
INTRODUCTION
Arsenic trioxide (ATO) was recently found to induce complete remission in relapsed or refractory acute promyelocytic leukemia (APL) patients, and the US Food and Drug Administration subsequently approved ATO as the frontline agent for APL treatment [1-3]. However, numerous clinical trials reported that chronic treatment with a therapeutic ATO concentration could cause severe cardiac arrhythmia, such as QT interval prolongation, T-U wave alternans, ST-T change, and torsades de pointes, or even sudden death [4-7]. This issue has limited the clinical usefulness of the drug. QT interval prolongation is the most common adverse effect, and is caused by delayed membrane repolarization in cardiomyocytes. Calcium current is the primary inward current in cellular membrane repolarization, and Ca2+ cycling regulates cardiac contraction. Increased [Ca2+]I is also a critical sign of cell apoptosis, and has been associated with a number of abnormalities in cardiac tissues, such as ventricular arrhythmias and contractile dysfunction [8]. ATO can augment ICa, L density, triggering intracellular calcium overload and disorder, and ultimately inducing arrhythmia and cell apoptosis [9-11].
The endoplasmic reticulum (ER) is the major intracellular Ca2+ reservoir. Ca2+-binding chaperones mediate proper folding of proteins in the ER lumen [12]. Previous studies showed that ATO is an ER stressor, and ATO-induced cardiac cell death is associated with ER stress-mediated apoptosis [13-15]. We recently demonstrated that ATO destroyed adult rat ventricular myocyte (ARVM) contractile functions and intracellular calcium homeostasis, inducing ER stress-mediated apoptosis [16]. Therefore, regulating calcium homeostasis imbalance and inhibiting ER stress may reduce ATO-induced myocardial injury.
The dried root of Salvia miltiorrhiza Bunge (also known as Danshen) is a popular traditional Chinese medicine, and has been used extensively in both Asian and Western countries to treat various diseases, including cerebrovascular diseases, coronary artery diseases, and myocardial infarction [17]. Salvianolic acid B (Sal B; Figure 1) is the most abundant and bioactive compound in Danshen, and appears to have anti-oxidant, anti-apoptosis, and anti-tumor activities [18]. Our previous studies showed that Sal B attenuates ATO-induced cardiac injury via PI3K/Akt signaling in H9c2 cells [19]. Sal B also alleviates ATO-induced loss of cardiac function and damage to cardiomyocytic structures in BALB/c mice, and can improve ATO-induced cytotoxicity and apoptosis in HepG2 and HeLa cells [20]. These data suggest that Sal B and ATO act synergistically, with reduced cardiovascular damage.
Figure 1: Sal B molecular structure. (A) and in vivo and in vitro experimental designs (B).
Previous studies confirmed that Sal B inhibits Ca2+ channels and Ca2+ influx in rat coronary artery vascular smooth muscle cells [21]. Sal B reduces cytoplasmic calcium by modulating intracellular calcium release and extracellular calcium influx [22]. We hypothesized that the Sal B cardioprotective effects may also be associated with calcium overload inhibition and subsequent ER stress-mediated apoptosis suppression. This study assessed the effects of Sal B on cardiac contraction and Ca2+ transients in ARVMs in real time using an IonOptix MyoCam system. We repeated previous BALB/c mouse model experiments to elucidate the Sal B cardioprotective mechanism. We then assessed sarcoplasmic reticulum Ca2+-ATPase (SERCA) activity, and Ca2+-handling- and ER stress signaling-related proteins in heart tissue and ARVMs. Thus, we assessed whether Sal B protects against ATO-induced cardiotoxicity via calcium homeostasis maintenance and ER stress inhibition.
RESULTS
Effect of Sal B on cardiomyocyte contractile function after ATO treatment
To determine whether Sal B protects sarcomeric contractile function against ATO-induced injury, ARVMs were treated with Sal B (1 μM) for 7 min after ATO treatment (100 μM) for 20 min. Sal B treatment alone did not affect resting cardiomyocyte contractile function compared with controls (Figure 2). However, Sal B and ATO-treated cells displayed normal sarcomere shortening amplitudes (Figure 2B), -dL/dt /+dL/dt (Figure 2C-2D), TR90 (Figure 2E), and TPS (Figure 2F), while ATO-treated cells (without Sal B) showed increased amplitudes. These data indicate that ATO perfusion severely impaired cardiomyocyte contractile function, and Sal B treatment eliminated this impairment.
