
INTRODUCTION
Lung cancer is the most lethal cancer in most developed countries. Around half of the patients diagnosed with lung cancer die within one year of diagnosis and the 5-year survival rates are less than 18% [1]. One of the main barriers to further progress in lung cancer therapy is the lack of effective biomarkers. However, research efforts are currently being diverted toward identifying new cancer biomarkers that would improve outcome in these patients. Growing research evidence have determined an unexpected relevant role of long non coding RNAs (lncRNAs) in tumor development [2, 3] that have opened new opportunities for the discovery of effective tumor biomarkers that can improve patient diagnosis and prognosis.
LncRNAs are transcripts with more than 200 nucleotides of length that have a limited or no protein-coding capacity [4-6]. Previous studies have demonstrated that lncRNAs have multifunctional roles in different cellular processes and in tumorigenesis by participating in both oncogenic and tumour suppressing pathways [7, 8]. Although the specific function of most lncRNAs is still in investigation, they can regulate a large fraction of targets working either in trans or in cis [9].
The FENDRR lncRNA (also named FOXF1-As1) and its neighbor protein-coding gene FOXF1 are located in the vicinity, with less than 2 kb of difference, at the long arm of chromosome 16, although they are transcribed from opposite strand [10]. FENDRR and FOXF1 are expressed in specific adult tissues as bladder, colon, esophagus, lung, prostate, stomach and small intestine [11]. Pioneer studies have assessed the impact of FENDRR in cancer. Initially, FENDRR was firstly reported to be downregulated in gastric cancer compared with normal gastric cells and the low FENDRR expression was related to poor prognosis [12]. Recently, in lung cancer, a lncRNA expression profiling identified to FENDRR as one of the lncRNAs were differentially expressed between cancer and the adjacent normal tissues [13]. Additionally, also in lung cancer, FENDRR expression was associated with tumour migration and metastasis [14]. However, the overall contributions of FENDRR to lung cancer remain to be investigated.
In this study, we examined the expression levels of FENDRR and FOXF1 in lung cancer patients through qRT-PCR. We found FENDRR and FOXF1 expression was downregulated in lung adenocarcinomas compared to adjacent normal lung tissues and that FENDRR mRNA expression levels correlated positively with FOXF1 expression. Interestingly, we found that FENDRR and FOXF1 had also a prognosis value since their high expression values were associated with a better clinical outcome in lung adenocarcinomas.
RESULTS
FENDRR and FOXF1 expression is lost in lung adenocarcinomas
FENDRR and FOXF1 expression was analyzed by qRT-PCR in 70 lung adenocacinomas (LUAD) and their adjacent normal lung tissues. As shown in Figure 1A, the expression of FENDRR and FOXF1 was significantly lower in the tumors than in adjacent matched normal tissue samples (p <0.001 in both cases). In almost all cases, FENDRR and FOXF1 expression levels in tumors were lower than those observed in normal samples, with a median in FENDRR expression levels of 0.008 and with a range value from 0,000034 to 0.273. While FOXF1 expression in tumor samples ranged from 0,00007 to 0.207, with a median of 0.0059. Interestingly, FENDRR expression levels showed a significant positive correlation with FOXF1 (Figure 1B).
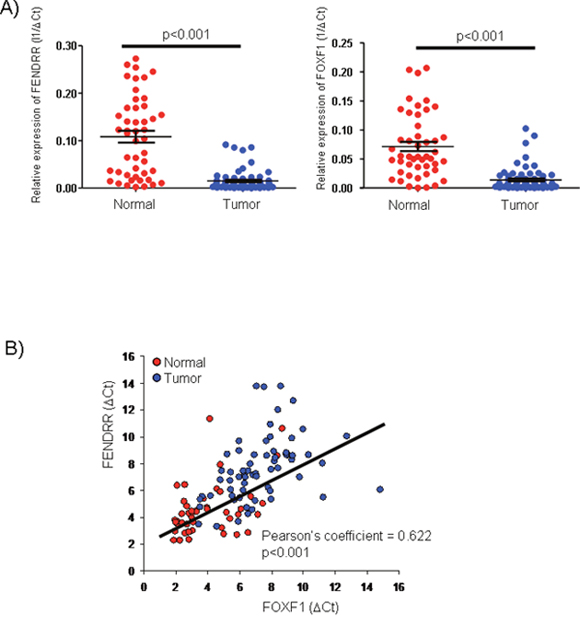
Figure 1: FENDRR and FOXF1 mRNAs are significant biomarkers of lung adenocarcinoma (LUAD). (A) FENDRR and FOXF1 expression was analyzed by qRT-PCR in 70 LUAD and 70 normal lung tissues. FENDRR and FOXF1 were significantly downregulated in lung adenocarcinomas versus their adjacent normal lung tissues. (B) The expression levels of FENDRR and FOXF1 was positive correlated in each other in lung tumor and adjacent normal lung tissues (Pearson’s coefficient = 0.622, p<0.001).
To corroborate these results, we used external datasets from GEO (Gene Expression Omnibus) and TCGA (http://methhc.mbc.nctu.edu.tw). Importantly, several and independent cancer datasets showed a significant positive correlation between FENDRR expression and FOXF1 RNA expression (Table 1).
