
INTRODUCTION
In the United States, breast cancer is the second leading cause of death among women (40,610 deaths and 252,710 new cancer cases are estimated in 2017) [1]. There are different therapies (surgery, radiotherapy, chemotherapy, endocrine therapies, or combination of these therapies) available which can treat primary tumor; however, most breast cancer-related deaths occur due to distance organ metastasis (mainly lung, brain and liver). Whereas, most epidemiological studies showed inconsistent finding in a correlation between serum androgen levels and breast cancer risk; but whether this association is consistent in post-menopausal women with breast cancers (who have high androgens levels and low 17β-estradiol) is not understood [2–11]. Previous studies have implicated a possible role for AR in breast cancers [12–14], and AR is found to be predominantly expressed in in-situ, invasive and metastatic breast cancers (~90% of primary tumors and 75% of metastases) [15–18]; however, the precise molecular mechanism(s) for AR's contribution to breast cancer is largely unknown. Hence, a detailed understanding of the potential role of AR in breast cancer is essential to design and develop a newer class of treatment option for the patients.
A meta-analysis of 19 studies that included 7693 patients showed that 74.8% of ER+ patients were concomitantly AR+ while 31.8% of ER- patients were AR+ [19], which suggests that AR expression is high in ER+ tumors compared to ER- tumors. In addition to this Grogg et al. showed that the expression of AR status is highly conserved during breast tumor progression and is expressed in metastasized breast tumors [20]. Although, ER found to regulate the transcription of genes that promote breast cancer cell proliferation, invasion, and survival [12], the role of AR in these processes is not known. Previous studies showed that AR reportedly decreased ER transcriptional activity [14], probably by competing for some of the same ER binding sites [21]. However, a recent ChIP-seq study demonstrated that treatment of MCF-7 breast cancer cells with the AR antagonists enzalutamide or MJC13 (which directly binds to the ligand-binding domain of AR with higher affinity than dihydrotestosterone) decreased the number of ER genomic binding sites by almost 50%, implicating a role for AR in ER genomic binding in response to 17β-estradiol (E2) [21]. This study also found that 75% of E2-induced AR-binding sites overlapped with ER-binding sites and surprisingly, when MCF-7 cells (breast cancer cell line) were treated with enzalutamide or MJC13, the genomic binding of ER was only inhibited at those overlapping sites [21], which implies that AR may facilitate ER binding at those loci. In addition to this, some ChIP-seq studies found that AR and ER interactions occur via a long-range enhancer and promoter chromatin interaction [22, 23]. These findings indicate that AR may preferentially regulate expression of a subset of ER-responsive genes based on the ERE sequences and that may induce metastasis.
The endocrine therapy is the most commonly used treatment for patients with ER+ breast tumors, which either blocks the E2 binding (tamoxifen) or stops the E2 production (aromatase inhibitors) [24, 25]. However, ~30-50% of initially responsive patients develop resistance to these endocrine treatments [26, 27], but the precise molecular mechanism of this process is currently unknown [28]. A study by Amicis et al. showed that AR overexpression induces tamoxifen (Tam) resistance in MCF-7 breast cancer cells [29], which suggests that AR may play a role in this process. On the other hand, most of the ER+ breast cancer found in post-menopausal women and these patients have very less E2 levels and high dihydrotestosterone (DHT) levels under treatment of aromatase inhibitors (AIs) [30, 31]. E2 can also be produced via peripheral conversion of androgens to E2 by CYP19 aromatase [32]. Although AIs like letrozole, exemestane, and anastrozole have been used for the treatment of these patients, the concentrations of androgens are not altered that much in patients [33–35]. Even though previous reports suggested that androgens levels play a significant role in resistance to this therapy (AIs) [36]; again the precise molecular mechanism(s) are yet to be investigated.
A recent study showed that the AR antagonist enzalutamide, which directly binds the ligand binding domain of AR with higher affinity than dihydrotestosterone (DHT), inhibited PT12 patient-derived xenograft (PDX) tumor growth. Enzalutamide also reduced the number of ‘metastatic’ tumors when injected intra-cardially in female NOD-SCID-IL2Rgc-/- mice strain [21]. ER+ breast cancer cell lines that grow as xenograft require exogenous E2 supplements [37] whereas, post-menopausal breast cancer patients have very low E2 in their blood (<18pg/mL) [38]. PDX tumors are more accurate, but they are very hard to establish from ER+ tumors [39] because the engraftment rate is very less (2.5%) [39]. Although these in vivo studies suggested that AR has a role in breast cancer metastasis, most of the current in vivo breast cancer model systems are not clinically relevant and have many limitations which hinder our understanding of the role of AR in breast cancer metastasis. A preclinical study also showed that the ratio of AR and ER is also an independent predictor of disease-free survival (HR = 4.04, 95% CI: 1.68, 9.69; p = 0.002) and disease-specific survival (HR = 2.75, 95% CI: 1.11, 6.86; p = 0.03) [40]. Anti-androgen drugs have been known for the treatment of triple-negative breast tumors, but there are very fewer data available on ER+ tumors. Thus, the range of molecular mechanisms of how AR involves in the regulation of ER transcriptome and what role AR plays in metastasis in ER+ tumors is completely unknown. In this review, we have highlighted multiple mechanisms on how AR is involved in breast cancer progression and its metastasis in ER+ breast tumors in post-menopausal women. We also emphasize the importance of prognostic implications of AR in breast cancers, potential therapeutic molecules, putative challenges, and related methodologies that deal with post-menopausal AR induced breast cancers. Furthermore, this understanding could also help in identifying novel biomarkers for the detection purposes.
Androgen receptor expression in different subtypes of breast tumors
Breast tumors are mainly classified into two subtypes based on cell types and molecular signatures: (1) luminal breast tumor, which expresses keratin 8/18 (a marker of luminal epithelial cells) and (2) basal breast tumor, which expresses keratin 5/6 (a marker of basal epithelial cells) [41]. However, some of the basal keratin can also be found to express in the luminal cells in the terminal duct lobular units (TDLUs) [42]. Based on gene expression profiling, breast tumors are also classified as luminal A, luminal B, human epidermal growth factor receptor 2 (HER2) and triple negative (Figure 1), and each of them has a different gene expression signature [43]. Luminal A (~40%) expresses ER and/or PR, but doesn’t express HER2, and has low levels of a proliferative marker, Ki67 expression [43]. Whereas luminal B (~20%) shows ER and/or PR, and either HER2 positive or HER2 negative and has high levels of a proliferative marker, Ki67 expression [43]. However, HER2 breast tumors don’t express ER and PR but express HER2. Breast tumors which express only ER, PR and HER2, are termed as triple-positive, while those that lack their expressions are classified as triple-negative breast cancer (TNBC) [44]. Among all tumors, the majority (~70%) of them express ER [45] and that a significant portion of metastatic tumors also retain their ER status when the primary tumor is ER+ [46, 47]. AR expression is lower in ERα- breast tumors and higher in ERα+ breast tumors [12, 19].
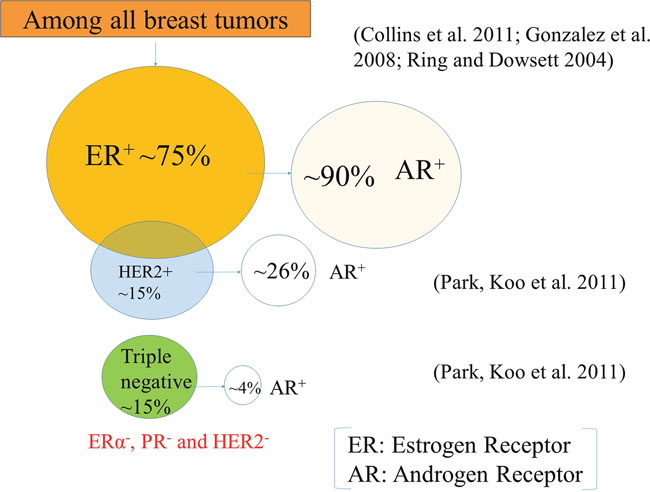
Figure 1: Breast tumor subtypes. Roughly 75% of the tumors express the estrogen receptor (ER), and ~15% of the tumors express the human epidermal growth factor receptor 2 (HER2), and almost 15% triple-negative breast cancers do not express ER, progesterone receptor (PR), and HER2. Interestingly, a significant portion (~90%) of ER-positive (ER+) breast tumors are also androgen receptor (AR) positive (AR+) ones. On the other hand, a very few percentage of HER2+ (26%) and TNBC (4%) express AR.
