 |
li:pe:ov:bo:lu |
SHANK1 |
pe:li:ov:ad:lu |
TNS1 |
ov:pe:ln:ad:lu |
ZFP36L1 |
li:ov:bo:ad:lu |
PITPNA |
li:ov:ln:bo:lu |
TNKS |
pe:li:ov:bo:ad |
CUL1 |
li:pe:ov:ad:lu |
AGL |
pe:li:ov:ln:lu |
PLS1 |
ov:pe:li:bo:ad |
POLR3E |
li:pe:ov:ad:lu |
CFTR |
pe:ov:li:ad:lu |
ZFYVE16 |
li:pe:ln:ad:lu |
CD9 |
li:pe:ln:ov:ad |
PPP1R10 |
pe:ov:bo:ad:lu |
ZFP64 |
pe:li:ov:ad:lu |
TBC1D22A |
ov:ln:bo:ad:lu |
HIF1A |
pe:li:ov:bo:ad |
PIGA |
li:pe:ov:bo:ad |
SLC35A3 |
pe:ov:li:ln:bo |
TMEM147 |
li:pe:ov:ln:lu |
XBP1 |
pe:ov:ln:bo:ad |
RREB1 |
li:pe:bo:ad:lu |
DERL1 |
li:ov:ln:bo:ad |
RENBP |
li:ln:ov:bo:ad |
ZDHHC14 |
pe:li:ov:bo:ad |
TAF1 |
pe:ov:ln:bo:ad |
PCSK1 |
li:pe:ov:bo:ad |
FAM135A |
li:pe:ov:ln:ad |
GLRB |
li:pe:ln:ad:lu |
CIRBP |
ov:ln:bo:ad:lu |
SDAD1 |
li:pe:ov:ln:bo |
SCD5 |
pe:li:bo:ad:lu |
IER3IP1 |
pe:ln:bo:ad:lu |
PRODH2 |
pe:ov:ln:bo:ad |
RPL35 |
li:pe:ov:ad:lu |
S100A1 |
ov:li:ln:bo:ad |
TSHB |
li:pe:ov:ad:lu |
PFDN2 |
pe:ov:li:ln:ad |
TEX2 |
pe:li:ov:bo:ad |
LEMD3 |
pe:ov:li:bo:ad |
GPR143 |
li:pe:ln:ov:bo |
TECTA |
li:ov:bo:ad:lu |
STK3 |
pe:li:bo:ad:lu |
CCRL2 |
pe:li:ov:bo:ad |
SHC1 |
li:pe:ov:ln:bo |
PNOC |
li:pe:ov:bo:ad |
RCN1 |
li:pe:ln:bo:ad |
PDPN |
pe:ov:bo:ad:lu |
PAPSS2 |
li:ln:bo:ad:lu |
USP4 |
pe:li:ln:bo:ad |
PRDM1 |
pe:li:ln:bo:ad |
GDPD3 |
li:pe:ov:ln:ad |
SLC38A4 |
pe:ov:bo:ad:lu |
CHN2 |
li:pe:ln:ad:lu |
SFI1 |
li:pe:ov:ln:ad |
SIL1 |
pe:ov:ln:bo:lu |
ABCC2 |
li:pe:ov:ln:ad |
SPAST |
pe:li:bo:ad:lu |
SNRK |
ov:li:ln:ad:lu |
IQSEC1 |
pe:ln:bo:ad:lu |
MYOZ2 |
li:pe:ln:ov:bo |
TULP2 |
pe:ov:li:ad:lu |
OXCT2 |
li:pe:ov:bo:ad |
TTYH1 |
li:ln:bo:ad:lu |
BCL2A1 |
li:ov:pe:ln:ad |
WNT1 |
li:ov:pe:ad:lu |
P4HA1 |
pe:li:ln:ad:lu |
SLC25A28 |
pe:ln:bo:ad:lu |
ASL |
li:pe:ov:ln:ad |
MKNK2 |
li:pe:ov:bo:ad |
DGKE |
pe:li:ov:bo:ad |
FZD6 |
ov:li:pe:ln:ad |
GTF2A2 |
pe:ov:li:bo:ad |
PLCD1 |
li:pe:ov:bo:ad |
PIGC |
li:pe:ov:ad:lu |
IFT57 |
li:pe:ov:bo:lu |
VPREB1 |
pe:li:ov:ad:lu |
WNT11 |
pe:li:ov:bo:ad |
SIVA1 |
li:pe:ov:bo:lu |
ZNF638 |
li:ov:bo:ad:lu |
CTSA |
li:pe:ln:ad:lu |
PGS1 |
pe:li:ov:bo:ad |
SYNE2 |
pe:ov:bo:ad:lu |
GRAP2 |
ov:li:ln:bo:ad |
XPNPEP2 |
li:ln:ov:bo:lu |
SPTLC1 |
pe:li:ov:bo:ad |
SQRDL |
li:pe:bo:ad:lu |
MRPL52 |
ov:li:ln:bo:ad |
SWAP70 |
li:pe:ov:bo:lu |
PLA2G2A |
li:pe:ov:bo:ad |
GPX7 |
pe:li:ov:ad:lu |
EXOSC2 |
pe:li:bo:ad:lu |
TAF2 |
pe:ov:ln:ad:lu |
SLC2A3 |
pe:ov:li:ln:ad |
VILL |
li:pe:ov:bo:ad |
ZSCAN16 |
li:pe:ln:ad:lu |
WT1 |
pe:ln:ov:bo:ad |
SPEN |
pe:li:ln:ad:lu |
SNRPD1 |
li:pe:ov:ad:lu |
LIF |
li:ov:ln:bo:ad |
ZNF281 |
pe:ov:ln:bo:ad |
NOX4 |
li:ov:ln:ad:lu |
TPM1 |
li:ov:bo:ad:lu |
RRH |
pe:li:ov:ln:bo |
ZYX |
pe:ov:bo:ad:lu |
UBAP1 |
li:pe:ov:bo:ad |
OBSCN |
pe:ov:li:ad:lu |
MFAP3L |
pe:ov:bo:ad:lu |
MPP1 |
li:pe:ov:bo:ad |
ADA |
li:pe:ov:bo:ad |
SH3TC1 |
ov:pe:li:bo:ad |
TTC22 |
li:pe:ln:ad:lu |
MRPS22 |
li:pe:ov:bo:ad |
TLE1 |
pe:li:ln:ad:lu |
GZMH |
pe:ov:li:bo:ad |
ADAM18 |
li:ov:ln:bo:ad |
LEFTY2 |
pe:ov:bo:ad:lu |
RARRES2 |
li:pe:ov:bo:ad |
EPN3 |
pe:ln:bo:ad:lu |
CLIP4 |
li:ov:pe:ad:lu |
CALML3 |
li:pe:ln:ad:lu |
TBCA |
li:pe:ov:bo:ad |
ABAT |
pe:li:ov:ln:bo |
SPAG1 |
li:pe:ov:ln:bo |
ZBED2 |
li:pe:ov:ln:bo |