Table 1: Data from different expression profiles of cancer available in GEO and TCGA database
Study |
Authors |
Tissue |
Type Cancer |
Nº Samples |
FENDRR expression |
p-value |
FOXF1 expression |
p-value |
|---|---|---|---|---|---|---|---|---|
GSE18842 |
Sanchez-Palencia, 2011 |
Lung |
LUSC |
45 |
-1.14 |
<0.001 |
-3.26 |
0.002 |
LUAD |
45 |
-2.52 |
0.015 |
|||||
TCGA-LUAD |
doi:10.7908/C18G8K47 |
Lung |
LUAD |
546 |
-2.31 |
<0.001 |
-2.02 |
<0.001 |
TCGA-LUSC |
doi:10.7908/C1XW4J7P |
Lung |
LUSC |
548 |
-2.26 |
<0.001 |
-2.13 |
<0.001 |
GSE19804 |
Lu TP, 2010 |
Lung |
NSCLC |
120 |
-1.16 |
<0.001 |
-2.01 |
<0.001 |
GSE31210 |
Okayama H, 2012 |
Lung |
LUAD |
246 |
-2.16 |
<0.001 |
-2.45 |
<0.001 |
GSE3268 |
Wachi S, 2005 |
Lung |
LUSC |
10 |
ND |
ND |
-1.85 |
<0.001 |
GSE55945 |
Arredouani MS, 2009 |
Prostate |
PCA |
21 |
-0.83 |
0.032 |
-1.08 |
0.021 |
GSE24514 |
Alhopuro P, 2010 |
Colon |
CRC |
49 |
ND |
ND |
-1.52 |
<0.001 |
The FENDRR and FOXF1 expression value of the tumors was normalized by the non-tumoral samples. CRC: Colorectal cancer; LUAD: Lung Adenocarcinoma; LUSC: Lung Squamous Cancer; NSCLC: Non Small Cell Lung Cancer; PCA: Prostate Cancer.
FENDRR and FOXF1 expression: lung cancer cell lines
FENDRR and FOXF1 expression levels were quantified by qRT-PCR in 5 human lung cancer cell lines (A549, H441, H661, H838 and H460), displaying similar trend than in primary tumors. Actually, four of them (A549, H441, H661 and H838) have lost most of the FENDRR and FOXF1 expression. Just H460, expressed FENDRR and FOXF1 at measurable levels by qPCR (Supplementary Figure 1). According to our results, the in silico analysis of FENDRR and FOXF1 expression from Cancer Cell Line Encyclopedia (CCLE) database [15] showed a significant positive correlation between FENDRR and FOXF1 in 1036 cancer cell lines (p<0.001) (Supplementary Figure 2A), and in particular 116 NSCLC cancer cell lines (p<0.00001). Interestingly, a human cell line derived from normal embryonic lung tissue (HLFA) displayed the highest levels FOXF1 and FENDRR (Supplementary Figure 2B).
FENDRR expression by RNA-FISH
To confirm its association with lung adenocarcinoma, using an independent method to qPCR, we examined FENDRR expression by RNA-FISH on 6 paraffin embedded LUAD and matched healthy tissues. When present, FENDRR staining was nuclear and cytoplasmic. RNA-FISH analysis showed that FENDRR was differently expressed in tumour and normal cells (Figure 2), and the qPCR analysis displayed their expression is lost in tumors.
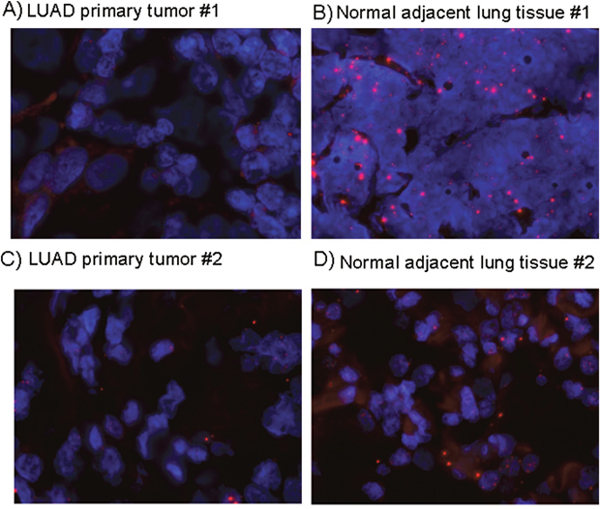
Figure 2: FENDRR expression detection by RNA-FISH in lung cancer and normal lung. RNA FENDRR fluorescence in situ hybridization (RNA-FISH) in two representative pairs of primary lung adenocarcinoma (A, C) and adjacent normal tissue (B, D). Red: FENDRR probe, bluet: DAPI.
Together, these data supported the conclusion that FENDRR could be a new biomarker for lung cancer.
High FENDRR and FOXF1 expression is associated with better clinical outcome in lung cancer
Because FENDRR and FOXF1 could influence outcome, we studied whether the expression levels of FENDRR and FOXF1 genes could predict the treatment response and/or survival of LUAD patients. No significant differences were found in the distribution of common clinical variables such as age, gender, pathological stage, lymph node status, type of treatment, relapse, and smoking in the groups of LUAD cases showing low or high expression of FENDRR or FOXF1 (Table 2). As show in Figure 3, Kaplan-Meier curves of cases in the combined high-FENDRR/FOXF1 expression group (n=64) showed a significant better survival times than patients with tumors with lower expression (p=0.01). These results are corroborated using larger dataset of lung cancer patients was in silico analyzed by the Kaplan–Meier software (KM plotter.com) [16]. Three microarray data sets were available from GEO under accession number GSE31210, GSE30219 and GSE3141, which contained 226, 142 and 76 samples of lung cancer patients respectively. Tumors were separated into two groups based on FENDRR expression. Low expression of FENDRR was associated with shorter survival of patients in these three datasets (hazard ratio [HR], 0.08-0.067; 95% CI, 0.01 to 0.96; p<0.05). The results were summarized in Supplementary Figure 5. We confirmed the association with overall survival for FENDRR and also found an association with survival probability.