Classical and non-classical pathway of AR in breast tumors
AR is a type I nuclear receptor which is present in the cytoplasm where it is sequestered by heat shock proteins (HSP-70 and HSP-90) in an inactive state, but when ligands such as androgens bind to its ligand binding domain (LBD) it undergoes a conformational change and is released from the HSPs [48]. Then due to the presence of an intrinsic nuclear localization signal (NLS), AR can translocate to the nucleus where it can bind to specific DNA motifs, and therein it recruits co-activators to regulate transcription of androgen-responsive genes as shown in Figure 2 [48]. In the presence of antagonists, it either recruits co-repressors or prevents co-activators recruitment to AR and represses the transcription of androgen-responsive genes [48]. Testosterone by itself can act as a ligand for AR, or it can also be converted to more potent from DHT via 5α reductase [48]. Testosterone can also get converted to E2 by the CYP19 aromatase as shown in Figure 2 [49]. Most of the previous in vitro studies on breast cancers have employed DHT (a non-aromatizable form of testosterone) or R1881 (a non-aromatizable synthetic analogue of testosterone) as an agonist for AR, but under normal physiological conditions body has other different androgens like dehydroepiandrosterone (DHEA), androstenedione, androstenediol (A5), androsterone, and DHT; and their binding capacities may different (Table 1). A study by Zava et al. showed that 10 nM concentration of DHT could not induce MCF-7 cell proliferation, whereas they found that 1μM concentration did induce proliferation [50]. Whereas, Bhattacharyya et al. showed that anti-estrogen fulvestrant could effectively suppress AR expression in several human prostate cancer cells (LNCaP, LAPC-4 LN95, and LN97 cells) [51].
Table 1: Summary of current literature review on human AR-agonist and antagonist
Agonists/antagonists |
Findings |
References |
|---|---|---|
Agonists: DHT, mibolerone, testosterone, and methyltrienolone |
At concentrations between 0.1 and 1 nM induce the AR NH2-terminal and carboxyl-terminal interaction more than 40-fold |
Kemppainen, Langley et al. 1999 [1] |
Agonists: anabolic steroids, oxandrolone and fluoxymesterone |
At concentrations between 10-100 nM induce the AR NH2-terminal and carboxyl-terminal interaction for up to 23-fold |
Kemppainen, Langley et al. 1999 [1] |
Antagonists: hydroxyflutamide, cyproterone acetate, RU56187 |
At concentrations up to 1 μM no interaction detected |
Kemppainen, Langley et al. 1999 [1] |
Inhibition androgen synthesis: ketoconazole and abiraterone |
A decline (50%) in prostate-specific antigen was observed in 28 (67%) of 42 phase II patients |
Attard, Reid et al. 2009 [2] |
Steroidal antiandrogens: cyproterone |
Several drugs toxicity like hepatotoxicity, interference with libido and potency, cardiovascular side effects were observed |
Jacobi, Altwein et al. 1980 [3] |
Non-steroidal antiandrogens: Flutamide |
No significant differences in efficacy between Flutamide and CPA monotherapy was detected |
Schroder, Whelan et al. 2004 [4] |
Non-steroidal antiandrogens: Bicalutamide |
At a daily oral dose of 25 mg/kg, bicalutamide effected a highly significant reduction in growth of Dunning R3227H transplantable rat prostate tumors |
Furr and Tucker 1996 [5] |
Non-steroidal antiandrogens: Enzalutamide |
It showed improvement in overall survival in metastatic castration-resistant prostate cancer post chemotherapy with docetaxel as a first line treatment before initiation of chemotherapy. |
Ramadan, Kabbara et al. 2015 [6] |
1. Kemppainen JA, Langley E, Wong CI, Bobseine K, Kelce WR, Wilson EM. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Mol Endocrinol. 1999; 13:440-54. https://doi.org/10.1210/mend.13.3.0255.
2. Attard G, Reid AH, A'Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009; 27:3742-8. https://doi.org/10.1200/jco.2008.20.0642.
3. Jacobi GH, Altwein JE, Kurth KH, Basting R, Hohenfellner R. Treatment of advanced prostatic cancer with parenteral cyproterone acetate: a phase III randomised trial. Br J Urol. 1980; 52:208-15.
4. Schroder FH, Whelan P, de Reijke TM, Kurth KH, Pavone-Macaluso M, Mattelaer J, van Velthoven RF, Debois M, Collette L. Metastatic prostate cancer treated by flutamide versus cyproterone acetate. Final analysis of the “European Organization for Research and Treatment of Cancer” (EORTC) Protocol 30892. Eur Urol. 2004; 45:457-64. https://doi.org/10.1016/j.eururo.2003.11.016.
5. Furr BJ, Tucker H. The preclinical development of bicalutamide: pharmacodynamics and mechanism of action. Urology. 1996; 47:13-25; discussion 9-32.
6. Ramadan WH, Kabbara WK, Al Basiouni Al Masri HS. Enzalutamide for patients with metastatic castration-resistant prostate cancer. Onco Targets Ther. 2015; 8:871-6. https://doi.org/10.2147/ott.s80488.
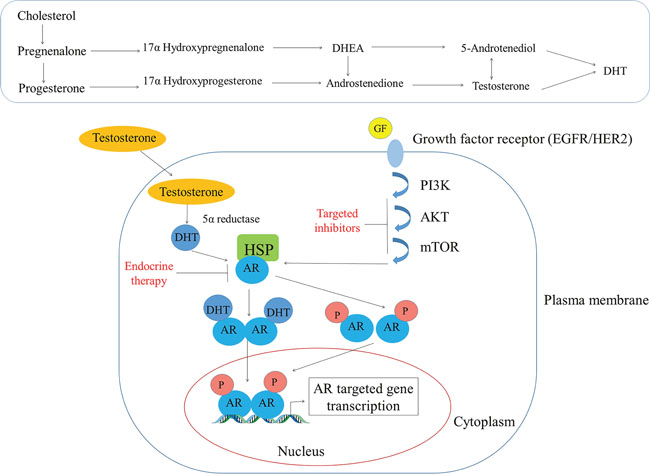
Figure 2: Androgen’s mode of action in the breast tissue. The top panel shows the conversion of cholesterol to different steroid hormones while the bottom panel depicts how testosterone diffuses through the plasma membrane and interacts with AR that is sequestered in the cytoplasm by the heat shock proteins (HSPs). Subsequently, AR undergoes a conformational change and is released from HSPs and then translocate to the nucleus due to its intrinsic nuclear localization domain. In the nucleus, AR binds to specific DNA motifs (AREs) wherein it recruits co-activators (not shown) to regulate the gene transcription activities.
AR can also be activated by non-genomic signaling pathways such as PI3kinase, Akt, and m-TOR [52] (Figure 2). Except these, a study showed that AR could also be regulated by ERK-signaling pathway [53, 54]. Targeting this pathway has been demonstrated to impair androgen-stimulated tumor cell growth, suggesting that targeting this signaling pathway could be a potential therapeutic approach to treat ER-/HER2+ breast cancers [55]. Another study showed bicalutamide; an AR antagonist can block the growth of MDA-MB-453 human breast cancer cells by blocking HER2/HER3 signaling and downstream effectors PI3K/AKT [56]. AR can also found to regulate gene expression through mitogen-activated protein kinase (MAPK) signaling pathway in ERα/PR-negative MCF-10A cells [57]. Some studies also reported that AR induces cell proliferation, migration, and apoptosis in prostate and breast cancer through PTEN cell-signaling pathway [58]. In addition to this, a similar study by this group also showed that AKT and PTEN negatively regulate each other via the transcription factor GATA2 [59]. However, more extensive studies need to be done to target specific pathways which could help to treat post-menopausal women with breast cancer.
