
Introduction
Polycystic ovary syndrome (PCOS) is the most common endocrinopathy, affecting about 10% of the reproductive-age female population [1], which has significant and diverse clinical symptoms including reproductive dysfunction (chronic anovulation, polycystic ovary, hyperandrogenism and hirsutism), metabolic disorders (insulin resistance, abdominal obesity, hypertension and dyslipidemia), and psychological distress (anxiety, depression, and sensitivity to stress) [1-4]. In addition to genetic predisposition, environmental and lifestyle factors contribute to the pathogenesis of PCOS [5]; an emerging body of evidence suggests that epigenetic events may in part be responsible for the development of PCOS [6-7]. In the past several years, genomics, proteomics and metabolomics studies have been performed with the goal of identifying factors linked to these disorders [5,8], and have provided reliable information about the molecular pathways involved in PCOS. However, to date, little is known about epigenomics, especially the DNA methylation profiles in the pathophysiology of PCOS. DNA methylation is an important epigenetic phenomenon that modulates gene expression without affecting the DNA sequence, to adapt to the environmental and lifestyle changes [9]. For this reason, the present study was undertaken to investigate comprehensive DNA methylation profiling, together with transcriptome analysis in ovaries from PCOS patients and normal ovaries, and to provide novel insight into the epigenetic-mediated dysfunction in the pathogenesis of PCOS.
Results
Differentially methylated and expressed profiling in PCOS
DNA methylation analysis showed 7929 differentially methylated CpG sites, about 3.2% of total sites in PCOS ovaries compared to normal ovaries (Fig. 1A and Supplementary Table 1); the hypermethylated and hypomethylated CpG sites were about 59.8% or 40.2%, respectively (Fig. 1B). Interestingly, there was a significant difference in methylated CpG content and neighborhood context (N_Shelf, N_Shore, Island, S_Shore, S_Shelf, Open Sea) between hypermethylated or hypomethylated CpG sites; in particular, less hypermethylated sites, but many more hypomethylated sites were residing in CpG islands and N_Shore (Fig. 1C and D). However, there was no significant difference in methylated CpG sites residing in chromosomal locations (Chr1-22, and XY), coding distribution (Coding RNA, Non-coding RNA, Other) and gene structure (TSS1500, TSS200, 5’UTR, First exon, Gene body, 3’UTR, Other) (Fig. 1C and D). As shown in Supplementary Table 2, expression profiling analysis showed 650 differential transcripts in PCOS ovaries compared to normal ovaries. Hierarchical clustering of the differentially methylated and expressed probes identified PCOS-specific gene clusters and similarities (Supplementary Fig. 1). Overall, these data suggest the existence of a marked difference in methylation and expression patterns, both of which are implicated in the development of PCOS.
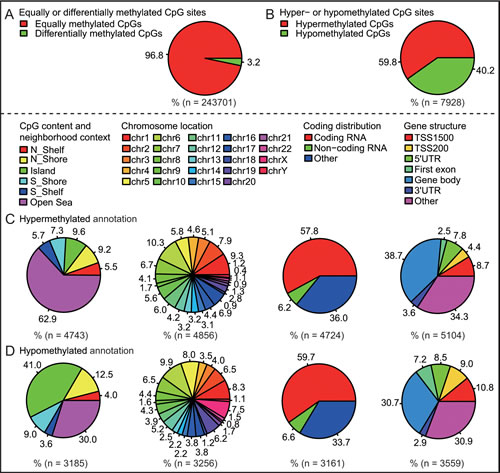
Fig 1: Characteristic methylation patterns between PCOS ovaries and normal ovaries. A, Percentage of equally or differentially methylated CpG sites. B, Percentage of hypermethylated or hypomethylated CpG sites. C, Hypermethylated annotation of differentially methylated CpG sites. D, Hypomethylated annotation of differentially methylated CpG sites.