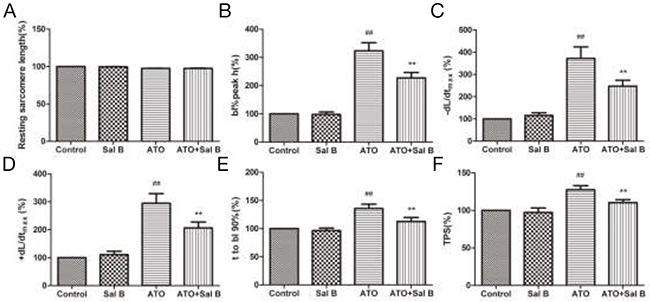
Figure 2: Effect of Sal B on cardiomyocyte contractile function after ATO treatment. Resting sarcomere length (A) Sarcomere shortening amplitude (B) Relengthening maximal velocity (-dL/dtmax) (C) Shortening maximal velocity (+dL/dtmax) (D) Time-to-90% relengthening (TR90) (E) Time-to-peak shortening (TPS) (F) n=30–40 cells from 3 rats per group; ##p<0.01 vs. control, **p<0.01 vs. ATO.
Effect of Sal B on intracellular Ca2+ transients after ATO treatment
We used intracellular fura-2 fluorescence to detect Ca2+ transients. As a calcium channel blocker [23], Sal B alone decreased the resting Ca2+ ratio and the amplitude of Ca2+ transients (Figure 3). Similar to previous findings [16], ATO treatment increased the resting Ca2+ ratio, amplitude, ±d [Ca2+]/dtmax, time-to-50% peak [Ca2+]i, and [Ca2+]i transients decay rate; Sal B treatment decreased those indexes (Figure 3A-3F). Sal B relieved Ca2+ overload and the intracellular calcium homeostasis imbalance induced by ATO in ARVMs.
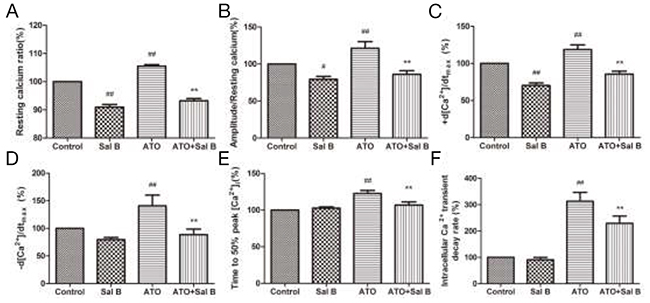
Figure 3: Effect of Sal B on ARVM intracellular Ca2+ transients after ATO treatment Resting calcium ratio (A) Amplitude/resting calcium (B) Ca2+ shortening maximal velocity (+d [Ca2+]/dtmax) (C) Ca2+ relaxation maximal velocity (-d [Ca2+]/dtmax) (D) Time-to-50% peak [Ca2+]i (E) Intracellular Ca2+ transient decay rate (F) n=30–40 cells from 3 rats per group; #p<0.05 vs. control, ##p<0.01 vs. control, **p<0.01 vs. ATO.
Effect of Sal B on SERCA activity after ATO treatment
SERCA regulates cardiac muscle Ca2+ homeostasis and contractility. To determine whether Sal B cardioprotection involves SERCA, we examined SERCA activity in sarcoplasmic reticulum (SR) vesicles extracted from heart tissue and ARVMs. ATO attenuated SERCA activity, whereas Sal B treatment effectively reversed SERCA activity compared with controls (Figure 4). These results indicate that Sal B protects against ATO-induced injury by enhancing SERCA activity.
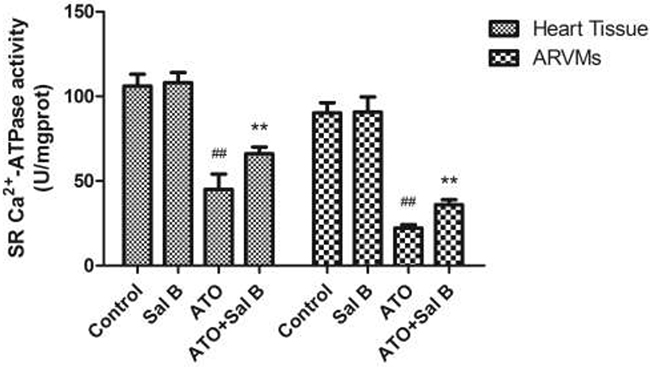
Figure 4: Effect of Sal B on SERCA activity after ATO treatment in heart tissue and ARVM. Experiments were performed three times. ##p<0.01 vs. control, **p<0.01 vs. ATO.