The role of AR in the resistance to endocrine therapies
The endocrine therapyis the most commonly used treatment for patients with ER+ breast tumors, which either blocks the E2 binding (tamoxifen) or stops the E2 production (AIs) [24, 25]. However, ~30-50% of initially responsive patients develop resistance to these endocrine treatments [26, 27], but precise molecular mechanisms of this process are currently unknown [28]. Previous studies showed that ER+ tumors which respond to neoadjuvant endocrine therapy has less AR mRNA levels, and tumors which don't respond to this treatment have shown a low level of AR mRNA levels [60, 61]. Similarly, another study demonstrated that resistance to Tam correlates with AR overexpression in both in vivo and in vitro experiment [29]. Since a majority (~70%) of breast tumors found to express ER and a significant portion (~90%) of ERα+ breast tumors are AR+ [62–66], but it’s still unknown whether AR plays any role in this process. A majority of ER+ tumors eventually develop into a more aggressive phenotype and in an E2 independent phenotype without undergoing an alteration of ER status [67]. Whereas, most of the ER+ breast tumors are found in post-menopausal women (~80%), and the normal range of DHT and E2 in postmenopausal women is 30-62pM/L and 55-220 pM/L, respectively, but at the same time DHT is increased, and E2 is decreased in ERα+ breast cancer patients on the AIs anastrozole [30, 31]. Additionally, androgens are found to associate with poor prognosis via inducing EGFR (epidermal growth factor receptor) signaling. In genomic pathway, ER is activated by E2, however as previously described androgen can also be converted to E2 by aromatase enzyme (CYP19) [33], which makes this interplay between these two hormones even more complicated. Most of the previous studies that have been done so far regarding the role of AR in metastasis of ER+ tumors used exogenous E2 and DHT stimuli [68]. So, it’s very important to determine whether AR promotes metastasis of ER+ breast cancer cells in the absence of E2 as a model of postmenopausal women with breast cancer (80% of ER+ breast tumors patients) treated with an aromatase inhibitor (AIs) [38] or in the presence of both E2 and DHT (where ER is fully active). An ER ChIP-seq study found that ERα-binding to chromatin still occurred in ERα+ Tam-resistant MCF-7 (MCF7-TamR) cells, implying that TamR was not due to loss of ER binding to DNA [69]. Moreover, genomic binding of ER was found to increase in metastatic tumors compared to primary tumors [69]. An enriched motif analysis showed that ER binding sites in EREs were increased in Tam-R MCF-7 cells and those were the same motifs observed in the ERα+ metastatic tumors (collected from breast cancer patients) [69]. As Enzalutamide reduced the E2-induced proliferation of MCF7-TamR cells and decreased the growth of MCF7-TamR cells in soft agar [21], suggesting that there is a good opportunity to use AR-antagonist as a therapeutic for patients who don't get a response to Tam. In future, we encourage more study to determine whether by blocking AR can prevent ER-regulated metastasis Tam-R ER+ breast cancer patients through a unique molecular mechanism other than anti-estrogen therapies.
The role of AR in ER+ breast tumors of post-menopausal women
Like AR, ER is a type 1 nuclear receptor, and upon its activation, through both genomic and non-genomic mechanisms, it induces transcription of estrogen-responsive genes [70]. There are two subtypes of ER, ERα and ERβ [70]. In healthy breast tissue, ERβ expression is found to be high, whereas during breast cancer ERα expression is increased [71]. In the genomic pathway, E2 acts as ERs ligand [70], however as previously described androgen can also be converted to E2 [33] (Figure 2) and can serve as an ER ligand. In a study, Szelei et al. showed that higher levels of AR expression are correlated with less proliferation of MCF-7 cell line [72], which suggests that AR may help in the progression of ER+ tumors. Similarly, another study showed that androgens induce apoptotic activity and also down-regulate proto-oncogenes bcl-2 in MCF-7, ZR75-1 and T47-D cells [8, 73]. A ChIP study demonstrated that ERα assembled on the AR gene promoter and this was associated with elevated basal expression of AR mRNA through EGFR/MAPK pathway in Tam-R MCF-7 cells [74]. Similarly, Kumar et al. showed that co-expression of ER and AR reduced the AR transcriptional activity by increasing concentrations of ER cDNA in the presence of both of DHT and E2 in QT6 cells [75]. AR can also bind to EREs, and down-regulate ERα transcriptional activity by competing for the same binding EREs [13]. A ChIP-seq and gene microarray analysis of the ZR-75-1 luminal breast cancer cell line proved that in the presence of higher concentration of one ligand (DHT versus estradiol) over the other could lead to either an increase or decrease in the ER/AR transcriptional activity [76]. For example, if AR binds to EREs it leads to an anti-proliferative effect rather than the proliferative effect of ERα binding to ERE and vice-versa for ERα binding to androgen response elements (AREs). Suzanne et al. showed that AR contributed to ERα transcriptional activity in ER+ MCF-7 cells by overexpressing aromatase (CYP19) and AR [77]. Another study by Richer et al. demonstrated that AR inhibitor Enzalutamide inhibited DHT- driven MCF-7 cells proliferation in vitro as well as tumor growth in both ER+ (MCF7) and ER- (MDA-MB-453) xenografts [40].
AR and ER cross-talk in breast cancers of post-menopausal women
Previous studies showed that ER and AR co-localized in ER+ malignant human breast epithelial cells [13], suggesting that they may interact in the same cell. Metastasis is a multistep process and requires cell proliferation, motility, invasion, and survival of breast cancer cells [78, 79]. Although ERα has been found to regulate the transcription of genes that promote breast cancer cell proliferation, motility, invasion, and survival [12], the role AR in these processes is controversial [13, 14, 57, 75, 80, 81], and may depend on the hormonal milieu. Although targeting of AR has been used in prostate cancer therapy, very less information is available on the therapy against breast cancer and its prognosis. Some recent studies did show that AR can modulate the genomic signaling of ER in MCF-7 cells [82]. Another study demonstrated that co-expression of AR with ERα decreased ERα transactivation by 74% and they also showed that amino-terminal domain of AR interacts with ligand-binding domain of the ERα [14]. Previous studies reported that AR could also bind to EREs, and shown to downregulate ERα transcriptional activity by competing for binding to the same EREs sequences [21]. However, a recent ChIP-seq study of ER binding sites demonstrated that treatment of MCF-7 breast cancer cells with the AR antagonist enzalutamide decreased the number of E2-induced ER genomic binding sites by 50%, implicating a role for AR in ER genomic binding [21]. This study also showed that 75% of the estradiol (E2)-induced AR-binding sites overlapped with ER-binding sites and enzalutamide inhibited ER binding only at those overlapping sites [21], which suggests that AR may facilitate genomic binding of ER at those particular genomic loci. AR binds to the consensus EREs in vitro and is recruited to the promoters of E2-regulated genes containing non-consensus EREs with DHT plus E2 treatment in T47D cells; however, AR-ER doesn't form a heterodimer complex [13]. Hence, one of the possible mechanisms may be that AR may induce some pioneer factor to increase the accessibility ER to the proximal regulatory regions of ER-responsive genes as shown in Figure 3. In addition to this, some ChIP-seq studies found that AR and ER interactions occur via a long-range enhancer along with promoter chromatin interaction [22, 23]. A study also showed that forkhead box (FOX) family member (FOXA1) binding in a subset of genes are the same as that of AR binding sites [83] and silencing of FoxA1 inhibits AR binding in MDA-MB-453 cells [84], which suggests that FOXA1 may act as a pioneer factor for AR. Some studies showed that FOXA1 binding helps H3K4 mono-methylation at enhancers, suggesting that AR may induce ER genomic binding via a long-range chromatin interaction as illustrated in Figure 3 [85–87]. These studies indicate that AR may regulate genomic binding of ER through recruiting some pioneer factors like FOXA1 and regulate transcription of a subset of genes, which may induce breast cancer in post-menopausal women.
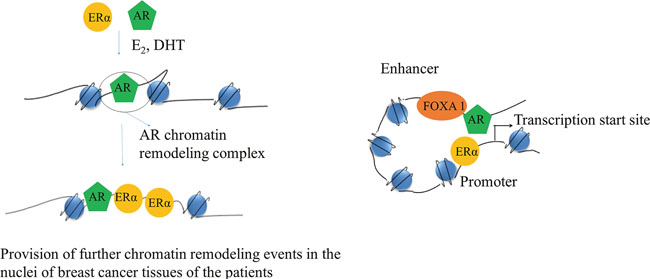
Figure 3: Schematic of a cartoon depicting AR and ERα interaction. On the left its showing how AR can increase accessibility of ERα to the proximal regulatory regions of ERα responsive genes. On the right side, it's showing how AR may also interact with ERα via long-range chromatin interaction.
Previous studies have already been reported that ER promotes transcription of genes which cause breast cancer and its metastasis [12], and the AR inhibitor Enzalutamide reduced ERα genomic binding by 50% [21], but how AR regulates ER transcriptome in ER+ breast cancer cells and whether it induces metastasis is not known. So it's essential to further study by a combined approach of microarray, RNA-seq, ChIP-seq, and ATAC-seq to determine which sets of genes regulated by AR in ER+ breast tumors. There are many potential genes which can be directly regulated by AR as shown in Figure 4, and which can induce breast cancer and its metastasis as listed in Table 2 (more genes can be found in MetaCore and ERGDB database). However, AR can also indirectly regulate ER-responsive genes. For example, HMG20B (transcribed from AR-responsive genes) can form a heterodimer with HMG20A, whereas HMG20A found to stabilize the ERα binding and has a role in EMT [88]. So these genes need to be checked by RNA-seq analysis between AR+ and AR knockout ER+ breast cancer cell line. Previously studies showed that in the presence of DHT membrane bound-AR activates AKT signaling, which subsequently activated ERα [89]. Therefore, AKT-phosphorylation status after treatment with AR ligands also needs to be checked. We also encourage further research to identify novel genes and pathways, which were not previously reported to induce breast cancer; and AR-regulated lincRNA, lncRNA and miRNAs (miRTarBase web tool) which may cause breast cancer in post-menopausal women.