FADD |
pe:ov:li:ln:ad |
LTF |
pe:li:ov:bo:ad |
RPS27L |
pe:ov:ln:bo:lu |
SSX2IP |
pe:ov:ln:bo:ad |
CACNG3 |
pe:ov:ln:ad:lu |
THUMPD2 |
li:ov:ln:ad:lu |
NXT2 |
li:pe:ov:bo:lu |
||
ABCA4 |
pe:li:ln:ad:lu |
FLNA |
li:pe:ov:bo:ad |
AUTS2 |
li:pe:ov:ln:ad |
||
YAP1 |
li:pe:ov:bo:ad |
RPS6KA3 |
pe:li:ov:ad:lu |
GK |
pe:ov:bo:ad:lu |
||
SLC2A11 |
li:ov:pe:ln:lu |
||||||
(Left panel) Genes significantly up-regulated (as described in Supplement 2) in the metastases over host tissue of at least 5 distinct target sites (of 7 sites total). Right panel) Genes significantly down-regulated in the metastases compared to host tissue of at least 5 distinct target sites (of 7). Liver = li, peritoneum = pe, ovary = ov, lymph node = ln, adrenal = ad, bone = bo, lung = lu.
To extend the meta-analysis, we also investigated an individual GEO data set (GSE2109) that contains metastases from various primary cancer specimens. A comparison of 47 gene expression signatures reveals distinct patterns among primary and metastatic sites. Not only do the colorectal cancers cluster, metastases from various primary tumors to the same organ also appear to share gene expression signatures. Most liver metastases cluster together, so do most ovarian metastases, whereas the clustering of lung metastases is less tight (Figure 3A). Metastatic signatures were initially identified by comparing the gene expression levels in primary colon cancers (Figure 3B) to the gene expression levels of colon cancer metastases in the livers (Figure 3C). The gene regulation profiles of the metastases were clearly distinct from the primary tumor. It was possible that contamination from the host tissue (liver) accounted for the altered gene expression profiles. When comparing the metastases to intact liver, it was apparent that some up-regulated genes were likely contributed by the host tissue (ASGR2, CRP, FGA, FGG, FGB, CYP2C9, ACSM2, ALB) (Figure 3D). However, the predominant expression profile in the target organ is very different from the expression profile in the residing metastases. This supports the notion of a specific signature that characterizes metastases (Supplement 5).
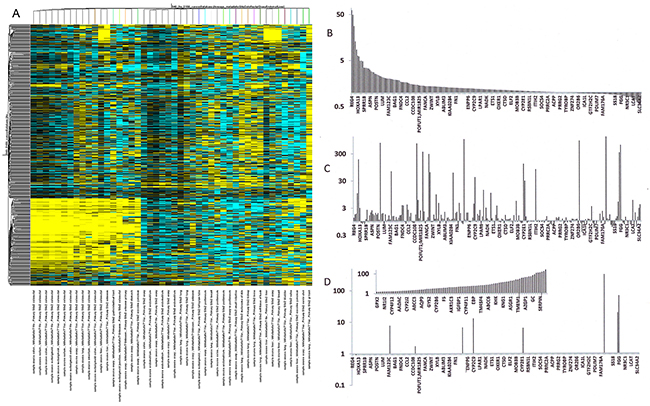
Figure 3: Comparison of the gene expression levels of primary colon cancers to colon cancer metastases in livers. (A) Gene expression profiles among 47 diverse cancers. In the heat map of GSE2109, the colorectal cancers cluster, metastases by various primary tumors to the same organ also appear to share gene expression signatures. (B, C) The gene list is organized from high to low expression in the primary tumor according to GSE2109. The gene expression signatures of the metastases (C) are clearly distinct from the primary tumor (B). (D) Some up-regulated genes in the metastases (ASGR2, CRP, FGA, FGG, FGB, CYP2C9, ACSM2, ALB) are also high in healthy liver tissue. They may have been contributed by the host. However, the major expression profile in the liver (insert shows highly expressed genes in increasing order) is clearly distinct from the metastases, supporting the notion of a cancer-specific signature.