Effect of Sal B on Ca2+ handling protein expression after ATO treatment
We investigated Sal B regulation of Ca2+ handling proteins in heart tissue and ARVMs using western blotting. Consistent with our previous study [16], ATO treatment decreased SERCA2a, PLB, and phosphorylated PLB (p-PLB) levels and increased NCX and p-CaMKII levels (Figure 5). Sal B treatment blocked these changes, suggesting that Sal B regulates Ca2+-handling protein expression to protect against ATO-induced intracellular calcium homeostasis imbalance.
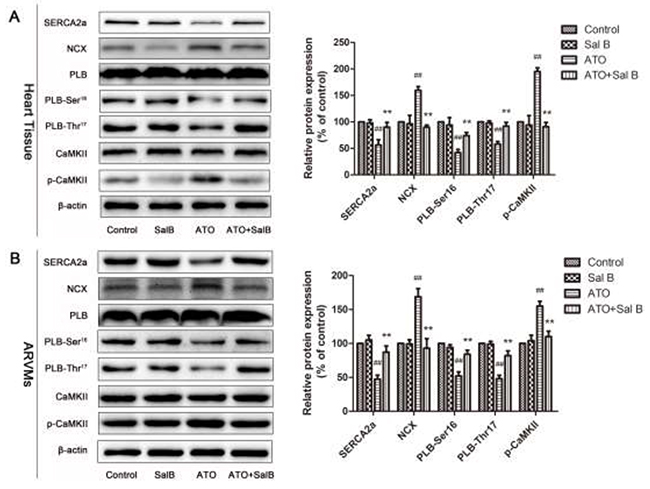
Figure 5: Effect of Sal B on Ca2+ handling protein levels after ATO treatment. Ca2+ handling protein levels in heart tissue (A) and ARVMs (B) Experiments were performed three times. ##p<0.01 vs. control, **p<0.01 vs. ATO.
Effect of Sal B on ER stress-related protein levels after ATO treatment
To confirm the involvement of ER stress in Sal B cardioprotection, we evaluated an ER stress-responsive marker (GRP78), ER stress sensors (protein kinase RNA-like ER kinase (PERK), activating transcription factor 6 (ATF6), inositol-requiring enzyme-1α (IRE1), and eukaryotic initiation factor 2 α (eIf2α), and ER stress-initiated proapoptotic factors (C/EBP homologous protein (CHOP) and Caspase12) in heart tissue and ARVMs via western blotting. GRP78, p-PERK, p-eIf2α, IRE1, and ATF6 were upregulated in the ATO group compared with controls (Figure 6). However, Sal B treatment reduced expression of GRP78 and ER stress sensors compared with the ATO group, and suppressed ATO-induced CHOP and caspase-12 upregulation. Together, these results show that ER stress was associated with ATO-induced cardiotoxicity via CHOP and caspase-12 activation through three ER stress sensors: ATF6, PERK, and IRE1. Sal B suppressed ATO-induced ER stress and apoptosis.
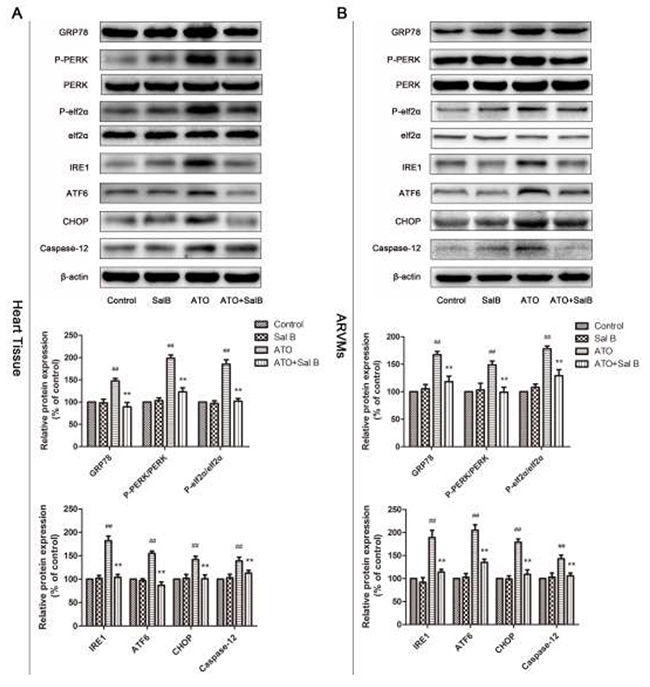
Figure 6: Effect of Sal B on ER stress-related protein levels after ATO treatment. Ca2+ handling protein levels in heart tissue (A) and ARVMs (B) Experiments were performed three times. ##p<0.01 vs. control, **p<0.01 vs. ATO.