Table 2: Association between FENDRR and FOXF1 expression and clinicopathological features (only available for 64 patients)
Clinicopathologic characteristics |
FENDRR p-value |
FOXF1 p-value |
|---|---|---|
Gender |
0.61 |
0.67 |
Male (45) |
||
Female (19) |
||
Age (y) |
0.42 |
0.38 |
<66 (36) |
||
>66 (28) |
||
Pathological stage (T) |
0.61 |
0.11 |
I (12) |
||
II (37) |
||
III (8) |
||
IV (1) |
||
Unknow (6) |
||
Regional lymph nodes (N) |
0.56 |
0.74 |
0 (29) |
||
1 (10) |
||
2 (7) |
||
Unknow (18) |
||
Adjuvant treatment |
0.65 |
0.12 |
None (29) |
||
Chemotherapy (18) |
||
Chemotherapy & Radiotherapy (11) |
||
Radiotherapy (3) |
||
Unknow (3) |
||
Relapse |
0.95 |
0.79 |
Yes (24) |
||
No (38) |
||
Unknow (2) |
||
Smoking at the time of the diagnosis |
0.50 |
0.09 |
Yes (56) |
||
No (5) |
||
Unknow (2) |
FENDRR and FOXF1 are considered as continues variable.
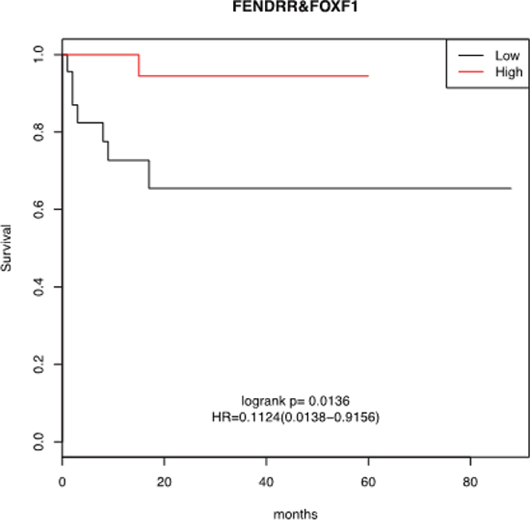
Figure 3: Kaplan-Meier survival analysis for the relationship between survival time and FENDRR and FOXF1 signature in lung adenocarcinomas. Higher FENDRR and FOXF1 expression was associated with a better overall survival for patients with lung cancer. The survival function is defined as the probability of surviving at least to the time determined in x-axis (in months).
Mutational analysis of FENDRR and FOXF1 in lung cancer
To gain additional insights into the inactivation mechanism of FENDRR and FOXF1, we decided to study the mutation status of the genes. For this purposed we sequenced these two genes in 27 pairs of matched tumor and normal of lung tissues. We found 49 and 18 single nucleotide variations in FENDRR and FOXF1 genes respectively. Since it is difficult to determine the functional repercussion of the mutations, especially in long non-coding genes, we filtered the data obtained following the analysis workflow described in the Figure 4A. None of these alterations were considered with as probable cause of the loss of expression of FENDR and FOXF1 we observed in LUAD because they were located in the introns, or in untranslated regions (in case of FOXF1), were reported as dbSNP, somatic or predicted as being (Figure 4B) described in detail.
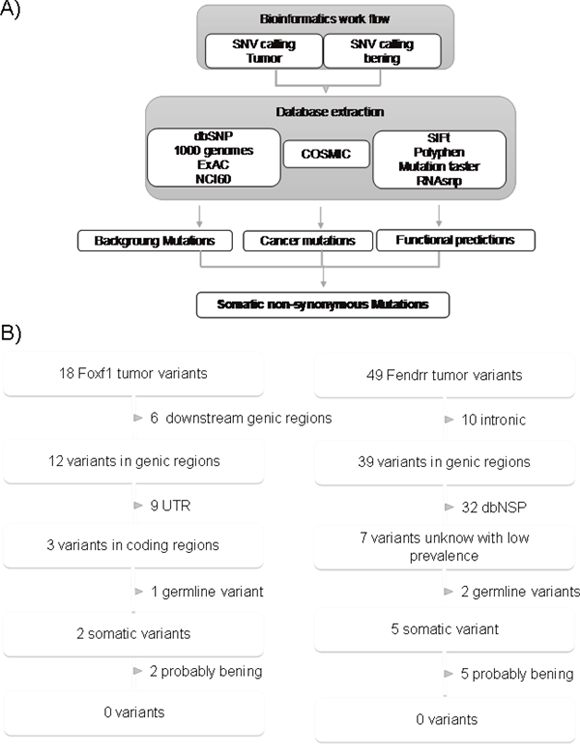
Figure 4: Identification process of somatic variants in FENDRR and FOXF1. (A) Schematic of bioinformatics SNV detection workflow. (B) Extraction of functionally relevant somatic mutations for FOXF1 and FENDRR in lung cancer. Variants were filtered for annotation in dbSNP, 1000genomes, ExAC, NCI60, somatic and functionally impairment. From dbSNP or the 1000 genomes variants with frequencies above 1% were excluded.