GO analysis of the differentially methylated and expressed genes associated with PCOS
To begin defining the functional significance of the extensive changes in DNA methylation and gene expression profiling of PCOS, GO analysis was performed, which revealed distinct functional categories for the PCOS-associated gene lists (Supplementary Table 3). As shown in Fig. 2A, these differentially methylated genes were widely associated with cell adhesion, the regulation of GTPase activity, neuron differentiation, synapse organization, skeletal system development and extracellular structure organization. In addition, differentially expressed genes were found to be highly involved in basal cellular processes like the regulation of transcription, cell death, apoptosis, cell proliferation, and response to stress, endogenous stimuli, hormone stimuli and organic substances (Fig. 2B).
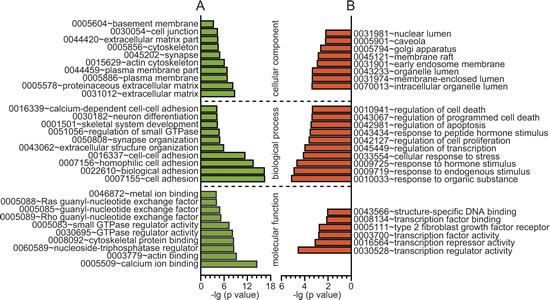
Fig 2: GO analysis of the differentially methylated and expressed genes. A, GO analysis of differential methylated genes (β > 0.1, P < 0.05). B, GO analysis of differential expressed genes (fold change > 1.5, P < 0.005). X axis, negative logarithm (-lg) of the p value; Y axis, GO category. The top 10 GO terms were shown if there were more than 10 terms.
KEGG analysis of the differentially methylated and expressed genes associated with PCOS
To further investigate key pathways linked to these distinct genes, the interaction of the significant pathways associated with PCOS was built according to the KEGG database; the distinct functional categories for the PCOS-associated gene lists are shown in Supplementary Table 4. Our analysis showed that these differentially methylated genes are widely involved in various cellular pathways; the top 10 are linked to type I diabetes mellitus, graft-versus-host disease, viral myocarditis, regulation of actin cytoskeleton, dorso-ventral axis formation, allograft rejection, focal adhesion, autoimmune thyroid disease, cell adhesion molecules and antigen processing and presentation (Fig. 3A), while these differentially expressed genes were only distributed in three significant pathways, including type I diabetes mellitus, the p53 signaling pathway and the NOD-like receptor signaling pathway (Fig. 3B). Notably, the co-existence of the type I diabetes mellitus pathway was observed in differentially expressed and methylated profiles.
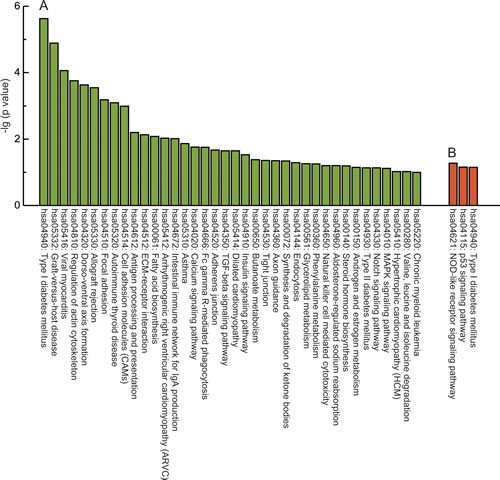
Fig 3: KEGG analysis of the differentially methylated and expressed genes. A, KEGG analysis of differential methylated genes (β > 0.1, P < 0.05). B, KEGG analysis of differential expressed genes (fold change > 1.5, P < 0.005). Y axis, negative logarithm (-lg) of the p value; X axis, KEGG category. The top 10 KEGG terms were shown if there were more than 10 terms.