Table 2: List of genes which are involved in breast cancer and its metastasis
Study type |
Methods |
Gene name-fold change (p value) |
References |
|---|---|---|---|
Highly modulated transcripts in ERα+ breast carcinomas |
This in vivo experiment was performed on 26 Stage I – Stage II invasive breast carcinomas (8 ERα- tumors and 18 ERα+). Serial Analysis of Gene Expression based on ERα status is reported. |
Cell proliferation related: |
Abba, Hu et al. 2005 [1] |
Genes altered in co-cultured cancer cells in response to estrogen; Fold change was consider on treatment with E2 (10−8 M) over untreated (EtOH) |
This in vitro study was performed on MDA-MB-231 (ER-negative human breast cancer cell line) cells stably transfected with ERα or ERβ. They used array-based expression profiling and quantitative RT-PCR to study the gene expression profiles. |
Fold changes in MDA-MB-231 cells stably transfected with ERα: |
Wang, Jarrett et al. 2007 [2] |
Genes involved in breast cancer metastatic to bone |
This in vivo study was performed on 107 primary breast tumors in patients. They used microarrays based approach to study the gene expression profile. |
Trefoil factor 1 (TFF1) 3.1 |
Smid, Wang et al. 2006 [3] |
Metastasis-associated protein 1 (MTA1) MTA1 overexpression correlates significantly with tumor grade and angiogenesis in human breast cancers. |
This in vivo experiment was done on 263 surgical specimens of breast cancer cases and IHC was performed to know the MTA1 expression level. |
MTA1 overexpression correlated significantly with higher tumor grade (grades 1 and 2 vs grade 3, P = 0.009). |
Jang, Paik et al. 2006 [4] |
Reduced expression of the breast cancer metastasis suppressor 1 (BRMS1) mRNA is correlated with poor progress in Breast Cancer. |
This in vivo experiment was performed on 161cases of invasive carcinoma of the breast and BRMS1 mRNA was assessed by Light Cycler quantitative real-time reverse transcription-PCR. |
They found that higher BRMS1 expression was correlated with better prognosis and overall disease-free survival in breast cancer. |
Zhang, Yamashita et al. 2006 [5] |
Genes differentially expressed between primary breast cancer and breast cancer brain metastasis |
This in vivo experiment was performed on 44 patients with brain metastasis from breast cancer and expression was measured on the NanoString nCounter Analysis System (NanoString Technologies, Seattle, WA, USA). |
MME (Membrane Metalloendopeptidase) |
Lee, Park et al. 2016 [6] |
1. Abba MC, Hu Y, Sun H, Drake JA, Gaddis S, Baggerly K, Sahin A, Aldaz CM. Gene expression signature of estrogen receptor alpha status in breast cancer. BMC Genomics. 2005; 6:37. https://doi.org/10.1186/1471-2164-6-37.
2. Wang J, Jarrett J, Huang CC, Satcher RL Jr, Levenson AS. Identification of estrogen-responsive genes involved in breast cancer metastases to the bone. Clin Exp Metastasis. 2007; 24:411-22. https://doi.org/10.1007/s10585-007-9078-6.
3. Smid M, Wang Y, Klijn JG, Sieuwerts AM, Zhang Y, Atkins D, Martens JW, Foekens JA. Genes associated with breast cancer metastatic to bone. J Clin Oncol. 2006; 24:2261-7. https://doi.org/10.1200/jco.2005.03.8802.
4. Jang KS, Paik SS, Chung H, Oh YH, Kong G. MTA1 overexpression correlates significantly with tumor grade and angiogenesis in human breast cancers. Cancer Sci. 2006; 97:374-9. https://doi.org/10.1111/j.1349-7006.2006.00186.x.
5. Zhang Z, Yamashita H, Toyama T, Yamamoto Y, Kawasoe T, Iwase H. Reduced expression of the breast cancer metastasis suppressor 1 mRNA is correlated with poor progress in breast cancer. Clin Cancer Res. 2006; 12:6410-4. https://doi.org/10.1158/1078-0432.ccr-06-1347.
6. Lee JY, Park K, Lee E, Ahn T, Jung HH, Lim SH, Hong M, Do IG, Cho EY, Kim DH, Kim JY, Ahn JS, Im YH, et al. Gene Expression Profiling of Breast Cancer Brain Metastasis. Sci Rep. 2016; 6:28623. https://doi.org/10.1038/srep28623.
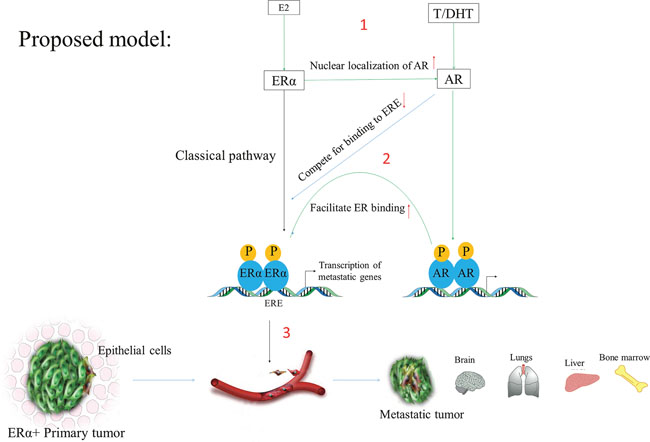
Figure 4: The classical pathway showing E2 and DHT action. In this scheme, AR induces metastasis by regulating ERα transcriptome via recruiting ERα to the promoters of the genes responsible for the metastatic phenotypes. (1) The left side indicates that when E2 binds to ERα it (ERα) it gets recruited to the promoter of ER-responsive genes (EREs). Similarly, DHT stimulates AR activation and its recruitment to the specific DNA motifs. (2) Since AR can also binds and compete to EREs and as well as it can facilitate ER binding via recruitment of other pioneer factors like FOXA1 or long-range promoter-enhancer interaction (need to be further research). (3) In this way, the associated gene expression changes may induce breast cancer phenotypes and its subsequent metastasis in post-menopausal women with breast cancer patients.
Androgen to estrogen dependence in breast cancers of post-menopausal women
It has been known that androgenic hormones (mainly testosterone, dihydrotestosterone and androstenedione) are the major circulating sex hormones in females, which are directly or metabolized into a different form and thereby acting as AR ligands. After menopause, the E2 level goes down to 10 folds, whereas androgens levels decrease only 1.5-fold [90]. Although these excess levels of androgens may reflect increased availability of androgenic ligands [91], breast tissue can metabolize androgens to E2 by enzyme CYP19 aromatase, which is one of the most potent ligands of ER [92, 93]. A study showed that when male mice treated with aromatase inhibitor, AR expression was increased compared to the control mice [94], suggesting that post-menopausal women may express more AR than pre-menopausal women or it could be that women after menopause express more aromatase for the production of E2, which may lead to breast cancers [95]. Similarly, previous studies showed that post-menopausal breast cancer patients have high testosterone levels and high expression of AR, which were significantly associated with the ER+ status of the tumors (OR 2.42; 95% CI, 1.22-4.82) [10, 11]. Although previous studies showed that patients with AR+ breast tumors showed prolonged survival and better response to hormone treatments, as androgens levels are positively associated with breast cancer risk in post-menopausal women, suggesting that It may be the AR expression that correlates with ER expression, but more studies are needed to prove this. Molecular apocrine tumors were known to express AR but not ER, however studies showed that these tumors have an expression profile like that of ER+ luminal breast tumors, and the likely mechanism could be through the AR signaling pathway [84]. Therefore, similar mechanisms may be responsible when ER+ breast cancer patients become resistant to endocrine therapies.
AR as a therapeutic target for breast cancer in post-menopausal women
Breast cancer patients have been treated with targeted therapies or endocrine therapies; however, many patients are not eligible for these treatments because their tumors lack expression of these receptors, and/or their tumors have become resistant to these therapeutic agents. In the first half of 20th century, androgen therapy has been used for breast cancer treatment in post-menopausal women; however, this treatment has been discontinued after some studies showed that androgen could be converted to estrogen [96]. A recent clinical study showed that among 116 cases of invasive breast cancer AR was expressed in 56% of the cases and AR expression was significantly associated with early stage (p < 0.03) of breast cancer [8]. Other clinical studies also showed AR expression is higher (n = 573, 85%) among all breast cancer, however, multivariable analysis for short-term follow-up indicated higher metastasis among patients with AR+ER- tumors (HR 3.5; 95% CI 1.4-9.1) than other (AR and ER combinations).