Methylation status of FENDRR and FOXF1
Another well-known mechanism of gene silencing is promoter hypermethylation. To determine the involvement this epigenetic mechanism of silencing for FENDRR and FOXF1, firstly we analyzed the in silico results from The Cancer Genome Atlas database [17] using the Wanderer web tool [18]. The methylation status of the promoter CpG islands of FENDRR and FOXF1 was significantly different in tumor versus normal tissue. Approximately 84% and 96% of CpG islands were differentially methylated for FENDRR and FOXF1 respectively although mean methylation level difference between tumor and normal was even sensibly higher in the FOXF1 case (Figure 5A). For this reason, we decided to analyze the methylation status of the FOXF1 promoter experimentally by pyrosequencing of bisulfite-modified DNA and MSP analysis in A549, H441, H661, H838 and H460 human lung cancer cell lines. Sequence analysis detected hypermethylation of FOXF1 promoter in 2 of 5 cell lines (H441 and H838) (Figure 5B), indicating that hypermethilation of FOXF1 promoter could mediate the downregulation of FOXF1 in lung cancer. Interestingly, treatment with demethylating agent 5-aza-deoxycytidine (5-Aza) of lung cancer cell lines didn’t produce any change in methylation of FOXF1 promoter (Supplementary Figure 3).
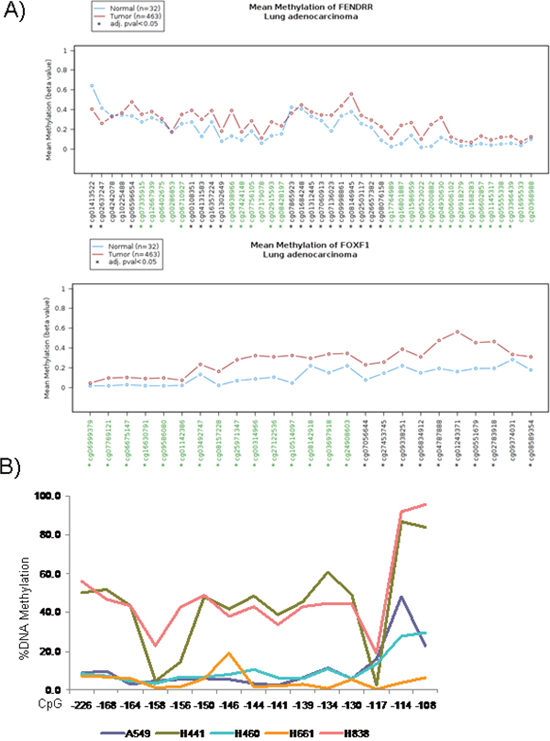
Figure 5: Epigenetic inactivation of FENDRR and FOXF1 in lung adenocarcinoma. (A) Graphic obtained from Wanderer data base (http://maplab.imppc.org/wanderer/) of all CpGs island of FENDRR and FOXF1 and their percentage of DNA methylation in lung cancer (medium of 463 samples, brown) and normal lung (medium of 32 samples, blue). In x-axis the cg# indicates the position of the CpG island. *: indicates differences statistically significant between normal and tumor samples. (B) Percentage of DNA Methylation in the FOXF1 promoter in 5 lung cancer cells lines by bisulphite sequencing. Hypermethylation of FOXF1 promoter was detected in 2 of 5 cell lines (H441 and H838).
Next, we searched for the presence of promoter hypermethylation by MSP analysis at FOXF1 in 10 lung adenocarcinoma samples and their adjacent normal tissues (Supplementary Figure 4), aberrant promoter hypermethylation was detected in 2 of 10 lung adenocarcinoma patients (2/10, 20%).
FENDRR and FOXF1 functional relationship
Recent studies have showed that many lncRNAs can alter gene expression via dsDNA:RNA triplex formation and recruit protein complexes [19-21]. To explore this possibility and analyze FENDRR and FOXF1 functional relationship we developed luciferase assays. Interestingly, after transfecting a plasmid expressing FENDRR in A549 cells we observed an 56% increment in the normalized luciferase expression when compared the reporter under FOXF1 promoter with the reporter under SV40 promoter that was used as reference (p=0.013) (Figure 6A). However, these differences where not appreciated when the FOXF1 promoter is methylated (Figure 6B, p=0.024) indicating that the binding of FENDRR to FOXF1 promoter could be sensitive to the methylation status of the DNA.
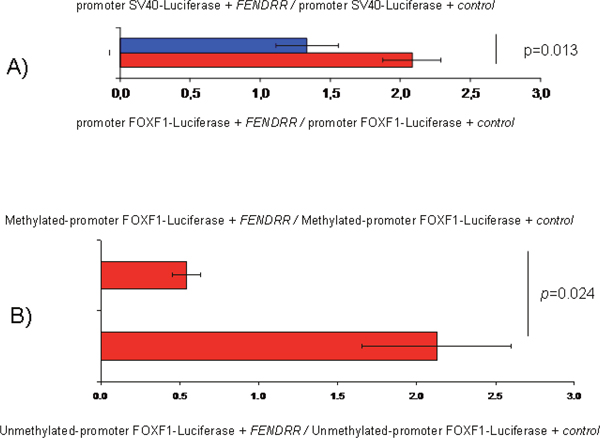
Figure 6: FENDRR and FOXF1 relationship determined by luciferase assays in the A549 cell line. (A) Normalized luciferase expression comparing the luciferase reporter under FOXF1 promoter with and without methylation after adding FENDRR. Error bars represent standard deviation. (B) Control: Normalized luciferase expression comparing the reporters under the FOXF1 promoters with and without methylation after FENDRR transfection. FENDRR: expression plasmid of FENDRR. Control: expression plasmid where FENDRR has been substituted by GFP.