Integrative analysis of differential DNA methylation and gene expression associated with PCOS
Accumulating evidence demonstrates that DNA methylation is an important epigenetic modification involved in regulating gene expression [10,11]. To date, mounting evidence indicates that DNA hypomethylation or hypermethylation is an alternative mechanism for gene activation or silencing, respectively. However, an emerging body of evidence suggests that DNA methylation is sometimes positively correlated with gene transcription [12,13]. As shown in Fig. 4 and Table 1, we identified 16 hypomethylated and up-regulated genes, 17 hypermethylated and down-regulated genes, 15 hypermethylated and up-regulated genes and 6 hypomethylated and down-regulated genes by integrating DNA methylation and gene expression data; the correlation coefficient data are shown in Supplementary Table 5. In addition, some of the differences were located in methylation patterns in promoter regions, which can modulate gene expression by affecting the binding of transcription factors. Therefore, as shown in Supplementary Table 6, all transcription factors which may bind the differentially methylated sites were predicted.
Table 1: Integrated analysis of DNA methylation and mRNA expression associated with PCOS
Gene_Symbol |
|
Hypomethylated and upregulated genes |
AIFM1, ALS2CR11, ANKRD11, CXorf36, DCAF12L1, FAM188B, FLJ44606, GEMIN8, HCG4, HLA-B, HSD17B1, ITGBL1, MGC16121, NR0B1, TMEM132A, TPRG1 |
Hypermethylated and downregulated genes |
AFAP1, CCL2, CREM, CYP4X1, DDB2, DIP2C, GALNTL2, GNG4, KIAA1683, MAFK, NOV, PRDM1, RASSF5, RGMB, SSBP2, WDR44, ZNF503 |
Hypermethylated and upregulated genes |
DNAJC5, FAHD1, FBN1, HLA-F, HRH1, KCNMA1, MAD1L1, MRVI1, NAV2, NBAS, PLXNC1, RNF213, TNIK, TRIM14, ZFAND3 |
Hypomethylated and downregulated genes |
INADL, NRK, ODZ2, PPAP2C, RAB21, RLIM |

Fig 4: Chromosome graph of differentially methylated genes correlated with gene transcription. 54 genes exhibited a significant correlation between DNA methylation and gene expression levels, which were located at different chromosome (chr) positions.
Discussion
Emerging evidence indicates that epigenetic mechanisms may in part be responsible for the PCOS phenotype [6,7,14]. However, to date, few studies have examined genome-wide DNA methylation profiling, especially together with gene expression profiling for PCOS ovaries. In this study, we reported for the first time that there were at least 3% differentially methylated sites and 650 differential transcripts between PCOS ovaries and normal ovaries.
Functional analysis of these genes associated with differentially methylated sites and expressed transcripts revealed strong enrichment in the biological process and molecular function. For gene expression profiling, these differentially expressed genes were found to be highly involved in basal cellular processes, such as transcriptional regulation, cell proliferation, apoptosis and death, and response to stress and stimulus. For DNA methylation profiling, these differentially methylated genes are especially associated with the regulation of cell adhesion and small GTPase activity. Similarly, several lines of evidence have demonstrated that intercellular and intracellular adhesion molecule-1, vascular cell adhesion molecule-1, and platelet endothelial cell adhesion molecule-1 all have an important role in PCOS [15-17]. Likewise, Small GTPases are a family of hydrolase enzymes that can bind and hydrolyze guanosine triphosphate. The Ras superfamily is the most common of small GTPases, which is further divided into subfamilies: RAS, RHO, RAB, RAP, ARF, RAN, RHEB, RAD and RIT. To date, RAS [18,19], RAC1 [20], RAB5B [21] and RAB8A [22] have been shown to be involved in the PCOS pathophysiology, but other GTPase-related members were not reported.