A previous study has demonstrated that androgen therapy was slightly less effective than estrogen-based therapy [97]. However, a few clinical trials have been conducted for the AR-directed therapy in breast cancers [98–100] and information of all drugs which were used in clinical trials are described in Table 3. Additionally, with the availability of more data regarding the presence of AR and the mechanism of its action regarding androgens in breast cancers, researchers now have opportunities to develop more efficacious and targeted therapies. On the other hand, in breast cancer patients where AR acts as an oncogenic driver, in that situation the anti-androgenic therapy would be more beneficial. Indeed, a drug called bicalutamide has been studied and is currently undergoing a phase II clinical trial (www.clinicaltrials.gov; identifier NCT00468715). Similarly, another drug known as abiraterone that blocks androgen production by inhibiting the cytochrome p-450 (CYP17) is also being studied in a clinical trial (Table 3). Again, more extensive studies are required to identify which patients would be treated most appropriately by these therapies.
Table 3: List of clinical trials that are currently underway for assessing the efficacy and safety of the AR targets
Drugs (Trail ID) |
Drugs details |
Study population |
Study phage (time) |
|---|---|---|---|
Alpelisib and Enzalutamide (NCT03207529) |
Drug name (Alternative name): Alpelisib (BYL-719; NVP-BYL-719) |
AR-positive and PTEN positive metastatic breast cancer patients |
Phase 1 |
Palbociclib |
Drug name (Alternative name): Palbociclib (Ibrance) |
AR-positive metastatic breast cancer |
Phase 2 |
Taselisib and Enzalutamide |
Drug name (Alternative name): Taselisib |
AR-positive metastatic TNBC |
Phase 2 |
Enzalutamide Trastuzumab (NCT02091960) |
Drug name (Alternative name): Trastuzumab (Herceptin) |
AR-positive, HER2 amplified metastatic or locally advanced breast cancer |
Phase 2 |
Enzalutamide and Taxol (NCT02689427) |
Drug name (Alternative name): Taxol (Paclitaxel) |
AR-positive TNBC, stage I–III breast cancer (neoadjuvant therapy) |
Phase 2 |
Enzalutamide (NCT02750358) |
Drug name (Alternative name): Enzalutamide (Xtandi) |
AR-positive TNBC, stage I–III breast cancer (adjuvant therapy) |
Phase 2 |
Abiraterone Acetate (NCT00755885) |
Drug name (Alternative name): Abiraterone Acetate |
Postmenopausal Women With Advanced or Metastatic Breast Cancer |
Phase 2 |
AZD8186 (NCT01884285) |
Drug name (Alternative name): AZD8186 |
Advanced TNBC |
Phase 1 |
Orteronel (NCT01990209) |
Drug name (Alternative name): Orteronel |
AR-positive metastatic breast cancer |
Phase 2 |
VT-464 (NCT02580448) |
Drug name (Alternative name): Seviteronel (VT-464) |
Advanced breast cancer. Phase I: TNBC or ER-positive, HER2 negative |
Phase 2 |
GTx-024 (NCT02368691) |
Drug name (Alternative name): Enobosarm (GTx-024) |
AR-positive advanced TNBC |
Phase 2 |
Pembrolizumab and Enobosarm (NCT02971761) |
Drug name (Alternative name): Pembrolizumab |
Androgen Receptor Positive Metastatic Triple Negative Breast Cancer patients |
Phase 2 |
DISCUSSION
In this review, we discussed the complex AR signaling pathway in ER+ breast tumor under the condition of hormonal levels of post-menopausal women. We noticed a lot of variability of results in vitro study conditions, may be due to the limitation of availability of limited AR+ cell lines and their different sub-clones; use of the various androgens, their concentrations, and duration of treatments may also interfere with the findings [101]. Some labs also used cells from fresh biopsy material of early stage or metastatic in origin of breast tumors. Again co-expression of AR on different subtypes of cancers (ER, HER2 or in TNBC) are not well studied. Apart from these, various studies used different cut-off values for AR (of ≥1%, ≥5% or ≥10%) in IHC to determine AR positivity. Hence due to this lack of guidelines, it’s difficult to compare the prognostic value of AR between different studies.
While reviewing studies, we observed that some subtypes of ER+ tumors showed that AR-agonist might be beneficial while other revealed that AR-antagonist has a positive effect on breast cancer. So, it's impotent to study further to determine which sub-types will be treated with which types of treatments. We also noticed there are inconsistency and need for the more relevant in vivo model system to study these effects. A previous in vivo study of AR function in metastasis of ER+ tumors used a breast cancer cell line xenograft and PDX models [21], which are very difficult to establish [39] and require non-physiological hormone supplements [37]. Therefore, we encourage researchers to test the AR-mediated metastatic growth of ER+ tumors in the mouse intraductal (MIND) model [38], which shows 30-100% engraftment rates and doesn’t require non-physiological hormone supplements [38]. It is also possible that expression of progesterone receptor (PR) and human epithelial growth factor receptor 2 (HER2) can interfere the findings. Hence, we also encourage to use triple-negative breast cancer cell line (which doesn't express PR, ER, and HER2) to know effect only AR in breast cancer. As androgen and estrogen are inter-convertible, we also encourage checking endogenous E2 and DHT level in serum these mice. A previous study also showed that absence of E2 MCF-7 cells up-regulates aromatase, which can metabolize androgens to estrogens [33]. To solve this problem, we suggest checking aromatase (CYP19) expression (western blot analysis); if it's expressed at high levels, then they can treat the MCF-7 cells with AIs along with these hormone treatments.
The role of AR in breast cancer is evolving rapidly; a recent study showed that AR has a role in breast cancer development and metastasis [102]. In this review article, we discussed most of the relevant studies which have been published regarding the pathophysiology of AR on breast cancer mainly in post-menopausal women. We also focused on the crosstalk of AR and ER in tumorigenesis and metastasis in post-menopausal women and some of the therapeutic targets along the AR pathway. This review will bring insight to a therapeutic opportunity for ER+ breast cancer patients (~80% of patients) by AR-antagonist that may reduce metastasis. This study will also provide evidence to determine whether AR-targeted immunohistochemistry of breast cancer samples is a useful diagnostic tool for early detection of metastasis breast cancer. It is also important to study whether by blocking AR can prevent ER-regulated metastasis TamR ER+ breast cancer patients through a unique molecular mechanism other than anti-estrogen therapy.
CONCLUSIONS
In this review, we discussed the effect of AR in breast cancer in post-menopausal women and the opportunities that lie ahead towards developing potential therapeutic strategies which either can be used as monotherapy or in combination with the existing treatment options. We also suggested that further studies in the area are urgently needed to use AR as a therapeutic target for post-menopausal women with breast cancers. Once the findings become available from the ongoing clinical trials with enobosarm and enzalutamide, we will have a better understanding of the pathway(s) that make post-menopausal women vulnerable to this dreaded oncogenic phenotype, and how to treat and diagnose them more effectively.
Abbreviations
AR: Androgen Receptor; ER: Estrogen Receptor; Tam: Tamoxifen; DHT: Dihydrotestosterone; E2: 17β-estradiol; ERE: Estrogen Response Elements; PDX: Patient-Derived Xenograft; ChIP-Seq: Chromatin Immunoprecipitation Sequencing; MIND: Mouse Intraductal; HER2: Human Epidermal Growth Factor Receptor 2; TNBC: Triple-Negative Breast Cancer.
Author contributions
AM wrote the manuscript, MS and SCT helped in assembling and editing of the final version before its submission.
ACKNOWLEDGMENTS
We are thankful to other lab members for their continued support and encouragement. We sincerely appreciate Dr. Carolyn M. Klinge for her critical inputs and thoughtful suggestions.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported, in part, by a grant from the National Institute of Health (Heart, Lung, and Blood Institute; No. HL- 74815).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7-30. https://doi.org/10.3322/caac.21387.
2. Kaaks R, Rinaldi S, Key TJ, Berrino F, Peeters PH, Biessy C, Dossus L, Lukanova A, Bingham S, Khaw KT, Allen NE, Bueno-de-Mesquita HB, van Gils CH, et al. Postmenopausal serum androgens, oestrogens and breast cancer risk: the European prospective investigation into cancer and nutrition. Endocr Relat Cancer. 2005; 12:1071-82. https://doi.org/10.1677/erc.1.01038.
3. Dorgan JF, Longcope C, Stephenson HE Jr, Falk RT, Miller R, Franz C, Kahle L, Campbell WS, Tangrea JA, Schatzkin A. Serum sex hormone levels are related to breast cancer risk in postmenopausal women. Environ Health Perspect. 1997; 105:583-5.
4. Wang DY, Allen DS, De Stavola BL, Fentiman IS, Brussen J, Bulbrook RD, Thomas BS, Hayward JL, Reed MJ. Urinary androgens and breast cancer risk: results from a long-term prospective study based in Guernsey. Br J Cancer. 2000; 82:1577-84. https://doi.org/10.1054/bjoc.1999.1180.
5. Kahan Z, Gardi J, Nyari T, Foldesi I, Hajnal-Papp R, Ormandi K, Lazar G, Thurzo L, Schally AV. Elevated levels of circulating insulin-like growth factor-I, IGF-binding globulin-3 and testosterone predict hormone-dependent breast cancer in postmenopausal women: a case-control study. Int J Oncol. 2006; 29:193-200.