DISCUSSION
Multiple lines of evidence increasingly link mutations and dysregulations of lncRNAs to diverse human diseases including cancer [22]. Recent progress suggests that the involvement of lncRNAs in human diseases could be far more prevalent than previously appreciated [23]. Therefore, the identification and investigation of cancer associated lncRNAs is critical for understanding the roles of lncRNAs in the carcinogenesis and may have clear clinical importance in lung cancer [23].
FENDRR and FOXF1 expression levels are relatively high in healthy lung tissue, where, with a median of 14 and 31 RPKM (Reads per kilo base per million of mapped reads) (n=320) respectively ranks between the top-5 tissues with more expression (Supplementary Figure 6) [11]. In the present study, we have investigated FENDRR and FOXF1 expression by qRT-PCR and RNA-FISH assays in lung adenocarcinoma patients. The results indicated that expression of FENDRR and FOXF1 in lung cancer tissues was significantly lower than those observed in adjacent normal tissues, suggesting the potential of using FENDRR and FOXF1 to successfully distinguish cancerous from normal tissues. Furthermore, we found FENDRR and FOXF1 significantly associated to survival. And patients with low levels FENDRR and FOXF1 expression had a significantly poorer overall survival compared with patients expressing high levels of these genes.
Based on these results, FENDRR and FOXF1 could be postulated to be as potential tumor suppressor genes. The mechanisms of inactivation of these genes in different tumor types have not been yet established. Alterations in the coding sequence have rarely been found in lung cancer [24, 25], indicating that mechanism of FENDRR and FOXF1 inactivation could be, among others, loss of heterozygosity (LOH) and promoter hypermethylation. In our dataset, the mutation is not frequently found in FENDRR or FOXF1 mutation. However, the methylation analysis showed a relatively high incidence of FOXF1 methylation among lung cancer patients and lung tumoral cell lines. Mitchell SM et al. [26] and PK Lo et al. [27] also found an important percentage of FOXF1 methylation in colorectal cancer and breast cancer. Our results, however, showed that FOXF1 expression could by silenced by promoter methylation also in lung cancer.
LncRNAs have been reported to affect the expression of neighboring genes positively or negatively. Based on mRNA expression analysis that revealed a positive correlation between FENDRR and FOXF1 for lung cancer, we explored the possible functional relationship between FENDRR and FOXF1 using luciferase assays. Our results indicated that FENDRR expression enhanced the activity of FOXF1 promoter significantly. This could be possible if FENDRR is able to bind to promoter region of FOXF1 directly and enhance FOXF1 transcription. Actually, using in silico bioinformatics analysis we suggested previously that FENDRR could regulate expression of FOXF1 forming a triplex-helix DNA:RNA structure [28]. Interestingly, we observed that DNA methylation abrogated the changes mediated by FENDRR suggesting that the binding of FENDRR to FOXF1 promoter could be sensitive to the methylation status of the promoter (Figure 6B).
We are still far to understand the roles of FOXF1 and FENDRR in the cancer development but there are suggestive functional data that can explain the tumor-suppressor features we have observed. FOXF1, a member of the forkhead box family of transcription factors that has been previously shown to be critical for lung development, homeostasis, and injury responses. It was observed that FOXF1 is induced upon DNA damage in a p53-dependent manner whereas the inactivation of FOXF1 stimulated cell invasion and migration [29]. More unknown is the role of FENDRR in tumor development. A recent report showed that mouse embryos lacking Fendrr displayed upregulation of several transcription factors controlling heart development [30]. The authors of this report showed that Fendrr binds to cancer related complexes like both PRC2 and TrxG/MLL, suggesting that it acts as modulator of chromatin signatures that define gene activity and to contribute to cell identity and differentiation, a feature lost during the tumor development.
In summary, this study identifies FENDRR and FOXF1 as novel potential tumour suppressor genes in lung cancer and that FENDRR acts by binding to unmethylated FOXF1 promoter. FENDRR and FOXF1 gene expression were predicted for overall survival, where patients with higher levels of FENDRR and FOXF1 expression had better prognosis. Thus, much more work is still required to determine the detailed mechanisms it functions in lung cancer and the potentiality of FENDRR and FOXF1 as therapeutic targets for lung cancer.
MATERIALS AND METHODS
Tissue collection
The study was approved by the Ethics Committee (CEI Granada), Department of Health, Government of Andalucía and from Basque Foundation for Health Innovation and Research, Spain. Participants provided written consent in accordance with institutional and national guidelines; consent procedure was also approved by the Ethics Committee. Seventy tumours samples from lung adenocarcinoma (LUAD) were taken from primary malignant lung tumours, as well as their adjacent non-tumoural tissue, based on macroscopic examination, by a trained pathologist.