Epidemiological studies have reported that individuals with type I diabetes have a higher prevalence of PCOS than the general population [23]. Consistent with this, KEGG analysis showed that the type I diabetes mellitus pathway was enriched in differentially expressed profiling. Intriguingly, the type I diabetes mellitus pathway was also significantly enriched in differentially methylated profiling. It seems that abnormal epigenetic and transcriptional regulation may be jointly involved in the progression of type I diabetes-related PCOS. In addition, the p53 signaling pathway is crucial for regulating proliferation, apoptosis, genomic stability and the inhibition of angiogenesis [24]. The NOD-like receptor signaling pathway is a critical regulator of innate and adaptive immune responses [25]. KEGG analysis of expression profiling suggested that the two pathways may potentially play a direct or indirect role in PCOS, although few reports have linked them to PCOS. Immune diseases (such as graft-versus-host disease, allograft rejection, autoimmune thyroid disease and intestinal immune network for IgA production) and inflammatory diseases (such as viral myocarditis and asthma) were significantly enriched in differentially methylated profiling. According to these results, it can be speculated that immune and inflammatory processes are a key regulator of PCOS. It should be noted that chronic inflammation and immune responses have been increasingly recognized as an important cause of endocrine, metabolic and reproductive disorders that characterize PCOS [26].
As shown in Table 1, 54 genes were identified in which the methylated levels were positively correlated with gene transcription. These data may help to identify several novel epigenetically regulated genes that are possibly involved in PCOS, and these aberrantly methylated and expressed genes may also be potential biomarkers of PCOS. Notably, there were several changes in DNA methylation which were located in gene coding regions, therefore not directly regulating gene transcription, but still having the capacity to influence gene expression via alternative splicing programs [27]. Until recent years, CCL2 [28,29] and FBN1 [30] have been suggested to contribute to PCOS, but other genes are still worthy of further investigation.
Taken together, our study demonstrated that differences in genome-wide DNA methylation and gene expression patterns exist between PCOS ovaries and normal ovaries, and understanding the epigenetic mechanisms involved in PCOS may yield new insight into the pathophysiology of the disorder.
Methods
Ethical Statements
The investigation was conducted in accordance with the ethical standards and according to the Helsinki Declaration of 1975.
Patients and tissue collection
This study was approved by the Institutional Review Board at China Medical University. Cervical cancer patients with normal ovaries and PCOS patients were enrolled between April 2012 and July 2012, and all patients gave informed consent. Fresh ovarian tissue was obtained from three cervical cancer patients (Control 1-3) at the follicular phase, with regular menstrual cycles (25–35 days) and younger than 40 years, who underwent radical hysterectomy and pelvic lymphadenectomy for cervical cancer. Fresh ovarian tissue was obtained from three PCOS patients (PCOS 1-3) at the follicular phase, with irregular menstrual cycle and younger than 40 years, who underwent laparoscopic wedge resection. PCOS was defined according to the 2003 Rotterdam criteria [4]. The subject’s characteristics are given in Supplementary Table 7.
DNA methylation profiling analysis
Genomic DNA was extracted from ovarian tissue with a DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). A total of 1.25 μg of genomic DNA was subjected to bisulfite conversion using the EZ DNA Methylation Gold Kit (Zymo Research, Irvine, CA). About 600 ng of the bisulfite-converted DNA was analyzed on an Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA) following the manufacturer’s guidelines. These Chips feature more than 450,000 methylation sites within and outside CpG islands. Methylation values for individual CpG sites were obtained as β-values, calculated as the ratio of the methylated signal intensity to the sum of both methylated and unmethylated signals after background subtraction. The β-values were reported as a DNA methylation score ranging from 0 (non-methylated) to 1 (completely methylated).
Gene expression profiling analysis
Total RNA was extracted from ovarian tissue with Trizol reagents (Invitrogen, Carlsbad, CA) and purified using an RNeasy minikit (Qiagen). RNA purity was assessed by measuring the A260/A280 ratio using a Nanodrop spectrophotometer (NanoDrop, Wilmington, DE), and RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA). Then, 1 μg of total RNA was used for amplification with the MessageAMP amplified RNA kit (Ambion, Foster City, CA). Fragmented and labeled RNA was hybridized to the Affymetrix Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA) according to the manufacturer’s protocol. Arrays were analyzed on a Hewlett-Packard Genearray scanner (Hewlett Packard, Palo Alto, CA) using the GeneChip software (Affymetrix).