Metastases are distinct from their originating primary tumors through shared gene expression signatures
While cancers have lists for site-specific gene deregulation in their metastatic growths compared to their originating tumors (manuscript in preparation), the expression of some genes is altered in the metastases of all types of cancer, suggesting that these genes are generally essential for conveying invasive potential to a primary tumor (Table 2). Two genes (MAGEA12, TRPC7) are up-regulated and one gene (FCGRT) is down-regulated in the metastases from the 4 cancer types with significant gene lists. Multiple genes are deregulated in the metastases derived from 3 types of cancer. It may be important to note that the gene list in Table 2 may represent necessary contributors for metastasis initiation, while the gene list in Table 1 is likely reflective of metastasis outgrowth.
Table 2: Up- and down-regulated common genes in metastases compared to source tumor
UP |
DOWN |
||||
|---|---|---|---|---|---|
gene symbol |
cancer source |
gene symbol |
cancer source |
gene symbol |
cancer source |
MAGEA12 |
ov:br:pr:ki: |
EIF3A |
ov:br:ki: |
FCGRT |
ov:br:pr:ki: |
TRPC7 |
ov:br:pr:ki: |
TAX1BP3 |
ov:br:ki: |
ERCC5 |
br:pr:ki: |
BBOX1 |
br:pr:ki: |
EYA3 |
br:pr:ki: |
EPN2 |
br:pr:ki: |
IPO9 |
br:pr:ki: |
CTSE |
br:pr:ki: |
C6orf15 |
br:pr:ki: |
TM6SF1 |
br:pr:ki: |
MMP26 |
br:pr:ki: |
NAALADL1 |
br:pr:ki: |
HP1BP3 |
ov:br:ki: |
LIG1 |
ov:br:ki: |
SAMM50 |
br:pr:ki: |
SUV420H1 |
br:st:ki: |
TRIM28 |
br:pr:ki: |
YIPF2 |
co:br:pr: |
ZNF667 |
br:pr:ki: |
CPS1 |
br:pr:ki: |
CCDC85B |
br:pr:ki: |
CEP164 |
br:pr:ki: |
ABCA2 |
ov:br:ki: |
EGR4 |
br:pr:ki: |
MAP2K4 |
br:pr:ki: |
SEMA4A |
br:pr:ki: |
FBXO34 |
br:pr:ki: |
POP4 |
br:pr:ki: |
CNNM2 |
br:pr:ki: |
CALCR |
br:pr:ki: |
SCGB1D2 |
br:pr:ki: |
TLR2 |
ov:br:ki: |
CAV2 |
br:pr:ki: |
FARS2 |
ov:br:pr: |
BRWD1 |
br:pr:ki: |
KRT35 |
br:pr:ki: |
DNAH7 |
ov:br:pr: |
HPN |
br:pr:ki: |
S100A7 |
ov:pr:ki: |
ZDHHC7 |
br:pr:ki: |
RAC1 |
br:pr:ki: |
TLE4 |
br:pr:ki: |
TAP2 |
ov:br:pr: |
DNAJB1 |
ov:br:pr: |
LMO1 |
br:pr:ki: |
GBE1 |
br:pr:ki: |
EFHD1 |
br:pr:ki: |
F9 |
br:pr:ki: |
ZFP36L1 |
br:pr:ki: |
DHX40 |
ov:pr:ki: |
POGK |
br:pr:ki: |
FOXI1 |
br:pr:ki: |
RAB26 |
br:pr:ki: |
TRPV2 |
br:pr:ki: |
ARMCX1 |
br:pr:ki: |
ZNF639 |
br:pr:ki: |
MLLT3 |
br:pr:ki: |
C15orf39 |
br:pr:ki: |
MYT1 |
br:pr:ki: |
ZBTB6 |
br:pr:ki: |
DDX17 |
br:pr:ki: |
VAPA |
ov:br:ki: |
PTBP1 |
br:pr:ki: |
DCHS1 |
br:pr:ki: |
VPS39 |
ov:br:ki: |
MCCC2 |
br:pr:ki: |
POLD1 |
br:pr:ki: |
SMPDL3A |
br:pr:ki: |
UBASH3A |
br:pr:ki: |
C1QA |
br:pr:ki: |
CRISP2 |
ov:br:pr: |
CD163 |
br:pr:ki: |
NARS2 |
ov:br:ki: |
LRRC47 |
br:pr:ki: |
GRIK1 |
br:pr:ki: |
TNPO2 |
br:pr:ki: |
NFIB |
br:pr:ki: |
SEC24C |
br:pr:ki: |
ATXN2L |
br:pr:ki: |
NCR1 |
br:pr:ki: |
CCL8 |
br:pr:ki: |
LYPLA1 |
ov:st:ki: |
CPNE3 |
br:pr:ki: |
ULBP2 |
br:pr:ki: |
HRH4 |
br:pr:ki: |
HECTD3 |
ov:br:ki: |
RPL18 |
br:pr:ki: |
NEIL3 |
br:pr:ki: |
BRSK2 |
ov:br:pr: |
COL17A1 |
br:pr:ki: |
DHDDS |
ov:br:ki: |
SSTR1 |
br:pr:ki: |
AOAH |
br:pr:st: |
GLP1R |
ov:pr:ki: |
COX11 |
br:pr:ki: |
ATP1B3 |
br:pr:ki: |
MVP |
br:pr:ki: |
CXCL6 |
br:pr:ki: |
HAPLN1 |
br:pr:ki: |
SCLY |
br:pr:ki: |
LTA |
br:pr:ki: |
MLC1 |
br:pr:ki: |
MAPK6 |
br:pr:ki: |
CASK |
br:pr:ki: |
ELOVL5 |
br:pr:ki: |
C1QB |
br:pr:ki: |
HIPK1 |
ov:br:pr: |
PARP16 |
br:pr:ki: |
ZNF34 |
br:pr:ki: |
FIGF |
br:pr:ki: |
||
SLC39A2 |
br:pr:ki: |
B3GALT2 |
br:pr:ki: |
||
ZFAND3 |
br:pr:ki: |
PDCD1 |
br:pr:ki: |
||
SNX24 |
ov:pr:ki: |
RGS17 |
br:pr:ki: |
||
TDO2 |
br:pr:ki: |
TCN2 |
br:pr:ki: |
||
ADCY7 |
ov:br:ki: |
RBM28 |
br:pr:ki: |
||
LPO |
br:pr:ki: |
ARHGAP11A |
br:pr:st: |
||
CCDC64 |
br:pr:ki: |
TGM2 |
br:pr:ki: |
||
KRT1 |
br:pr:ki: |
SEMA3E |
br:pr:ki: |
||
SDAD1 |
br:pr:ki: |
TNFSF10 |
br:pr:ki: |
||
MSMB |
br:pr:ki: |
CASD1 |
br:pr:ki: |
||
GDAP1L1 |
br:pr:ki: |
SF3A2 |
br:pr:ki: |
||
SULT1C2 |
br:pr:ki: |
TPM1 |
br:pr:ki: |
||
KCND3 |
br:pr:ki: |
SLC18A2 |
br:pr:st: |
||
ENO2 |
br:pr:ki: |
CACNG3 |
br:pr:ki: |
||
BMP2 |
ov:br:ki: |
MICB |
br:pr:ki: |
||
ZBTB24 |
ov:br:pr: |
DNAJB9 |
br:pr:ki: |
||
FHOD3 |
br:pr:ki: |
WDHD1 |
br:pr:ki: |
||
DMXL2 |
ov:br:ki: |
PSMB1 |
br:pr:ki: |
||
EID1 |
br:pr:ki: |
CAPN7 |
br:pr:ki: |
||