6. Helzlsouer KJ, Gordon GB, Alberg AJ, Bush TL, Comstock GW. Relationship of prediagnostic serum levels of dehydroepiandrosterone and dehydroepiandrosterone sulfate to the risk of developing premenopausal breast cancer. Cancer Res. 1992; 52:1-4.
7. Missmer SA, Eliassen AH, Barbieri RL, Hankinson SE. Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among postmenopausal women. J Natl Cancer Inst. 2004; 96:1856-65. https://doi.org/10.1093/jnci/djh336.
8. Shen G, Ma X. High Levels of 17beta-Estradiol Are Associated with Increased Matrix Metalloproteinase-2 and Metalloproteinase-9 Activity in Tears of Postmenopausal Women with Dry Eye. J Ophthalmol. 2016; 2016:2415867. https://doi.org/10.1155/2016/2415867.
9. Thomas HV, Key TJ, Allen DS, Moore JW, Dowsett M, Fentiman IS, Wang DY. A prospective study of endogenous serum hormone concentrations and breast cancer risk in premenopausal women on the island of Guernsey. Br J Cancer. 1997; 75:1075-9.
10. Secreto G, Venturelli E, Meneghini E, Greco M, Ferraris C, Gion M, Zancan M, Fabricio AS, Berrino F, Cavalleri A, Micheli A. Testosterone and biological characteristics of breast cancers in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2009; 18:2942-8. https://doi.org/10.1158/1055-9965.epi-09-0540.
11. Secreto G, Meneghini E, Venturelli E, Cogliati P, Agresti R, Ferraris C, Gion M, Zancan M, Fabricio AS, Berrino F, Cavalleri A, Micheli A. Circulating sex hormones and tumor characteristics in postmenopausal breast cancer patients. A cross-sectional study. Int J Biol Markers. 2011; 26:241-6. https://doi.org/10.5301/jbm.2011.8883.
12. Cicatiello L, Mutarelli M, Grober OM, Paris O, Ferraro L, Ravo M, Tarallo R, Luo S, Schroth GP, Seifert M, Zinser C, Chiusano ML, Traini A, et al. Estrogen receptor alpha controls a gene network in luminal-like breast cancer cells comprising multiple transcription factors and microRNAs. Am J Pathol. 2010; 176:2113-30. https://doi.org/10.2353/ajpath.2010.090837.
13. Peters AA, Buchanan G, Ricciardelli C, Bianco-Miotto T, Centenera MM, Harris JM, Jindal S, Segara D, Jia L, Moore NL, Henshall SM, Birrell SN, Coetzee GA, et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009; 69:6131-40. https://doi.org/10.1158/0008-5472.can-09-0452.
14. Panet-Raymond V, Gottlieb B, Beitel LK, Pinsky L, Trifiro MA. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol Cell Endocrinol. 2000; 167:139-50.
15. Lea OA, Kvinnsland S, Thorsen T. Improved measurement of androgen receptors in human breast cancer. Cancer Res. 1989; 49:7162-7.
16. Kuenen-Boumeester V, Van der Kwast TH, van Putten WL, Claassen C, van Ooijen B, Henzen-Logmans SC. Immunohistochemical determination of androgen receptors in relation to oestrogen and progesterone receptors in female breast cancer. Int J Cancer. 1992; 52:581-4.
17. Isola JJ. Immunohistochemical demonstration of androgen receptor in breast cancer and its relationship to other prognostic factors. J Pathol. 1993; 170:31-5. https://doi.org/10.1002/path.1711700106.
18. Hall RE, Aspinall JO, Horsfall DJ, Birrell SN, Bentel JM, Sutherland RL, Tilley WD. Expression of the androgen receptor and an androgen-responsive protein, apolipoprotein D, in human breast cancer. Br J Cancer. 1996; 74:1175-80.
19. Vera-Badillo FE, Templeton AJ, de Gouveia P, Diaz-Padilla I, Bedard PL, Al-Mubarak M, Seruga B, Tannock IF, Ocana A, Amir E. Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J Natl Cancer Inst. 2014; 106:djt319. https://doi.org/10.1093/jnci/djt319.
20. Grogg A, Trippel M, Pfaltz K, Ladrach C, Droeser RA, Cihoric N, Salhia B, Zweifel M, Tapia C. Androgen receptor status is highly conserved during tumor progression of breast cancer. BMC Cancer. 2015; 15:872. https://doi.org/10.1186/s12885-015-1897-2.
21. D'Amato NC, Gordon MA, Babbs B, Spoelstra NS, Carson Butterfield KT, Torkko KC, Phan VT, Barton VN, Rogers TJ, Sartorius CA, Elias A, Gertz J, Jacobsen BM, et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol Cancer Res. 2016; 14:1054-67. https://doi.org/10.1158/1541-7786.mcr-16-0167.
22. Biddie SC, John S, Hager GL. Genome-wide mechanisms of nuclear receptor action. Trends Endocrinol Metab. 2010; 21:3-9. https://doi.org/10.1016/j.tem.2009.08.006.
23. Tang Q, Chen Y, Meyer C, Geistlinger T, Lupien M, Wang Q, Liu T, Zhang Y, Brown M, Liu XS. A comprehensive view of nuclear receptor cancer cistromes. Cancer Res. 2011; 71:6940-7. https://doi.org/10.1158/0008-5472.can-11-2091.
24. Criscitiello C, Fumagalli D, Saini KS, Loi S. Tamoxifen in early-stage estrogen receptor-positive breast cancer: overview of clinical use and molecular biomarkers for patient selection. Onco Targets Ther. 2010; 4:1-11. https://doi.org/10.2147/ott.s10155.
25. Nabholtz JM. Long-term safety of aromatase inhibitors in the treatment of breast cancer. Ther Clin Risk Manag. 2008; 4:189-204.
26. Bergh J, Jonsson PE, Lidbrink EK, Trudeau M, Eiermann W, Brattstrom D, Lindemann JP, Wiklund F, Henriksson R. FACT: an open-label randomized phase III study of fulvestrant and anastrozole in combination compared with anastrozole alone as first-line therapy for patients with receptor-positive postmenopausal breast cancer. J Clin Oncol. 2012; 30:1919-25. https://doi.org/10.1200/jco.2011.38.1095.
27. Mouridsen H, Gershanovich M, Sun Y, Perez-Carrion R, Boni C, Monnier A, Apffelstaedt J, Smith R, Sleeboom HP, Jaenicke F, Pluzanska A, Dank M, Becquart D, et al. Phase III study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: analysis of survival and update of efficacy from the International Letrozole Breast Cancer Group. J Clin Oncol. 2003; 21:2101-9. https://doi.org/10.1200/jco.2003.04.194.
28. Clarke R, Shajahan AN, Riggins RB, Cho Y, Crawford A, Xuan J, Wang Y, Zwart A, Nehra R, Liu MC. Gene network signaling in hormone responsiveness modifies apoptosis and autophagy in breast cancer cells. J Steroid Biochem Mol Biol. 2009; 114:8-20.
29. De Amicis F, Thirugnansampanthan J, Cui Y, Selever J, Beyer A, Parra I, Weigel NL, Herynk MH, Tsimelzon A, Lewis MT, Chamness GC, Hilsenbeck SG, Ando S, et al. Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res Treat. 2010; 121:1-11. https://doi.org/10.1007/s10549-009-0436-8.
30. Bajetta E, Martinetti A, Zilembo N, Pozzi P, La Torre I, Ferrari L, Seregni E, Longarini R, Salvucci G, Bombardieri E. Biological activity of anastrozole in postmenopausal patients with advanced breast cancer: effects on estrogens and bone metabolism. Ann Oncol. 2002; 13:1059-66.
31. Dimitrakakis C, Bondy C. Androgens and the breast. Breast Cancer Res. 2009; 11:212. https://doi.org/10.1186/bcr2413.
32. Bagnoli F, Oliveira VM, Silva MA, Taromaru GC, Rinaldi JF, Aoki T. The interaction between aromatase, metalloproteinase 2,9 and CD44 in breast cancer. Rev Assoc Med Bras (1992). 2010; 56:472-7.
33. Sikora MJ, Cordero KE, Larios JM, Johnson MD, Lippman ME, Rae JM. The androgen metabolite 5alpha-androstane-3beta, 17beta-diol (3betaAdiol) induces breast cancer growth via estrogen receptor: implications for aromatase inhibitor resistance. Breast Cancer Res Treat. 2009; 115:289-96. https://doi.org/10.1007/s10549-008-0080-8.
34. Iveson TJ, Smith IE, Ahern J, Smithers DA, Trunet PF, Dowsett M. Phase I study of the oral nonsteroidal aromatase inhibitor CGS 20267 in healthy postmenopausal women. J Clin Endocrinol Metab. 1993; 77:324-31. https://doi.org/10.1210/jcem.77.2.8345035.