DISCUSSION
ATO is a potent anticancer agent that is highly effective against APL. However, many patients are prevented from receiving ATO due to its cardiotoxicity [24]. In this study, we detected real-time changes in sarcomere shortening and cytoplasmic Ca2+ transients in ARVMs. In agreement with our previous findings [16], ATO treatment impaired cell contractility and Ca2+ homeostasis. However, Sal B effectively attenuated or improved ATO-induced cardiac contractile and intracellular Ca2+ dysfunctions. Sal B treatment also increased SERCA activity in heart tissue and ARVMs. Our results also implicated ER stress-associated apoptosis inhibition in the Sal B cardioprotective mechanism. Combined with our previous report [19, 20], our findings demonstrated that Sal B protects against ATO-induced cardiotoxicity not only through its anti-oxidative and anti-apoptotic capacities, but also by modulating calcium homeostasis and suppressing ER stress.
Ca2+ is a crucial secondary messenger in ventricular myocytes [25]. The [Ca2+]i transients decay rate is commonly used to characterize the speed of intracellular Ca2+ clearing in the cytoplasm [26]. SERCA is responsible for calcium uptake from the cytosol into the ER lumen after contraction. We found that Sal B treatment reversed the ATO-induced decline in [Ca2+]i transients decay rate, indicating that Sal B likely enhanced SERCA activity to maintain calcium homeostasis. SERCA is controlled by a closely associated SR membrane protein, phospholamban (PLB), which has two phosphorylation sites, Ser16 and Thr17 [27]. Dephosphorylated PLB can restrain SERCA activity, and PLB phosphorylation eliminates this inhibition. In rat ventricular myocytes, SERCA and NCX are responsible for >90% and ~7% of Ca2+ removal, respectively [28]. Calcium/calmodulin-dependent protein kinase II (CaMK II) regulates intracellular Ca2+ transport and modulates myocardial cell contractile and electrical activities [29]. A recent study associated CaMKII with arrhythmia occurrence. Increased CaMKII activity could lead to Ca2+ overload in cells and trigger malignant ventricular arrhythmia [30, 31]. We found that Sal B upregulated ATO-decreased SERCA and p-PLB levels, and simultaneously downregulated ATO-increased p-CaMK II and NCX levels in heart tissue and ARVMs. Therefore, Sal B ameliorated ATO-induced Ca2+ overload in ARVMs by regulating SERCA expression and activity.
ATO treatment can induce ATP depletion, oxidative stress, and calcium imbalance, leading to accumulation and aggregation of unfolded proteins in the ER lumen, and ultimately the unfolded protein response (UPR) and ER stress [32, 33]. Glucose-regulated proteins 78 (GRP78) are ER chaperones and serve as markers of ER stress [34]. UPR is characterized by the actions of three stress sensors: PERK, ATF6, and IRE1 [35-37]. When ER stress is not mitigated and homeostasis is not restored, the endogenous ER stress response transitions from promoting cell survival to apoptosis through CHOP-, c-Jun NH2-terminal kinase (JNK)-, and caspase-12-dependent pathways [38-41].
Although the significance of ER stress in the ATO anticancer effect has been studied, few studies focused on alleviating ATO cardiotoxicity by suppressing ER stress, and whether ATO-induces ER stress in the heart requires further study [13]. Our data showed that ATO enhanced expression of the ER stress-responsive marker, GRP78, the ER stress sensors, p-ATF6, p-PERK, eIf2α, and IRE1, and the downstream apoptosis proteins, CHOP and caspase-12. This indicated that ATO activated ER stress and, therefore, apoptosis. However, Sal B suppressed these ATO-induced processes in heart tissue and ARVMs. GRP78, p-PERK, ATF6, IRE1, CHOP, and caspase-12 were all downregulated in Sal B-treated versus ATO-treated cells. Thus, Sal B directly inhibited ATO-induced ER stress.