Reverse transcription
RNA obtained was reverse transcribed in the presence of 5 mM MgCl2, 1X PCR Buffer II, 1 mM dNTPs, 25 ul MuLV Reverse Transcriptase, 1 ul RNA Ribonuclease inhibitor, 2.5 μM Random hexamers in a final reaction volume of 20 μl. DNA-ase was using during the protocol to avoid gDNA amplificacion. Reactions were carried out at 42°C for 30 minutes in a Gene Amp PCR system 9600 (PE Applied Biosystems), followed by a 10-minute step at 99°C to denature the enzyme, and then by cooling to 4°C.
Quantitative RT-PCR analysis
A SYBR Green quantitative real-time PCR (qRT-PCR) was carried out to quantify the expression levels of FENDRR lncRNA and FOXF1 protein-coding RNA. All PCRs were performed using the ABI prism 7900 system (Applied Biosystems) under the conditions recommended by the manufacturers. All experiments were performed in triplicate and the mean of triplicates was used. The gene expression was determined using comparative threshold cycle (Ct). Afterward, the mean threshold cycle value of GAPDH as a reference gene was subtracted from the mean threshold cycle value of the target genes (FENDRR and FOXF1) to obtain ΔCt, and ΔΔCt values of each sample were calculated from the corresponding Ct values. qPCR primers are indicated in the Supplementary Table 1. Finally, target/internal control gene was calculated using the formula expression ratio R = 2^-DCt. Differences in gene expression groups were estimated using Student’s t-tests.
RNA fluorescence in situ hybridization (RNA-FISH)
FENDRR FISH was performed on thin (approximately 4 μm thick) tissue sections mounted on positively charged slides (SuperFrost, Thermo Scientific). The QuantiGene ViewRNA Assays (Affimetrix) was used following the manufacturer’s instructions. Briefly, the method consists of sample permeabilization in protease to allow target accessibility, followed by target hybridization with a specific FENDRR probe. After the hybridization, the RNA signals are amplified via a series of sequential hybridization steps. Finally, the slides are washed in cold 2xSSC, 1xSSC and 0.5xSSC and mounted in Vectashield with 1 ug/ml DAPI (Vector) prior to microscopy analyses. FENDRR was visualized using a standard fluorescence microscope and images were captured using a CCD camera (Photometrics SenSys camera) connected to a PC running the Zytovision image analysis system (Applied Imaging Ltd., UK) with focus motor and Z stack software. All slides were examined for FENDRR ISH signals in morphologically intact cells and scored manually by S.R.
Mutational status of FENDRR and FOXF1
A custom capture gene panel with FENDRR and FOXF1 coding sequences and 150 bp of upstream and downstream flanking sequences was designed using NimbleGen’s SeqCap EZ Choice Library capture system (Roche NimbleGen, Inc, Madison, WI). Libraries were prepared using 300 ng of genomic DNA. After appropriate quality controls, libraries were pooled and captured according to NimbleGen’s SeqCap EZ Choice Library capture protocols. Captured libraries were multiplexed twenty per cartridge and sequenced to generate 2x150 bp paired-end reads using NextSeq 500/550 Mid Output v2 kit (300 cycles) on a NextSeq 500 sequencer (Illumina, San Diego, CA).
Cell lines and culture conditions
Cell lines A549, H441, H661, H838 and H460 were purchased from American Type Culture Collection (ATCC) and were cultured based on the conditions suggested by ATCC. They were cultivated at 37ºC and a 5% CO2 atmosphere in RPMI medium supplemented with 10% fetal bovine serum, penicillin, streptomycin and amphotericin B. Cell line cultures were tested regularly for mycoplasma infection using Venor® GeM-qEP qPCR kit. Cells were used to low passes after defrosting.
Methylation analysis
100 ng to 500 ng of genomic DNA was treated with sodium bisulfite using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. The samples were eluted in 20 ul of M-Elution Buffer, and 1 ul to 3 ul were used for each PCR reaction. Both bisulfite conversion and subsequent pyrosequencing analysis were done at the DNA Methylation Analysis Core, The University of Texas M.D. Anderson Cancer Center.
PCR primers for pyrosequencing methylation analysis of the genomic area proximal to the transcription start site of FOXF1 were designed using the Pyromark Assay Design SW 1.0 software (Qiagen, Hilden, Germany). Pyrosequencing and MSP primers are indicated in the Supplementary Table 1. In brief, a sequencing primer was identified within 1 to 5 base pairs near the CpG sites of interest, with an annealing temperature of 40±5 ºC. After that, forward and reverse primers are identified uspstream and downstream to the sequencing primer. Optimal annealing temperatures for each of these primers were tested using gradient PCR. Controls for high methylation (SssI-treated DNA), low methylation (WGA-amplified DNA) and no-DNA template were included in each reaction. PCR reactions were performed in a total volume of 15 μl, and the entire volume was used for each pyrosequencing reaction as previously described [31]. Briefly, PCR product purification was done with streptavidin-sepharose high-performance beads (GE Healthcare Life Sciences, Piscataway, NJ), and co-denaturation of the biotinylated PCR products and sequencing primer (3.6 pmol/reaction) was conducted following the PSQ96 sample preparation guide. Sequencing was performed on a PSQ HS 96 system (Biotage AB, Uppsala, Sweden) with the PyroMark Gold Q96 CDT Reagents (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The degree of methylation was calculated using the Pyro-Q CpG 1.0.9v software (Biotage AB, Uppsala, Sweden). The DNA obtained from FFPE tissues was investigated using methylation-specific PCR according to standard protocols, with minor modifications [32].