(Left panel) Genes significantly upregulated (as described in Supplement 3) in the metastases over primary tumors of at least 3 distinct types of cancer. Right panel) Genes significantly downregulated in the metastases over primary tumors of at least 3 distinct types of cancer. Ovary = ov, breast = br, prostate = pr, kidney = ki, stomach = st, colon = co.
A murine metastasis model corroborates the meta-analysis results
We tested the meta-analysis results experimentally, using the cell line B16-F10. After tail vein injection, it is metastatic to multiple sites, and the subcutaneous injection approximates a primary tumor. The comparison of B16-F10 metastases to host tissue and primary tumor reveals groups of up- or down-regulated genes in all pairwise comparisons (Figure 4A). A network map shows the importance of metabolism, vascularization and extracellular matrix interactions (Figure 4B). As had been the case in the meta-analysis of human data, the GO categories for biological function by the murine metastases displayed the frequent representation of skewed metabolism, matrix interaction/migration, vascularization, and alteration in ionic homeostasis – for the comparison of metastases to their cognate host site as well as metastases to the primary tumor (Supplement 6). When comparing to the core metastasis signature identified in the meta-analysis (genes up- or down-regulated in most or all metastases in relation to either their primary tumor or the target host tissue) a large proportion of the identified human metastasis genes also display similar expression changes in the murine metastases.
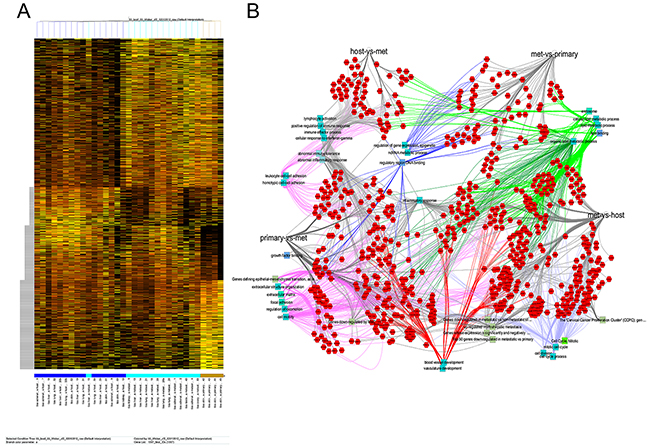
Figure 4: Murine metastases. B16-F10 cells were injected intravenously and disseminated colonies were retrieved after two weeks for cell sorting and ensuing RNA extraction. Site- and tissue-specific gene expression was assessed by RNAseq. (A) Heat map of gene expression comparing host tissue to metastases to primary (undisseminated) tumor. (B) Network map (generated with Cytoscape 3.5.1) indicating the relationships among differentially expressed genes in metastases, cognate host tissue and cutaneous melanoma. The corners contain the comparisons of host versus metastases (top left), primary tumor versus metastases (bottom left), metastases versus host (lower right), and metastases versus primary tumor (top right). Connectivity related to metabolism (top right) is shown with dark or light green edges, vascularization (bottom) has red edges, and cell motility/cell adhesion (lower left) are connected via lilac edges.
DISCUSSION
The genetic core program of metastasis encodes four functional entities, which are activated over the primary tumor and are maintained in the target site. Firstly, the gene expression analysis corroborates recent findings that metastasis genes alter the metabolism of cancer cells to increase energy production [35]. This adjustment conveys anchorage-independent survival [36, 37], a feature that may still be required in the foreign micro-environment of a metastatic target organ [38]. Secondly, angiogenesis and tissue remodeling is also reflected in the gene expression profiles. Tissue remodeling is consistent with our recognition that metastasis genes are stress response genes [7, 8]. A third component is involved in reducing extracellular matrix interactions and enhancing cell motility. This reflects the feature of cancer cell homing. As the number of genes down-regulated in metastases compared to primary tumors is substantial, the inactivation of metastasis suppressor genes may be as important for cancer dissemination as the up-regulation of metastasis genes [34, 39, 40]. Down-regulation was previously observed for bone marrow micro-metastases of breast cancers [41]. Fourthly, we found in a prior study that anchorage-independence is associated with genetic reprogramming for the homeostasis of inorganic ions [42]. This phenomenon seems to play a continued and substantial role in the destination sites of dissemination. Inorganic molecules, including copper and potassium, have been associated with tumor progression and angiogenesis [43-45]. The molecular connections, patho-physiologic effects and therapeutic targeting potential of the metastasis-associated modifications in ionic balance remain to be fully elucidated.