In conclusion, Sal B treatment protected against ATO-induced cardiotoxicity by improving contractile recovery and maintaining calcium homeostasis. We found that Ca2+-handling protein regulation and ER stress-associated apoptosis inhibition are important in Sal B cardioprotection in vivo and in vitro. Sal B treatment blocked Ca2+ overload and upregulated SERCA activity and expression, ameliorating ATO-induced calcium homeostasis imbalance. Finally, Sal B inhibited ATO-induced ER stress by downregulating GRP78, ERS, PERK, ATF6, and IRE1. Our results suggest that Sal B may prevent ATO cardiotoxicity during clinical application. However, our data do not fully explain the direct relationship between calcium homeostasis imbalance and ER stress. Therefore, the exact mechanisms of Sal B cardioprotective activity require further study.
MATERIALS AND METHODS
Materials
Sal B (≥98%) was purchased from Shanghai Winherb Medical S&T Development Co., Ltd. (Shanghai, China). ATO was acquired from Harbin YI-DA Pharmaceutical Ltd. (Harbin, China). Collagenase Type II and Fura-2/AM were purchased from Life Technologies Corporation (Carlsbad, CA, USA). The sarcoplasmic reticulum Ca2+-ATPase (SERCA) activity detection kit was acquired from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All antibodies were obtained from Santa Cruz Biotechnology except for SERCA and NCX, which were purchased from Abcam (Cambridge, UK). All chemicals were obtained from Sigma Chemical Co., Ltd. (St. Louis, MO, USA).
Animals and treatments
Sixty male BALB/c mice weighing 18–20 g each (Vital River Laboratories, Beijing, China) were used in the experiment. All procedures were approved by the local animal committee. All animal care procedures and interventions were performed in accordance with the Guidelines and Policies for Animal Surgery provided by the Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China, and were approved by the Institutional Animal Use and Care Committee (registration number: #IMPLAD2015090216). Experimental protocols were previously described [20]. In brief, rats were randomly divided into four groups (n=15/group): (I) the control group (10 mL/kg saline), (II) Sal B-treated group (2 mg/kg Sal B), (III) ATO-treated group (1 mg/kg ATO), and (IV) ATO + Sal B group (2 mg/kg Sal B 1 h before ATO administration). All treatments were injected once a day via the tail vein for 2 weeks. The heart was excised, and myocardial homogenates were prepared for western blotting and SERCA detection.
Adult rat ventricular myocyte isolation
ARVMs were isolated using a modified enzymatic method as previously described [42]. Briefly, hearts were immediately excised from adult male Sprague-Dawley rats (180–200 g each) under pentobarbital sodium anesthesia (140 mg/kg intraperitoneally). Hearts were mounted on a Langendorff perfusion apparatus and perfused with Ca2+-containing Tyrode’s solution equilibrated with O2 (NaCl, 137; KCl, 5.4; MgCl2, 1.2; HEPES, 10; glucose, 10; and CaCl2, 1.2 [in mM]) for 2 min at a rate of 6 mL/min. Hearts were then perfused with Ca2+-free Tyrode’s solution for 5 min along with the components mentioned above, except for CaCl2. Hearts were then perfused with Ca2+-free Tyrode’s solution with collagenase Type II (210.00 units/mg) and 0.7 g/l bovine serum albumin until they became flaccid. The digested tissues were separated and filtered through a nylon mesh (300 mm) after perfusion. The filtered cell suspension was rinsed several times with Ca2+-containing Tyrode’s solution to progressively increase extracellular Ca2+ to 1.2 mM. Only viabilities over 85% and rod-shaped ARVMs with clear edges were used in this study.
Treatment and measurement of sarcomere shortening and Ca2+ transients in ARVMs
Sarcomere shortening and Ca2+ transients were assessed simultaneously upon field stimulation (0.5 Hz with 2 ms duration, 16 V) using a video-based sarcomere contractility and calcium-recording module in a SoftEdge MyoCam system (IonOptix Corporation, Milton, MA, USA). To measure intracellular Ca2+, freshly isolated myocytes were incubated with Fura-2/AM (2 μM) for 15 min at 37°C and then washed twice with Ca2+-containing Tyrode’s solution to remove residual Fura-2/AM. ARVMs were then placed in a Warner chamber mounted on the stage of an inverted microscope (Olympus, IX-70) and superfused with the following: (I) Ca2+-containing Tyrode’s solution for 27 min (control group), (II) Ca2+-containing Tyrode’s solution with Sal B (1 μM) for 7 min (Sal B group), (III) Ca2+-containing Tyrode’s solution with ATO (100 μM) for 20 min (ATO group), and (IV) Ca2+-containing Tyrode’s solution with ATO (100 μM) for 20 min before Ca2+-containing Tyrode’s solution with Sal B (1 μM) for 7min (ATO + Sal B group) at a rate of 1.5 mL/min. Myocytes (n=30–40 cells from 5 rats per group) were allowed to equilibrate for about 10 min before measurement. Data were recorded and analyzed using IonWizard software (version 6.2.0.59) [43].