35. Lipton A, Demers LM, Harvey HA, Kambic KB, Grossberg H, Brady C, Adlercruetz H, Trunet PF, Santen RJ. Letrozole (CGS 20267). A phase I study of a new potent oral aromatase inhibitor of breast cancer. Cancer. 1995; 75:2132-8.
36. Macedo LF, Guo Z, Tilghman SL, Sabnis GJ, Qiu Y, Brodie A. Role of androgens on MCF-7 breast cancer cell growth and on the inhibitory effect of letrozole. Cancer Res. 2006; 66:7775-82. https://doi.org/10.1158/0008-5472.can-05-3984.
37. Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007; 7:659-72. https://doi.org/10.1038/nrc2193.
38. Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, Raffoul W, Delaloye JF, Treboux A, Fiche M, Vilo J, Ayyanan A, Brisken C. A Preclinical Model for ERalpha-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell. 2016; 29:407-22. https://doi.org/10.1016/j.ccell.2016.02.002.
39. Cottu P, Marangoni E, Assayag F, de Cremoux P, Vincent-Salomon A, Guyader C, de Plater L, Elbaz C, Karboul N, Fontaine JJ, Chateau-Joubert S, Boudou-Rouquette P, Alran S, et al. Modeling of response to endocrine therapy in a panel of human luminal breast cancer xenografts. Breast Cancer Res Treat. 2012; 133:595-606. https://doi.org/10.1007/s10549-011-1815-5.
40. Cochrane DR, Bernales S, Jacobsen BM, Cittelly DM, Howe EN, D'Amato NC, Spoelstra NS, Edgerton SM, Jean A, Guerrero J, Gomez F, Medicherla S, Alfaro IE, et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014; 16:R7. https://doi.org/10.1186/bcr3599.
41. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, et al. Molecular portraits of human breast tumours. Nature. 2000; 406:747-52. https://doi.org/10.1038/35021093.
42. Gusterson B. Do 'basal-like' breast cancers really exist? Nat Rev Cancer. 2009; 9:128-34. https://doi.org/10.1038/nrc2571.
43. Inic Z, Zegarac M, Inic M, Markovic I, Kozomara Z, Djurisic I, Inic I, Pupic G, Jancic S. Difference between Luminal A and Luminal B Subtypes According to Ki-67, Tumor Size, and Progesterone Receptor Negativity Providing Prognostic Information. Clin Med Insights Oncol. 2014; 8:107-11. https://doi.org/10.4137/cmo.s18006.
44. Yam C, Mani SA, Moulder SL. Targeting the Molecular Subtypes of Triple Negative Breast Cancer: Understanding the Diversity to Progress the Field. Oncologist. 2017. https://doi.org/10.1634/theoncologist.2017-0095.
45. Reese JM, Suman VJ, Subramaniam M, Wu X, Negron V, Gingery A, Pitel KS, Shah SS, Cunliffe HE, McCullough AE, Pockaj BA, Couch FJ, Olson JE, et al. ERbeta1: characterization, prognosis, and evaluation of treatment strategies in ERalpha-positive and -negative breast cancer. BMC Cancer. 2014; 14:749. https://doi.org/10.1186/1471-2407-14-749.
46. Harrell JC, Dye WW, Allred DC, Jedlicka P, Spoelstra NS, Sartorius CA, Horwitz KB. Estrogen receptor positive breast cancer metastasis: altered hormonal sensitivity and tumor aggressiveness in lymphatic vessels and lymph nodes. Cancer Res. 2006; 66:9308-15. https://doi.org/10.1158/0008-5472.can-06-1769.
47. Zheng WQ, Lu J, Zheng JM, Hu FX, Ni CR. Variation of ER status between primary and metastatic breast cancer and relationship to p53 expression*. Steroids. 2001; 66:905-10.
48. Gao W, Bohl CE, Dalton JT. Chemistry and structural biology of androgen receptor. Chem Rev. 2005; 105:3352-70. https://doi.org/10.1021/cr020456u.
49. Gillies GE, McArthur S. Estrogen actions in the brain and the basis for differential action in men and women: a case for sex-specific medicines. Pharmacol Rev. 2010; 62:155-98. https://doi.org/10.1124/pr.109.002071.
50. Zava DT, McGuire WL. Androgen action through estrogen receptor in a human breast cancer cell line. Endocrinology. 1978; 103:624-31. https://doi.org/10.1210/endo-103-2-624.
51. Bhattacharyya RS, Krishnan AV, Swami S, Feldman D. Fulvestrant (ICI 182,780) down-regulates androgen receptor expression and diminishes androgenic responses in LNCaP human prostate cancer cells. Mol Cancer Ther. 2006; 5:1539-49. https://doi.org/10.1158/1535-7163.mct-06-0065.
52. Foradori CD, Weiser MJ, Handa RJ. Non-genomic actions of androgens. Front Neuroendocrinol. 2008; 29:169-81. https://doi.org/10.1016/j.yfrne.2007.10.005.
53. Chia KM, Liu J, Francis GD, Naderi A. A feedback loop between androgen receptor and ERK signaling in estrogen receptor-negative breast cancer. Neoplasia. 2011; 13:154-66.
54. Naderi A, Chia KM, Liu J. Synergy between inhibitors of androgen receptor and MEK has therapeutic implications in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011; 13:R36. https://doi.org/10.1186/bcr2858.
55. Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, Rimm DL, Liu XS, Brown M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011; 20:119-31. https://doi.org/10.1016/j.ccr.2011.05.026.
56. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011; 121:2750-67. https://doi.org/10.1172/jci45014.
57. Garay JP, Karakas B, Abukhdeir AM, Cosgrove DP, Gustin JP, Higgins MJ, Konishi H, Konishi Y, Lauring J, Mohseni M, Wang GM, Jelovac D, Weeraratna A, et al. The growth response to androgen receptor signaling in ERalpha-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res. 2012; 14:R27. https://doi.org/10.1186/bcr3112.
58. Wang Y, Romigh T, He X, Tan MH, Orloff MS, Silverman RH, Heston WD, Eng C. Differential regulation of PTEN expression by androgen receptor in prostate and breast cancers. Oncogene. 2011; 30:4327-38. https://doi.org/10.1038/onc.2011.144.
59. Wang Y, He X, Ngeow J, Eng C. GATA2 negatively regulates PTEN by preventing nuclear translocation of androgen receptor and by androgen-independent suppression of PTEN transcription in breast cancer. Hum Mol Genet. 2012; 21:569-76. https://doi.org/10.1093/hmg/ddr491.
60. Harvell DM, Spoelstra NS, Singh M, McManaman JL, Finlayson C, Phang T, Trapp S, Hunter L, Dye WW, Borges VF, Elias A, Horwitz KB, Richer JK. Molecular signatures of neoadjuvant endocrine therapy for breast cancer: characteristics of response or intrinsic resistance. Breast Cancer Res Treat. 2008; 112:475-88. https://doi.org/10.1007/s10549-008-9897-4.
61. Harvell DM, Richer JK, Singh M, Spoelstra N, Finlayson C, Borges VF, Elias AD, Horwitz KB. Estrogen regulated gene expression in response to neoadjuvant endocrine therapy of breast cancers: tamoxifen agonist effects dominate in the presence of an aromatase inhibitor. Breast Cancer Res Treat. 2008; 112:489-501. https://doi.org/10.1007/s10549-008-9923-6.
62. Collins LC, Cole KS, Marotti JD, Hu R, Schnitt SJ, Tamimi RM. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Mod Pathol. 2011; 24:924-31. https://doi.org/10.1038/modpathol.2011.54.
63. Niemeier LA, Dabbs DJ, Beriwal S, Striebel JM, Bhargava R. Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod Pathol. 2010; 23:205-12. https://doi.org/10.1038/modpathol.2009.159.
64. Guedj M, Marisa L, de Reynies A, Orsetti B, Schiappa R, Bibeau F, MacGrogan G, Lerebours F, Finetti P, Longy M, Bertheau P, Bertrand F, Bonnet F, et al. A refined molecular taxonomy of breast cancer. Oncogene. 2012; 31:1196-206. https://doi.org/10.1038/onc.2011.301.
65. Kuenen-Boumeester V, Van der Kwast TH, Claassen CC, Look MP, Liem GS, Klijn JG, Henzen-Logmans SC. The clinical significance of androgen receptors in breast cancer and their relation to histological and cell biological parameters. Eur J Cancer. 1996; 32a:1560-5.
66. Moinfar F, Okcu M, Tsybrovskyy O, Regitnig P, Lax SF, Weybora W, Ratschek M, Tavassoli FA, Denk H. Androgen receptors frequently are expressed in breast carcinomas: potential relevance to new therapeutic strategies. Cancer. 2003; 98:703-11. https://doi.org/10.1002/cncr.11532.