In various target organs, the four core modules are activated through overlapping, but topologically distinct genetically encoded programs. Beside the four required functional entities, adjustments to the particular host tissue micro-environment, which has been colonized, are also important for the outgrowth of clinically relevant cancer metastases. These adaptations reflect the site-specific component of the genetic metastasis signature. The gene expression profile of metastasis is characterized by a core program component that increases motility/metabolism/vascularization/ionic balance plus a specific program component that adjusts to the target site.
Cancer dissemination is associated with alterations in gene expression within the cancer cells. The metastasis gene deregulation is not specific for any primary tumor. This has been known for individual metastasis genes, such as osteopontin, which is associated with the progression of about 30 different cancers [46, 47]. We now expand this paradigm to entire gene expression profiles, as we have found common signatures for metastases that are not active in the original type of cancer (see Table 2) and signatures that are shared in metastases to the same organ (see Table 1).
Gene expression analysis cannot discern whether the RNA profile of a metastasizing cancer cell is altered at the onset of dissemination, and thus is the determinant of organ preference, or is shaped in the metastatic micro-environment, and thus is a consequence of having arrived at a specific target organ (the question is under active discussion [48-50]). Frequently, studies into cancer aggressiveness have focused on biomarkers in the primary tumors. The over-expression of specific genes has been associated with risk for progression in numerous types of cancer. Remarkably, our analysis finds several genes with such attributions, including metalloproteinases (specifically MMP1), chemokines, and POSTN to be suppressed in disseminated growths. This suggests that some of the gene products for tumor progression are only required for the early stages of cancer spread, not for the outgrowth of distant metastases after the transformed cells have reached their target organs. The disappointing clinical trial results with MMP inhibitors may well indicate that the drugs were given too late in disease progression [51].
The meta-analysis approach to the evaluation of actual patient data escapes the compromises associated with small local cohorts in original studies or with in vivo experimental models for cancer, but it is limited by the correlative nature of the results obtained. The robustness of the findings is also dependent on quality and quantity of the input data sets, which vary with source and target sites. Derived from increasingly available computing power and cross-platform normalization algorithms, the ability to assemble multi-gene lists for distinguishing metastases from their primary growths and mapping them to their target organs has identified gene regulation pathways, some of which have thus far escaped laboratory research. Such pathways may be detectable only in their actual molecular pathophysiology context, but not in experimental research models that selectively target individual genes. While requiring confirmation through additional research approaches, the investigation of 8723 genes in 653 gene expression profiles (close to 5.7 million data points) has generated novel insights. Confirmation has been obtained in a murine model of metastasis.
The treatment of metastatic cancer has historically been guided by the originating primary tumor, such that liver metastases from colorectal cancer have received chemotherapy regimens deemed appropriate for primary colorectal cancer whereas liver metastases from pancreatic cancers have been treated with the drug combinations found suitable for cancers originating in the pancreas. Because the gene expression signatures of cancer metastases change substantially from their primary tumors and because metastases from various anatomical sites of origin to the same target organ converge in their gene expression patterns, a more promising strategy could be to focus the choice of combination chemotherapy on the target organ for metastases. This would imply that it may be more efficacious to treat all liver metastases with similar drug regimens that target the genetic core program of metastasis or the site-specific genetic program of liver metastasis, regardless of their organ of origin.
MATERIALS AND METHODS
Meta-analysis
Source data
We investigated the gene expression arrays from a search for solid tumor metastases, comprising a total of 653 metastases and their controls. All array data and their annotations were downloaded from NCBI’s GEO database [52]. Of these 653 mined experiments, 449 had been performed using the Affymetrix platform, and the remaining 204 had utilized the Agilent platform. These samples belong to 8 “same target site” groups and 6 “same source site” groups, where the former refer to specimens derived from different primary tumor sites but metastasized to the same target locations, while the latter refer to specimens with the same primary tumor locations and metastases into various target organs. The “same target site” group includes 294 samples. The “same source site” group includes 551 samples. 192 samples are shared between the “same target site” and the “same source site” groups (Supplement 1). The target site “chestwall” was not sufficiently defined for matching host tissue to be identified, so it was included only in the “same source site” analysis. The meta-analysis was conducted by AccuraScience.
Informatics reconciliation among array platforms and versions
The entire set of array data was derived from 5 different versions of Affymetrix arrays and 1 version of Agilent arrays. Because this study focused on coding RNA and aimed at covering all GEO sets identified in the search, all platforms were included. The source heterogeneity posed two primary challenges for an integrated analysis of array data across different platforms and their multiple versions. The first challenge was an informatics task involving the integration and reconciliation of annotation data from the diverse array platforms and versions. This was handled with a Perl program (Supplement 1). The second challenge was to identify a proper method for removing the batch effect, and to perform proper cross-platform normalization of these array data from various origins. YuGene [53] uses a cumulative proportion transform. Let Pi denote the expression of a gene, and P(i) denote the expression of the same gene, but in descending order. The YuGene transformed value for the gene is
For most data sets, this process achieved excellent normalization (see Supplement 1).