Measurement of SERCA activity
As previously described [44], SERCA activity measurements were completed using a commercially available kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Western blot analysis
After exposure to different treatment solutions, ARVM and heart tissue homogenates were lysed on ice with tissue or cell protein extraction reagent containing 1% phenylmethylsulfonyl fluoride (CoWin Bioscience Co., Ltd., Beijing, China). Supernatants were collected after centrifuging at 12,000 rpm at 4°C for 20 min. Protein concentrations were determined using a BCA kit (Pierce Corporation, Rockford, USA). Equal amounts of protein were separated via 8–12% SDS-PAGE and then transferred to nitrocellulose membranes (Millipore Corporation, USA) in a Tris-glycine buffer at 100 V for 1 h in an icebox. Membranes were blocked with 5% (w/v) non-fat milk powder in Tris-buffered saline containing 0.1% (v/v) Tween-20 (TBST) for 3 h at room temperature, and then incubated overnight with appropriate primary antibodies (1:200) at 4°C. Membranes were washed with TBST three times, incubated with corresponding secondary HRP-conjugated antibodies (1:1000) for 2 h on a shaking table, and then washed again three times with TBST. Membranes were developed using an enhanced chemiluminescence solution. Protein levels were visualized with Image Lab Software (Bio-Rad, USA). Western blot analysis was performed as previously described [45].
Statistical analysis
Data were expressed as means ± SD from at least three independent experiments. Statistical comparisons between different groups were determined by one-way ANOVA or the Student-Newman-Keuls method with GraphPad Prism 5.0 software (SPAA Inc., Chicago). P<0.05 was considered statistically significant.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
The present work was supported by the Major Scientific and Technological Special Project for “Significant New Drugs Formulation” (Grant No. 2017ZX09301069), the National Natural Science Foundation of China (No. 81374011), the Special Project for the National Traditional Chinese Medicine Industry of China (Grant no. 201507004), the PUMC Youth Fund (Grant no. 3332015049), and the Fundamental Research Funds for the Central Universities.
REFERENCES
1. Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997; 89:3354-3360.
2. Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J, Scheinberg DA, Pandolfi PP, Warrell RJ. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998; 339:1341-1348.
3. Fox E, Razzouk BI, Widemann BC, Xiao S, O’Brien M, Goodspeed W, Reaman GH, Blaney SM, Murgo AJ, Balis FM, Adamson PC. Phase 1 trial and pharmacokinetic study of arsenic trioxide in children and adolescents with refractory or relapsed acute leukemia, including acute promyelocytic leukemia or lymphoma. Blood. 2008; 111:566-573.
4. Soignet SL, Frankel SR, Douer D, Tallman MS, Kantarjian H, Calleja E, Stone RM, Kalaycio M, Scheinberg DA, Steinherz P, Sievers EL, Coutre S, Dahlberg S, et al. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol. 2001; 19:3852-3860.
5. Huang SY, Chang CS, Tang JL, Tien HF, Kuo TL, Huang SF, Yao YT, Chou WC, Chung CY, Wang CH, Shen MC, Chen YC. Acute and chronic arsenic poisoning associated with treatment of acute promyelocytic leukaemia. Br J Haematol. 1998; 103:1092-1095.
6. Little RE, Kay GN, Cavender JB, Epstein AE, Plumb VJ. Torsade de pointes and T-U wave alternant associated with arsenic poisoning. Pacing Clin Electrophysiol. 1990; 13:164-170.
7. Unnikrishnan D, Dutcher JP, Varshneya N, Lucariello R, Api M, Garl S, Wiernik PH, Chiaramida S. Torsades de pointes in 3 patients with leukemia treated with arsenic trioxide. Blood. 2001; 97:1514-1516.