Data analysis
DNA methylation profiling data were processed by the Methylation Module of GenomeStudio v1.9 software using default parameters (Illumina). In each individual, probes with a detection P > 0.05 were removed, and β-values in PCOS cases were compared against controls using two-sided Student’s t-tests. Statistical differences were considered significant at β > 0.1. Expression profiling data were assessed using the Affymetrix GeneChip Operating Software (Affymetrix). Statistical differences were considered significant at fold change > 1.5, P < 0.005. DAVID (Database for Annotation, Visualization, and Integrated Discovery) was used to identify functional groups of differential genes and biological processes which are associated with PCOS; Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations were also made.
Conflict of interest statement
The authors declare that they have no competing interests.
Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 81402130) and Doctoral Start-up Foundation of Liaoning Province (No. 20141045). We are grateful to Qi Wang (Shanghai Biotechnology Corporation, Shanghai, China) for data analysis.
Editorial note:
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011; 7:219-231.
2. Teede H, Deeks A, Moran L. Polycystic ovary syndrome: a complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC Med. 2010; 8:41.
3. Witchel SF, Tena-Sempere M. The Kiss1 system and polycystic ovary syndrome: lessons from physiology and putative pathophysiologic implications. Fertil Steril. 2013; 100:12-22.
4. Rotterdam EA-SPCWG. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004; 81:19-25.
5. Insenser M, Montes-Nieto R, Murri M, Escobar-Morreale HF. Proteomic and metabolomic approaches to the study of polycystic ovary syndrome. Mol Cell Endocrinol. 2013; 370:65-77.
6. Qu F, Wang FF, Yin R, Ding GL, El-Prince M, Gao Q, Shi BW, Pan HH, Huang YT, Jin M, Leung PC, Sheng JZ, Huang HF. A molecular mechanism underlying ovarian dysfunction of polycystic ovary syndrome: hyperandrogenism induces epigenetic alterations in the granulosa cells. J Mol Med. 2012; 90:911-923.
7. Wang P, Zhao H, Li T, Zhang W, Wu K, Li M, Bian Y, Liu H, Ning Y, Li G, Chen ZJ. Hypomethylation of the LH/choriogonadotropin receptor promoter region is a potential mechanism underlying susceptibility to polycystic ovary syndrome. Endocrinology. 2014; 155:1445-1452.
8. Escobar-Morreale HF, Insenser M, Corton M, San Millan JL, Peral B. Proteomics and genomics: A hypothesis-free approach to the study of the role of visceral adiposity in the pathogenesis of the polycystic ovary syndrome. Proteomics Clin Appl. 2008; 2:444-455.
9. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004; 22:4632-4642.
10. Li D, Bi FF, Cao JM, Cao C, Liu B, Yang Q. Regulation of DNA methyltransferase 1 transcription in BRCA1-mutated breast cancer: a novel crosstalk between E2F1 motif hypermethylation and loss of histone H3 lysine 9 acetylation. Mol Cancer. 2014; 13:26.
11. Li D, Bi FF, Chen NN, Cao JM, Sun WP, Zhou YM, Cao C, Li CY, Yang Q. Epigenetic repression of phosphatidylethanolamine, N-methyltransferase (PEMT) in BRCA1-mutated breast cancer. Oncotarget. 2014; 5:1315-1325.
12. Kulis M, Queiros AC, Beekman R, Martin-Subero JI. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta. 2013; 1829:1161-1174.
13. Shenker N, Flanagan JM. Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br J Cancer. 2012; 106:248-253.
14. Hickey TE, Legro RS, Norman RJ. Epigenetic modification of the X chromosome influences susceptibility to polycystic ovary syndrome. J Clin Endocrinol Metab. 2006; 91:2789-2791.
15. Moran LJ, Noakes M, Wittert GA, Clifton PM, Norman RJ. Weight loss and vascular inflammatory markers in overweight women with and without polycystic ovary syndrome. Reprod Biomed Online. 2012; 25:500-503.