Principal component analysis
Principal component analysis (PCA) allows all samples to be examined on a low-dimensional space, representing the first few principal components. It also offers an effective way to evaluate and compare normalization methods. PCA was carried out using the function pca() in the R (version 3.2.1) package FactoMineR (version 1.31.4) [54], using all genes shared among all array platforms/versions. Configuration: scale.unit = TRUE, ncp = 5, ind.sup = NULL, quanti.sup = NULL, quali.sup = NULL, row.w = NULL, col.w = NULL, graph = TRUE, axes = c(1,2). PCA was done separately for the gene expression profiles in the “same target site” groups and the “same source site” groups, following YuGene-based normalization. The results allowed for a few clearly distinguishable clusters to be identified.
Differential expression analysis
Differential expression analysis of the array data was accomplished using the limma R package [55]. For each of the “same source site” groups, comparison was made between the primary tumor growth (representing the source site), and all metastases (regardless of their target sites) lumped together. For each of the “same target site” samples, comparison was made between the healthy host organ (the target site) and tissue-specific metastases to that site from all source tumors lumped together. Multiple stringency cut-offs (FDR < 0.05, FDR < 0.2 and P value < 0.05) were used to select significantly up- and down-regulated genes.
A second approach conducted pair-wise differential expression analysis between each of the metastatic sites and the primary source tumor, or between the healthy target site and each of the metastases from various primary tumors to that site. Following a non-stringent cut-off, the obtained gene lists were included in the pathway enrichment analyses. This procedure of pairwise evaluation resulted in identical gene lists and GO category lists to the lumped analysis, differing only in stringency (the number of members on the lists). Whether the source sites were lumped or compared pairwise to the metastatic target sites, the smaller of the resulting gene lists/GO categories was always contained in the larger list (with 0-7% of the entries in the shorter list diverging from the longer list, not shown). Hence, we performed the study with lumping together metastases in “same target site” or primary tumors in “same source site” investigations.
Pathway enrichment analysis
Differentially expressed genes were used as input for the pathway enrichment analysis, which identifies biological pathways (denoted as Gene Ontology terms or GO categories) that are associated with the up-regulated or the down-regulated genes. The function enrichDAVID() in the R (version 3.2.1) package clusterProfiler (version 3.0.1) [56] was utilized. Because the maximal number of genes that clusterProfiler can accept as input is 2000, when the number of up- and down-regulated genes was greater only 2000 genes were randomly selected. An FDR cut-off of 0.05 was used in selecting significant pathways. Enriched categories (GO terms) were evaluated with the understanding that these results are highly susceptible to many factors. The P-value of a GO term was produced using Fisher’s exact test (a hypergeometric distribution-based test), which is very sensitive to the numbers of genes in the up- or down-regulated gene list.
Individual data sets
To identify metastatic signatures, we compared the gene expression levels in primary colon cancers with the gene expression levels in colon cancer metastases to the liver. The source data comprise a human data set from GEO (GSE2109). An assessment of 47 signatures, derived from various specimens, reflected on gene expression patterns among primary and metastatic sites. It also allowed the comparison between metastases and healthy target tissues. To identify the gene expression signature of metastases to the same organ from various primary tumors, we analyzed the expression profiles of liver metastases (from cancers of the breast, colorectum, exocrine pancreas, ovary, vulva, and unknown site; each corrected for contamination by the host tissue) for genes with the least variation in expression. This approach was based on the rationale that the highly consistently expressed genes are most likely to be functionally important in organ-specific metastasis, whereas large variations in expression levels may reflect noise generated by the various organs of origin of the primary tumors or by inter-individual differences among patients. Approaches that consider signal-to-noise metrics have been used successfully to analyze metastases of diverse origins [33].
We analyzed a human breast cancer Agilent data set in GEO (GSE26338-GPL1390) [57]. From 22576 genes, we identified 687 with location-specific expression patterns by using ANOVA (with equal or non-equal variance assumptions) with Benjamini-Hochberg FDR < 0.05, then combined the total of 1146 genes to seek expression differences above 25%. The results were clustered and divided into 11 k-means groups for the identification of differentially expressed genes. The data were treated with rows (genes, those with few probes averaged to one value) and columns aligned. This first run had grouped the columns according to breast tumor and then the different sites of metastasis.
Multi-pathway analysis
We have brought into production a new web server, http://metastatic.cchmc.org, in which we have gathered and combined a great deal of current reference knowledge and data derived from the literature and from published genomic data sets. The system includes the ability to search for metastatic cancer-related information derived from disease descriptions in OMIM, UMLSKS, mouse knockout and transgenic allele phenotypes, gene ontology and pathway data sources, and gene expression, epigenetic, and chemical biology-based genomic data sets that relate to metastatic experimental models, phenotypes, or observational profiling studies. The tool allows for subsequent integration of this information and the construction of biological networks [58]. Using a database constructed from gene expression microarray data of cancer samples (http://gataca.cchmc.org/gataca/metastatic), we now analyze differential gene expression in distinct metastatic tumors and metastatic sites.
Murine metastasis model
The in vivo experiments were conducted under an IACUC-approved protocol, which followed the standards of the ARRIVE guidelines [59]. The B16-F10 cells were obtained from ATCC. Cells were kept in culture for no more than 4-6 weeks before replacing with a fresh batch. Mycoplasma tests were conducted on a regular basis (every 4-8 weeks). We injected C57Bl/6 TG [UBC-GFP] mice with 0.2 x 106 B16-F10 dsRed melanoma cells via the tail vein. This allowed the formation of disseminated foci without the presence of large primary tumors requiring early termination of the experiment. While pulmonary dissemination was predominant, metastases were found in multiple organ sites. Subcutaneously injected cells (rear flank) served as model for the primary tumor. After 14 days, total necropsy identified metastases as black spots on the surface of target organs (lungs, liver, adrenals, kidney, ovaries, testes). The spots were excised with a narrow margin, single cell suspensions were generated by grinding between frosted glass slides, followed by passing through a strainer. The cells were sorted according to forward scatter, side scatter, red and green fluorescence in order to separate metastatic cells from host cells.