8. Li P, Song Q, Liu T, Wu Z, Chu X, Zhang X, Zhang Y, Gao Y, Zhang J, Chu L. Inhibitory effect of cinobufagin on L-type Ca2+ currents, contractility, and Ca2+ homeostasis of isolated adult rat ventricular myocytes. ScientificWorldJournal. 2014; 2014:496705.
9. Chen X, Shan H, Zhao J, Hong Y, Bai Y, Sun I, Pan Z, Zhang Y, Yang B, Du Z. L-type calcium current (ICa, L) and inward rectifier potassium current (IK1) are involved in QT prolongation induced by arsenic trioxide in rat. Cell Physiol Biochem. 2010; 26:967-974.
10. Vineetha VP, Girija S, Soumya RS, Raghu KG. Polyphenol-rich apple (Malus domestica L.) peel extract attenuates arsenic trioxide induced cardiotoxicity in H9c2 cells via its antioxidant activity. Food Funct. 2014; 5:502-511.
11. Zhao XY, Li GY, Liu Y, Chai LM, Chen JX, Zhang Y, Du ZM, Lu YJ, Yang BF. Resveratrol protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Br J Pharmacol. 2008; 154:105-113.
12. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013; 1833:3460-3470.
13. Miao X, Tang Z, Wang Y, Su G, Sun W, Wei W, Li W, Miao L, Cai L, Tan Y, Liu Q. Metallothionein prevention of arsenic trioxide-induced cardiac cell death is associated with its inhibition of mitogen-activated protein kinases activation in vitro and in vivo. Toxicol Lett. 2013; 220:277-285.
14. Binet F, Chiasson S, Girard D. Arsenic trioxide induces endoplasmic reticulum stress-related events in neutrophils. Int Immunopharmacol. 2010; 10:508-512.
15. Tang CH, Chiu YC, Huang CF, Chen YW, Chen PC. Arsenic induces cell apoptosis in cultured osteoblasts through endoplasmic reticulum stress. Toxicol Appl Pharmacol. 2009; 241:173-181.
16. Zhang JY, Sun GB, Wang M, Liao P, Du YY, Yang K, Sun XB. Arsenic trioxide triggered calcium homeostasisimbalance and induced endoplasmic reticulum stress-mediated apoptosis in adult rat ventricularmyocytes. Toxicol. Res. 2016; 5: 682-688.
17. Yagi A, Fujimoto K, Tanonaka K, Hirai K, Takeo S. Possible active components of tan-shen (Salvia miltiorrhiza) for protection of the myocardium against ischemia-induced derangements. Planta Med. 1989; 55:51-54.
18. Lv H, Wang L, Shen J, Hao S, Ming A, Wang X, Su F, Zhang Z. Salvianolic acid B attenuates apoptosis and inflammation via SIRT1 activation in experimental stroke rats. Brain Res Bull. 2015; 115:30-36.
19. Wang M, Sun G, Sun X, Wang H, Meng X, Qin M, Sun J, Luo Y, Sun X. Cardioprotective effect of salvianolic acid B against arsenic trioxide-induced injury in cardiac H9c2 cells via the PI3K/Akt signal pathway. Toxicol Lett. 2013; 216:100-107.
20. Wang M, Sun G, Wu P, Chen R, Yao F, Qin M, Luo Y, Sun H, Zhang Q, Dong X, Sun X. Salvianolic Acid B prevents arsenic trioxide-induced cardiotoxicity in vivo and enhances its anticancer activity in vitro. Evid Based Complement Alternat Med. 2013; 2013:759483.
21. Lam FF, Yeung JH, Kwan YW, Chan KM, Or PM. Salvianolic acid B, an aqueous component of danshen (Salvia miltiorrhiza), relaxes rat coronary artery by inhibition of calcium channels. Eur J Pharmacol. 2006; 553:240-245.
22. Shou Q, Pan Y, Xu X, Xu J, Wang D, Ling Y, Chen M. Salvianolic acid B possesses vasodilation potential through NO and its related signals in rabbit thoracic aortic rings. Eur J Pharmacol. 2012; 697:81-87.
23. Lam FF, Yeung JH, Kwan YW, Chan KM, Or PM. Salvianolic acid B, an aqueous component of danshen (Salvia miltiorrhiza), relaxes rat coronary artery by inhibition of calcium channels. Eur J Pharmacol. 2006; 553:240-245.