16. Pepene CE. Soluble platelet/endothelial cell adhesion molecule (sPECAM)-1 is increased in polycystic ovary syndrome and related to endothelial dysfunction. Gynecol Endocrinol. 2012; 28:370-374.
17. Seow KM, Juan CC, Wang PH, Ho LT, Hwang JL. Expression levels of vascular cell adhesion molecule-1 in young and nonobese women with polycystic ovary syndrome. Gynecol Obstet Invest. 2012; 73:236-241.
18. Corbould A, Zhao H, Mirzoeva S, Aird F, Dunaif A. Enhanced mitogenic signaling in skeletal muscle of women with polycystic ovary syndrome. Diabetes. 2006; 55:751-759.
19. Villavicencio A, Goyeneche A, Telleria C, Bacallao K, Gabler F, Fuentes A, Vega M. Involvement of Akt, Ras and cell cycle regulators in the potential development of endometrial hyperplasia in women with polycystic ovarian syndrome. Gynecol Oncol. 2009; 115:102-107.
20. Maurya VK, Sangappa C, Kumar V, Mahfooz S, Singh A, Rajender S, Jha RK. Expression and activity of Rac1 is negatively affected in the dehydroepiandrosterone induced polycystic ovary of mouse. J Ovarian Res. 2014; 7:32.
21. Louwers YV, Stolk L, Uitterlinden AG, Laven JS. Cross-ethnic meta-analysis of genetic variants for polycystic ovary syndrome. J Clin Endocrinol Metab. 2013; 98:E2006-2012.
22. Rosas C, Gabler F, Vantman D, Romero C, Vega M. Levels of Rabs and WAVE family proteins associated with translocation of GLUT4 to the cell surface in endometria from hyperinsulinemic PCOS women. Hum Reprod. 2010; 25:2870-2877.
23. Amato MC, Guarnotta V, Ciresi A, Modica R, Panto F, Giordano C. No phenotypic differences for polycystic ovary syndrome (PCOS) between women with and without type 1 diabetes mellitus. J Clin Endocrinol Metab. 2014; 99:203-211.
24. Stegh AH. Targeting the p53 signaling pathway in cancer therapy - the promises, challenges and perils. Expert Opin Ther Targets. 2012; 16:67-83.
25. Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. 2013; 39:432-441.
26. Ojeda-Ojeda M, Murri M, Insenser M, Escobar-Morreale HF. Mediators of low-grade chronic inflammation in polycystic ovary syndrome (PCOS). Curr Pharm Des. 2013; 19:5775-5791.
27. Li-Byarlay H, Li Y, Stroud H, Feng S, Newman TC, Kaneda M, Hou KK, Worley KC, Elsik CG, Wickline SA, Jacobsen SE, Ma J, Robinson GE. RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc Natl Acad Sci U S A. 2013; 110:12750-12755.
28. Piltonen TT, Chen J, Erikson DW, Spitzer TL, Barragan F, Rabban JT, Huddleston H, Irwin JC, Giudice LC. Mesenchymal stem/progenitors and other endometrial cell types from women with polycystic ovary syndrome (PCOS) display inflammatory and oncogenic potential. J Clin Endocrinol Metab. 2013; 98:3765-3775.
29. Schmidt J, Weijdegard B, Mikkelsen AL, Lindenberg S, Nilsson L, Brannstrom M. Differential expression of inflammation-related genes in the ovarian stroma and granulosa cells of PCOS women. Mol Hum Reprod. 2014; 20:49-58.
30. Prodoehl MJ, Hatzirodos N, Irving-Rodgers HF, Zhao ZZ, Painter JN, Hickey TE, Gibson MA, Rainey WE, Carr BR, Mason HD, Norman RJ, Montgomery GW, Rodgers RJ. Genetic and gene expression analyses of the polycystic ovary syndrome candidate gene fibrillin-3 and other fibrillin family members in human ovaries. Mol Hum Reprod. 2009; 15:829-841.