Treatment with 5-aza-deoxycytidine
5-aza-CdR was dissolved in water to a final concentration of 10 mg/ml and stored in aliquots at −80°C. The 5-aza-decoxycytidine treatment was optimized to establish a working concentration, using a range from 1 to 50 μM. The cells were exposed to 5-aza-CdR for 2 days to allow the drug to be incorporated into DNA at 10 μM concentration. Tissue culture medium was changed every day for both control and treated cells, to maintain the drug stability during treatment.
Luciferase reporter assay
The dual-luciferase Reporter Assay System (Promega) was used to monitor the interaction of FENDRR on FOXF1 promoter. A cloned segment of 1.7 kb DNA from the region between FENDRR and FOXF1 was introduced in vector pGL3-control vector. All SV40 coding sequences are removed. Transfections on A549 lung cancer cell line were performed using TransIT-X2® Dynamic Delivery System (Mirus Bio LLC) per triplicate.
Generation of lncRNA FENDRR plasmid
We purchased the clone HLUNG2006614 (pME18SFL3 vector) from Biological Resource Center (NBRC), National Institute of Technology and Evaluation (NITE). Next, it was subcloned into the viral vector pLVC-AcGFP-N1-PGK-PURO removing AcGFP region.
CpG methylation of the plasmid vector
pGL3-promoter vector was incubated 8 hours at 37 °C with and without methyl donor S-adenosylmethionine 10 uM (NEB) and M.SssI (30 unit). DNA was purified by using a gel extraction kit and eluted in 30 μl of H2O.
Statistical analysis
Student t-test was used to compare means of continuous variables, and Chi-square or 2-sided Fisher exact test were chosen for categorical variables. Correlations of gene expression levels with factors were performed using two-sample Welch or Wilcoxon rank sum tests. Data analyses were carried out with the SPSS statistical software, version 15.0 (SPSS Inc., Chicago, IL) and R. Using Anderson-Darling normality test implemented in the “nortest” R package we determined that FENDERR and FOXF1 datasets, are normaly distributed.
For survival analysis, high or low expression of FENDRR and FOXF1 was established for each patient if their expression levels were higher or lower than the median, respectively. Overall survival of patients with high and low expression was then compared using Kaplan-Meier and log-rank test.
Relative risks and their 95% CI were estimated by Cox proportional hazards regression.
Abbreviations
lncRNA: Long non-coding RNA; qRT-PCR: Quantitative real-time polymerase chain reaction, RNA-FISH: RNA in situ hybridization; LUAD: Lung adenocarcinoma; Ct: Threshold cycle; SSC: Saline-sodium citrate buffer; UTR: Untranslated region; ATCC: American Type Culture Collection; RPMI: Roswell Park Memorial Institute medium; 5-Aza: 5-aza-deoxycytidine; NBRC: Biological Resource Center; NITE: National Institute of Technology and Evaluation; FFPE: Formalin-fixed paraffin-embedded; LUSC: lung cell squamous cancer; CCLE: Cancer Cell Line Encyclopedia; TCGA: The Cancer Genome Atlas; MSP: Methylation-Specific PCR; GFP: Green fluorescent protein; OS: Overall survival; HR: Hazard ratio.
Author contributions
AHM, MC and PPM designed all experiments and wrote article. MIR performed the qPCR of human patients. SRP performed RNA-FISH on paraffin tissues. RT performed FENDRR cloning experiments. MRE performed MSP and CpG pyrosequencing. LC and MS provided samples and obtained the clinicopathological data. PC performed statistical and data analysis.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
FUNDING
PM’s lab is funded by the Ministry of Economy of Spain (SAF2015-67919-R), Junta de Andalucía (P12-BIO-1655), the Deutsche José Carreras Leukämie-Stiftung, BBVA Foundation and Eduardo-Gallego Grant 2015 from Francisco Cobos Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
1. Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016; 5:288–300. https://doi.org/10.21037/tlcr.2016.06.07. [PubMed].
2. Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015; 21:1253–61. https://doi.org/10.1038/nm.3981. [PubMed].
3. Peinado P, Herrea A, Baliñas C, Martin J, Boyero L, Cuadros M, Coira IF, Rodriguez MI, Zurita F, Rufino E, Lupiañez JA, Medina PP. LncRNA as cancer biomarker. Cancer and Noncoding RNAs. 2017.
4. Mattick JS. The genetic signatures of noncoding RNAs. PLoS Genet. 2009; 5:e1000459. https://doi.org/10.1371/journal.pgen.1000459. [PubMed].
5. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011; 43:904–14. https://doi.org/10.1016/j.molcel.2011.08.018. [PubMed].
6. Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, Yang X, Amit I, Meissner A, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011; 477:295–300. https://doi.org/10.1038/nature10398. [PubMed].
7. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010; 329:689–93. https://doi.org/10.1126/science.1192002. [PubMed].
8. Qiu MT, Hu JW, Yin R, Xu L. Long noncoding RNA: an emerging paradigm of cancer research. Tumour Biol. 2013; 34:613–20. https://doi.org/10.1007/s13277-013-0658-6. [PubMed].
9. Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013; 11:59. https://doi.org/10.1186/1741-7007-11-59. [PubMed].
10. Villegas VE, Zaphiropoulos PG. Neighboring gene regulation by antisense long non-coding RNAs. Int J Mol Sci. 2015; 16:3251–66. https://doi.org/10.3390/ijms16023251. [PubMed].
11. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, et al, and GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013; 45:580–85. https://doi.org/10.1038/ng.2653. [PubMed].
12. Xu TP, Huang MD, Xia R, Liu XX, Sun M, Yin L, Chen WM, Han L, Zhang EB, Kong R, De W, Shu YQ. Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J Hematol Oncol. 2014; 7:63. https://doi.org/10.1186/s13045-014-0063-7. [PubMed].
13. Li Q, Wu C, Song G, Zhang H, Shan B, Duan Y, Wang Y. Genome-Wide Analysis of Long Noncoding RNA Expression Profiles in Human Xuanwei Lung Cancer. Clin Lab. 2015; 61:1515–23. https://doi.org/10.7754/Clin.Lab.2015.150323. [PubMed].
14. Miao L, Huang Z, Zengli Z, Li H, Chen Q, Yao C, Cai H, Xiao Y, Xia H, Wang Y. Loss of long noncoding RNA FOXF1-AS1 regulates epithelial-mesenchymal transition, stemness and metastasis of non-small cell lung cancer cells. Oncotarget. 2016; 7:68339–49. https://doi.org/10.18632/oncotarget.11630. [PubMed].
15. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–07. https://doi.org/10.1038/nature11003. [PubMed].
16. Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013; 8:e82241. https://doi.org/10.1371/journal.pone.0082241. [PubMed].
17. Gutman DA, Cobb J, Somanna D, Park Y, Wang F, Kurc T, Saltz JH, Brat DJ, Cooper LA, Kong J. Cancer Digital Slide Archive: an informatics resource to support integrated in silico analysis of TCGA pathology data. J Am Med Inform Assoc. 2013; 20:1091–98. https://doi.org/10.1136/amiajnl-2012-001469. [PubMed].
18. Díez-Villanueva A, Mallona I, Peinado MA. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin. 2015; 8:22. https://doi.org/10.1186/s13072-015-0014-8. [PubMed].
19. Grote P, Herrmann BG. The long non-coding RNA Fendrr links epigenetic control mechanisms to gene regulatory networks in mammalian embryogenesis. RNA Biol. 2013; 10:1579–85. https://doi.org/10.4161/rna.26165. [PubMed].
20. Davidovich C, Zheng L, Goodrich KJ, Cech TR. Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol. 2013; 20:1250–57. https://doi.org/10.1038/nsmb.2679. [PubMed].
21. Cifuentes-Rojas C, Hernandez AJ, Sarma K, Lee JT. Regulatory interactions between RNA and polycomb repressive complex 2. Mol Cell. 2014; 55:171–85. https://doi.org/10.1016/j.molcel.2014.05.009. [PubMed].
22. Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011; 21:354–61. https://doi.org/10.1016/j.tcb.2011.04.001. [PubMed].
23. Lalevée S, Feil R. Long noncoding RNAs in human disease: emerging mechanisms and therapeutic strategies. Epigenomics. 2015; 7:877–79. https://doi.org/10.2217/epi.15.55. [PubMed].
24. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088. [PubMed].
25. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–04. https://doi.org/10.1158/2159-8290.CD-12-0095. [PubMed].
26. Mitchell SM, Ross JP, Drew HR, Ho T, Brown GS, Saunders NF, Duesing KR, Buckley MJ, Dunne R, Beetson I, Rand KN, McEvoy A, Thomas ML, et al. A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer. 2014; 14:54. https://doi.org/10.1186/1471-2407-14-54. [PubMed].
27. Lo PK, Lee JS, Liang X, Han L, Mori T, Fackler MJ, Sadik H, Argani P, Pandita TK, Sukumar S. Epigenetic inactivation of the potential tumor suppressor gene FOXF1 in breast cancer. Cancer Res. 2010; 70:6047–58. https://doi.org/10.1158/0008-5472.CAN-10-1576. [PubMed].
28. Navarro C, Cano C, Cuadros M, Herrera-Merchan A, Molina M, Blanco A. A Mechanistic Study of lncRNA Fendrr Regulation of FoxF1 Lung Cancer Tumor Supressor in: Bioinformatics and Biomedical Engineering. Proceedings of the 4th International Conference. 2016; 781–9.
29. Tamura M, Sasaki Y, Koyama R, Takeda K, Idogawa M, Tokino T. Forkhead transcription factor FOXF1 is a novel target gene of the p53 family and regulates cancer cell migration and invasiveness. Oncogene. 2014; 33:4837–46. https://doi.org/10.1038/onc.2013.427. [PubMed].
30. Grote P, Wittler L, Hendrix D, Koch F, Währisch S, Beisaw A, Macura K, Bläss G, Kellis M, Werber M, Herrmann BG. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. 2013; 24:206–14. https://doi.org/10.1016/j.devcel.2012.12.012. [PubMed].
31. Estécio MR, Yan PS, Ibrahim AE, Tellez CS, Shen L, Huang TH, Issa JP. High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res. 2007; 17:1529–36. https://doi.org/10.1101/gr.6417007. [PubMed].
32. Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996; 93:9821–26. https://doi.org/10.1073/pnas.93.18.9821. [PubMed].