RNA was extracted using the Qiagen RNAeasy kit. An initial amplification step comprised the use of the Ovation RNA-Seq System v2 (NuGEN) to create double-stranded cDNA from 0.5 ng total RNA. The concentrations were calculated using the Qubit dsDNA BR assay. An Agilent RNA Nano 6000 LabChip analyzed the size distribution, and the samples confirmed the expected size distribution traces. Nextgen sequencing was performed on SE100 at 20 million reads per sample, covering 36,000 genes.
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
FUNDING
This research was supported by DOD grant PR094070 to GFW and BA. The breast cancer portion also received support from the Marlene Harris-Ride Cincinnati/Pilot Program to GFW.
REFERENCES
1. Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889; 133:571-573.
2. Ewing J. Neoplastic Diseases. A Treatise on Tumors. Philadelphia/London: W.B. Saunders Co. 1928.
3. Fidler IJ. Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res. 1975;35: 218-224.
4. Matrisian LM, Bowden GT, Krieg P, Fürstenberger G, Briand JP, Leroy P, Breathnach R. The mRNA coding for the secreted protease transin is expressed more abundantly in malignant than in benign tumors. Proc Natl Acad Sci USA 1986;83:9413-9417.
5. Senger DR, Perruzzi CA, Papadopoulos A. Elevated expression of secreted phosphoprotein I (osteopontin, 2ar) as a consequence of neoplastic transformation. Anticancer Res. 1989;9:1291-1299.
6. Günthert U, Hofmann M, Rudy W, Reber S, Zöller M, Hausmann I, Matzku S, Wenzel A, Ponta H, Herrlich P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 1991;65:13-24.
7. Weber GF, Ashkar S. Stress response genes - the genes that make cancer metastasize. J Mol Med. 2000;78:404-408.
8. Weber GF. Molecular mechanisms of metastasis. Cancer Lett. 2008;270:181-190.
9. Steeg PS, Bevilacqua G, Kopper L. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst. 1988;80:200-204.
10. Alvarez OA, Carmichael DF, DeClerck YA. Inhibition of collagenolytic activity and metastasis of tumor cells by a recombinant human tissue inhibitor of metalloproteinases. J Natl Cancer Inst. 1990;82:589-595.
11. Lee JH, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, Welch DR. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J. Natl. Cancer Inst. 1996;88:1731-1737.
12. Stange DE, Engel F, Longerich T, Koo BK, Koch M, Delhomme N, Aigner M, Toedt G, Schirmacher P, Lichter P, Weitz J, Radlwimmer B. Expression of an ASCL2 related stem cell signature and IGF2 in colorectal cancer liver metastases with 11p15.5 gain. Gut 2010;59:1236-1244.
13. Mekenkamp LJ, Haan JC, Koopman M, Vink-Börger ME, Israeli D, Teerenstra S, Ylstra B, Meijer GA, Punt CJ, Nagtegaal ID. Chromosome 20p11 gains are associated with liver-specific metastasis in patients with colorectal cancer. Gut. 2013;62:94-101.
14. Cho YB, Lee WY, Choi SJ, Kim J, Hong HK, Kim SH, Choi YL, Kim HC, Yun SH, Chun HK, Lee KU. CC chemokine ligand 7 expression in liver metastasis of colorectal cancer. Oncol. Rep. 2012;28:689-694.
15. Sun L, Hu H, Peng L, Zhou Z, Zhao X, Pan J, Sun L, Yang Z, Ran Y. P-cadherin promotes liver metastasis and is associated with poor prognosis in colon cancer. Am J Pathol. 2011;179:380-390.
16. Wong JC, Chan SK, Schaeffer DF, Sagaert X, Lim HJ, Kennecke H, Owen DA, Suh KW, Kim YB, Tai IT. Absence of MMP2 expression correlates with poor clinical outcomes in rectal cancer, and is distinct from MMP1-related outcomes in colon cancer. Clin Cancer Res. 2011;17:4167-4176.
17. Kim SH, Choi SJ, Park JS, Lee J, Cho YB, Kang MW, Lee WY, Choi YS, Kim HK, Han J, Chun HK, Kim J. Tropism between hepatic and pulmonary metastases in colorectal cancers. Oncol Rep. 2012;28:459-464.
18. Kim SH, Choi SJ, Cho YB, Kang MW, Lee J, Lee WY, Chun HK, Choi YS, Kim HK, Han J, Kim J. Differential gene expression during colon-to-lung metastasis. Oncol Rep. 2011;25:629-636.
19. Tan F, Liu F, Liu H, Hu Y, Liu D, Li G. CTHRC1 is associated with peritoneal carcinomatosis in colorectal cancer: a new predictor for prognosis. Med Oncol. 2013;30:473.
20. Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, Iinuma H, Kanazawa T, Tanaka T, Konishi T, Ikeuchi H, Eshima K, Muto T, Nagawa H. Gene expression of mesenchyme forkhead 1 (FOXC2) significantly correlates with the degree of lymph node metastasis in colorectal cancer. Int Surg. 2011;96:207-216.
21. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400-404.
22. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70.
23. Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, Shlien A, Cooke SL, Hinton J, et al. The life history of 21 breast cancers. Cell. 2012;149:994-1007.
24. Sänger N, Effenberger KE, Riethdorf S, Van Haasteren V, Gauwerky J, Wiegratz I, Strebhardt K, Kaufmann M, Pantel K. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int. J. Cancer. 2011;129:2522-2526.
25. Wu JM, Fackler MJ, Halushka MK, Molavi DW, Taylor ME, Teo WW, Griffin C, Fetting J, Davidson NE, De Marzo AM, Hicks JL, Chitale D, Ladanyi M, et al. Heterogeneity of breast cancer metastases: comparison of therapeutic target expression and promoter methylation between primary tumors and their multifocal metastases. Clin Cancer Res. 2008;14:1938-1946.
26. Weber GF. Molecular Mechanisms of Cancer. Dordrecht: Springer. 2007.
27. Streeter PR, Berg EL, Rouse BT, Bargatze RF, Butcher EC. A tissue-specific endothelial cell molecule involved in lymphocyte homing. Nature. 1988;331:41-46.
28. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-56.
29. Ruoslahti E. Vascular zip codes in angiogenesis and metastasis. Biochem Soc Transactions. 2004;32:397-402.
30. Nishimori H, Yasoshima T, Hata F, Denno R, Yanai Y, Nomura H, Tanaka H, Kamiguchi K, Sato N, Hirata K. A novel nude mouse model of liver metastasis and peritoneal dissemination from the same human pancreatic cancer line. Pancreas. 2002;24:242-250.
31. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537-549.
32. Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, Ponomarev V, Gerald WL, Blasberg R, Massagué J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44-55.
33. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49-54.
34. Daves MH, Hilsenbeck SG, Lau CC, Man TK. Meta-analysis of multiple microarray datasets reveals a common gene signature of metastasis in solid tumors. BMC Med Genomics. 2011;4:56.
35. Weber GF. Metabolism in cancer metastasis. Int J Cancer. 2016;138:2061-2066.
36. Shi Z, Mirza M, Wang B, Kennedy MA, Weber GF. Osteopontin-a alters glucose homeostasis in anchorage independent breast cancer cells. Cancer Lett. 2014;344:47-53.
37. Shi Z, Wang B, Chihanga T, Kennedy MA, Weber GF. Energy metabolism during anchorage independence. Induction by osteopontin-c. PLos One. 2014;9:e105675.
38. Kim HM, Jung WH, Koo JS. Site-specific metabolic phenotypes in metastatic breast cancer. J Transl Med. 2014;12:354.
39. Yoshida BA, Sokoloff MM, Welch DR, Rinker-Schaeffer CW. Metastasis-suppressor genes: a review and perspective on an emerging field. J Natl Cancer Inst. 2000;92:1717-1730.
40. Shevde LA, Welch DR. Metastasis suppressor pathways--an evolving paradigm. Cancer Lett. 2003;198:1-20.
41. Woelfle U, Cloos J, Sauter G, Riethdorf L, Jänicke F, van Diest P, Brakenhoff R, Pantel K. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 2003;63:5679-5684.
42. He B, Mirza M, Weber GF. An osteopontin splice variant induces anchorage independence in human breast cancer. Oncogene. 2006;25:2192-2202.
43. Garber K. Targeting copper to treat breast cancer. Science. 2015;349:128-129.
44. Pardo LA, Stühmer W. The roles of K+ channels in cancer. Nat Rev Cancer. 2014;14:39-48.
45. Hu Z, Fan C, Livasy C, He X, Oh DS, Ewend MG, Carey LA, Subramanian S, West R, Ikpatt F, Olopade OI, van de Rijn M, Perou CM. A compact VEGF signature associated with distant metastases and poor outcomes. BMC Med. 2009;7:9.
46. Weber GF, Lett GS, Haubein NC. Osteopontin is a marker for cancer aggressiveness and patient survival. Brit J Cancer. 2010;103:861-869.
47. Weber GF, Lett GS, Haubein NC. Meta-analysis of Osteopontin as a clinical cancer marker. Oncology Rep. 2011;25:433-441.
48. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009;9:302-312.
49. Klein CA. Selection and adaptation during metastatic cancer progression. Nature. 2013;501:365-372.
50. Rinker-Schaeffer CW, Hickson JA. Stopping cancer before it colonizes. Nature Med. 2006;12:887-888.
51. Weber GF. Why does cancer therapy lack effective anti-metastasis drugs? Cancer Lett. 2012;328:207-211.
52. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41:D991-995.
53. Lê Cao KA, Rohart F, McHugh L, Korn O, Wells CA. YuGene: A simple approach to scale gene expression data derived from different platforms for integrated analyses. Genomics. 2014;103:239-251.
54. Lê S, Josse J, Husson F. FactoMineR: an R package for multivariate analysis. J Statistical Software. 2008;25:1-18.
55. Smyth GK. Limma: Linear Models for Microarray Data. in: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S (eds.). Bioinformatics and computational biology solutions using R and Bioconductor: Springer. 2005;397-420.
56. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics : J Integrative Biol. 2012;16:284-287.
57. Harrell JC, Prat A, Parker JS, Fan C, He X, Carey L, Anders C, Ewend M, Perou CM. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2012;132:523-535.
58. Karns R, Tabar S, Bardes EE, Jegga AG, Aronow BJ. How Do Bioinformatics Approaches Apply to the Analysis and Understanding of Disease Pathology? In: McManus LM, Mitchell RN (Eds.). Pathobiology of Human Disease. A Dynamic Encyclopedia of Disease Mechanisms (Reference Module in Biomedical Sciences). 2014;4140-4157.
59. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412.