24. Amini-Khoei H, Hosseini MJ, Momeny M, Rahimi-Balaei M, Amiri S, Haj-Mirzaian A, Khedri M, Jahanabadi S, Mohammadi-Asl A, Mehr SE, Dehpour AR. Morphine attenuated the cytotoxicity induced by arsenic trioxide in H9c2 cardiomyocytes. Biol Trace Elem Res. 2016.
25. Bernardi P, Rasola A. Calcium and cell death: the mitochondrial connection. Subcell Biochem. 2007; 45:481-506.
26. Wang M, Xu X, Xu H, Wen F, Zhang X, Sun H, Yao F, Sun G, Sun X. Effect of the total saponins of Aralia elata (Miq) Seem on cardiac contractile function and intracellular calcium cycling regulation. J Ethnopharmacol. 2014; 155:240-247.
27. Hagemann D, Xiao RP. Dual site phospholamban phosphorylation and its physiological relevance in the heart. Trends Cardiovasc Med. 2002; 12:51-56.
28. Hove-Madsen L, Bers DM. Sarcoplasmic reticulum Ca2+ uptake and thapsigargin sensitivity in permeabilized rabbit and rat ventricular myocytes. Circ Res. 1993; 73:820-828.
29. Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007; 73:631-640.
30. Kirchhof P, Fabritz L, Kilic A, Begrow F, Breithardt G, Kuhn M. Ventricular arrhythmias, increased cardiac calmodulin kinase II expression, and altered repolarization kinetics in ANP receptor deficient mice. J Mol Cell Cardiol. 2004; 36:691-700.
31. Khoo MS, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, Mendes L, Ni G, Madu EC, et al. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circulation. 2006; 114:1352-1359.
32. Prell T, Lautenschlager J, Weidemann L, Ruhmer J, Witte OW, Grosskreutz J. Endoplasmic reticulum stress is accompanied by activation of NF-kappaB in amyotrophic lateral sclerosis. J Neuroimmunol. 2014; 270:29-36.
33. Yu Y, Sun G, Luo Y, Wang M, Chen R, Zhang J, Ai Q, Xing N, Sun X. Cardioprotective effects of Notoginsenoside R1 against ischemia/reperfusion injuries by regulating oxidative stress- and endoplasmic reticulum stress- related signaling pathways. Sci Rep. 2016; 6:21730.
34. Gorman AM, Healy SJ, Jager R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther. 2012; 134:306-316.
35. Shen X, Zhang K, Kaufman RJ. The unfolded protein response--a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat. 2004; 28:79-92.
36. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007; 8:519-529.
37. Hoozemans JJ, Scheper W. Endoplasmic reticulum: the unfolded protein response is tangled in neurodegeneration. Int J Biochem Cell Biol. 2012; 44:1295-1298.
38. Kadowaki H, Nishitoh H, Ichijo H. Survival and apoptosis signals in ER stress: the role of protein kinases. J Chem Neuroanat. 2004; 28:93-100.
39. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008; 7:1013-1030.
40. Yu Y, Sun G, Luo Y, Wang M, Chen R, Zhang J, Ai Q, Xing N, Sun X. Cardioprotective effects of Notoginsenoside R1 against ischemia/reperfusion injuries by regulating oxidative stress- and endoplasmic reticulum stress- related signaling pathways. Sci Rep. 2016; 6:21730.
41. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005; 115:2656-2664.
42. Ren J, Wold LE. Measurement of cardiac mechanical function in isolated ventricular myocytes from rats and mice by computerized video-based imaging. Biol Proced Online. 2001; 3:43-53.
43. Wang M, Xu X, Xu H, Wen F, Zhang X, Sun H, Yao F, Sun G, Sun X. Effect of the total saponins of Aralia elata (Miq) Seem on cardiac contractile function and intracellular calcium cycling regulation. J Ethnopharmacol. 2014; 155:240-247.
44. Wang M, Sun G, Zhang J, Luo Y, Yu Y, Xu X, Meng X, Zhang M, Lin W, Sun X. Elatoside C protects the heart from ischaemia/reperfusion injury through the modulation of oxidative stress and intracellular Ca2+ homeostasis. Int J Cardiol. 2015; 185:167-176.
45. Luo Y, Sun G, Dong X, Wang M, Qin M, Yu Y, Sun X. Isorhamnetin attenuates atherosclerosis by inhibiting macrophage apoptosis via PI3K/AKT activation and HO-1 induction. PLoS One. 2015; 10:e120259.