67. Afratis NA, Bouris P, Skandalis SS, Multhaupt HA, Couchman JR, Theocharis AD, Karamanos NK. IGF-IR cooperates with ERalpha to inhibit breast cancer cell aggressiveness by regulating the expression and localisation of ECM molecules. Sci Rep. 2017; 7:40138. https://doi.org/10.1038/srep40138.
68. Birrell SN, Hall RE, Tilley WD. Role of the androgen receptor in human breast cancer. J Mammary Gland Biol Neoplasia. 1998; 3:95-103.
69. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481:389-93. https://doi.org/10.1038/nature10730.
70. Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics. 2006; 7:497-508.
71. Rondon-Lagos M, Villegas VE, Rangel N, Sanchez MC, Zaphiropoulos PG. Tamoxifen Resistance: Emerging Molecular Targets. Int J Mol Sci. 2016; 17. https://doi.org/10.3390/ijms17081357.
72. Szelei J, Jimenez J, Soto AM, Luizzi MF, Sonnenschein C. Androgen-induced inhibition of proliferation in human breast cancer MCF7 cells transfected with androgen receptor. Endocrinology. 1997; 138:1406-12. https://doi.org/10.1210/endo.138.4.5047.
73. Kandouz M, Lombet A, Perrot JY, Jacob D, Carvajal S, Kazem A, Rostene W, Therwath A, Gompel A. Proapoptotic effects of antiestrogens, progestins and androgen in breast cancer cells. J Steroid Biochem Mol Biol. 1999; 69:463-71.
74. Britton DJ, Hutcheson IR, Knowlden JM, Barrow D, Giles M, McClelland RA, Gee JM, Nicholson RI. Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat. 2006; 96:131-46. https://doi.org/10.1007/s10549-005-9070-2.
75. Kumar MV, Leo ME, Tindall DJ. Modulation of androgen receptor transcriptional activity by the estrogen receptor. J Androl. 1994; 15:534-42.
76. Need EF, Selth LA, Harris TJ, Birrell SN, Tilley WD, Buchanan G. Research resource: interplay between the genomic and transcriptional networks of androgen receptor and estrogen receptor alpha in luminal breast cancer cells. Mol Endocrinol. 2012; 26:1941-52. https://doi.org/10.1210/me.2011-1314.
77. Rechoum Y, Rovito D, Iacopetta D, Barone I, Ando S, Weigel NL, O'Malley BW, Brown PH, Fuqua SA. AR collaborates with ERalpha in aromatase inhibitor-resistant breast cancer. Breast Cancer Res Treat. 2014; 147:473-85. https://doi.org/10.1007/s10549-014-3082-8.
78. Singh M, Kumar V. Transgenic mouse models of hepatitis B virus-associated hepatocellular carcinoma. Rev Med Virol. 2003; 13:243-53. https://doi.org/10.1002/rmv.392.
79. White DE, Muller WJ. Multifaceted roles of integrins in breast cancer metastasis. J Mammary Gland Biol Neoplasia. 2007; 12:135-42. https://doi.org/10.1007/s10911-007-9045-5.
80. Fioretti FM, Sita-Lumsden A, Bevan CL, Brooke GN. Revising the role of the androgen receptor in breast cancer. J Mol Endocrinol. 2014; 52:R257-65. https://doi.org/10.1530/jme-14-0030.
81. Greeve MA, Allan RK, Harvey JM, Bentel JM. Inhibition of MCF-7 breast cancer cell proliferation by 5alpha-dihydrotestosterone; a role for p21(Cip1/Waf1). J Mol Endocrinol. 2004; 32:793-810.
82. Ando S, De Amicis F, Rago V, Carpino A, Maggiolini M, Panno ML, Lanzino M. Breast cancer: from estrogen to androgen receptor. Mol Cell Endocrinol. 2002; 193:121-8.
83. Zaret KS, Lerner J, Iwafuchi-Doi M. Chromatin Scanning by Dynamic Binding of Pioneer Factors. Mol Cell. 2016; 62:665-7. https://doi.org/10.1016/j.molcel.2016.05.024.
84. Robinson JL, Macarthur S, Ross-Innes CS, Tilley WD, Neal DE, Mills IG, Carroll JS. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. Embo j. 2011; 30:3019-27. https://doi.org/10.1038/emboj.2011.216.
85. Augello MA, Hickey TE, Knudsen KE. FOXA1: master of steroid receptor function in cancer. Embo j. 2011; 30:3885-94. https://doi.org/10.1038/emboj.2011.340.
86. Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep. 2016; 17:2715-23. https://doi.org/10.1016/j.celrep.2016.11.028.
87. Cai M, Kim S, Wang K, Farnham PJ, Coetzee GA, Lu W. 4C-seq revealed long-range interactions of a functional enhancer at the 8q24 prostate cancer risk locus. Sci Rep. 2016; 6:22462. https://doi.org/10.1038/srep22462.
88. Rivero S, Ceballos-Chavez M, Bhattacharya SS, Reyes JC. HMG20A is required for SNAI1-mediated epithelial to mesenchymal transition. Oncogene. 2015; 34:5264-76. https://doi.org/10.1038/onc.2014.446.
89. Paliouras M, Diamandis EP. Androgens act synergistically to enhance estrogen-induced upregulation of human tissue kallikreins 10, 11, and 14 in breast cancer cells via a membrane bound androgen receptor. Mol Oncol. 2008; 1:413-24. https://doi.org/10.1016/j.molonc.2008.01.001.
90. Rothman MS, Carlson NE, Xu M, Wang C, Swerdloff R, Lee P, Goh VH, Ridgway EC, Wierman ME. Reexamination of testosterone, dihydrotestosterone, estradiol and estrone levels across the menstrual cycle and in postmenopausal women measured by liquid chromatography-tandem mass spectrometry. Steroids. 2011; 76:177-82. https://doi.org/10.1016/j.steroids.2010.10.010.
91. Suzuki T, Darnel AD, Akahira JI, Ariga N, Ogawa S, Kaneko C, Takeyama J, Moriya T, Sasano H. 5alpha-reductases in human breast carcinoma: possible modulator of in situ androgenic actions. J Clin Endocrinol Metab. 2001; 86:2250-7. https://doi.org/10.1210/jcem.86.5.7444.
92. Talley RW, Haines CR, Waters MN, Goldenberg IS, Olson KB, Bisel HF. A dose-response evaluation of androgens in the treatment of metastatic breast cancer. Cancer. 1973; 32:315-20.
93. McNamara KM, Harwood DT, Simanainen U, Walters KA, Jimenez M, Handelsman DJ. Measurement of sex steroids in murine blood and reproductive tissues by liquid chromatography-tandem mass spectrometry. J Steroid Biochem Mol Biol. 2010; 121:611-8. https://doi.org/10.1016/j.jsbmb.2010.02.001.
94. Li X, Strauss L, Makela S, Streng T, Huhtaniemi I, Santti R, Poutanen M. Multiple structural and functional abnormalities in the p450 aromatase expressing transgenic male mice are ameliorated by a p450 aromatase inhibitor. Am J Pathol. 2004; 164:1039-48. https://doi.org/10.1016/s0002-9440(10)63191-4.
95. Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A. History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev. 2009; 30:343-75. https://doi.org/10.1210/er.2008-0016.
96. West CD, Damast BL, Sarro SD, Pearson OH. Conversion of testosterone to estrogens in castrated, adrenalectomized human females. J Biol Chem. 1956; 218:409-18.
97. Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971; 25:270-5.
98. Goldenberg IS, Waters N, Ravdin RS, Ansfield FJ, Segaloff A. Androgenic therapy for advanced breast cancer in women. A report of the cooperative breast cancer group. Jama. 1973; 223:1267-8.
99. Manni A, Arafah BM, Pearson OH. Androgen-induced remissions after antiestrogen and hypophysectomy in stage IV breast cancer. Cancer. 1981; 48:2507-9.
100. Ingle JN, Twito DI, Schaid DJ, Cullinan SA, Krook JE, Mailliard JA, Tschetter LK, Long HJ, Gerstner JG, Windschitl HE, Levitt R, Pfeifle DM. Combination hormonal therapy with tamoxifen plus fluoxymesterone versus tamoxifen alone in postmenopausal women with metastatic breast cancer. An updated analysis. Cancer. 1991; 67:886-91.
101. Burdall SE, Hanby AM, Lansdown MR, Speirs V. Breast cancer cell lines: friend or foe? Breast Cancer Res. 2003; 5:89-95.
102. Feng J, Li L, Zhang N, Liu J, Zhang L, Gao H, Wang G, Li Y, Zhang Y, Li X, Liu D, Lu J, Huang B. Androgen and AR contribute to breast cancer development and metastasis: an insight of mechanisms. Oncogene. 2017; 36:2775-90. https://doi.org/10.1038/onc.2016.432.