
Introduction
Mutational status of the tumor protein p53 (TP53) gene has been one of the most important prognostic molecular markers in B-chronic lymphocytic leukemia (B-CLL) for a long time [1-7], although more recently additional promising novel candidate genes have been described in B-CLL patients as results of whole-exome and/or whole-genome sequencing [8-18]. These technologies provide a novel opportunity to analyze the clonal heterogeneity of the B-CLL genome, with the potential for sensitive detection of mutations restricted to a small fraction of the total tumor cell population. In particular, besides TP53, two of the most extensively studied genes in B-CLL involved in p53-pathway are ataxia telangiectasia mutated (ATM) [8-11] and NOTCH1 [12-18]. ATM mutation has been reported in up to 16% of B-CLL patients and may be particularly relevant in the setting of del11q, which invariably results in the deletion of one ATM allele. Anyhow, sole 11q deletion does not result in ATM inactivation by contrast to biallelic defects involving mutations. Interestingly, it has been shown that ATM dysfunction leads to impaired CDKN1A/p21 induction after doxorubicin exposure [11]. Mutations in NOTCH1 have been described in 8-12% of newly diagnosed B-CLL patients with increasing frequencies (>20%) in advanced disease stages [12-18]. Patients with NOTCH1 mutations have shorter time to treatment (TTT) and overall survival (OS) independent of other prognostic factors. In this contest, we have previously demonstrated that in vitro exposure to the small molecule non-genotoxic activator of the p53 pathway Nutlin-3 activates the NOTCH1 pathway in B-CLL [19]. This finding envisions a negative feed-back loop between p53 and NOTCH1 [19], because a potential mechanism of action of NOTCH1 as oncogene is the suppression of p53-mediated apoptosis through the regulation of p53 stability [20].
Nutlin-3 is a small molecule able to specifically target the p53/MDM2 interaction, leading to the increment of p53 protein levels, transcriptional activation of the p53 molecular targets and, subsequently, to the promotion of cell-cycle arrest and apoptosis induction in a variety of tumor cells [21-24]. Of note, Nutlin-3 and its derivatives have therapeutic perspectives in hematological malignancies [25].
On these bases, a cohort of B-CLL patient samples (n=108) was characterized for the presence of mutations in TP53 and in a subset of genes related to the p53-pathway, and then analyzed for the transcriptional response to the in vitro Nutlin-3 treatment, with particular attention to the induction of CDKN1A/p21, which accurately predict the therapeutic response in B-CLL [26].
Results
Targeted deep sequencing analysis of B-CLL
By using a next generation sequencing (NGS) approach, we have performed a mutation screening for TP53 and for a subset of TP53 related genes on peripheral CD19+ cells obtained from 108 B-CLL patients. As summarized in Table 1, the study population included patients at different disease stage and characterized by different clinical prognostic markers. A total of 216 libraries (2 libraries per B-CLL sample) were sequenced on Ion Chips 316 and 318; quality control and reads alignment to the reference genomic target regions reported an average reads aligned on target at 90.2% with a average depth over 600-fold per sample. Overall, a total of 44 variants were detected in five of the targeted genes (ATM, CDKN1A, NOTCH1, NOTCH2, TP53) and were validated in 38 patients (Table 2). These included 31 nucleotide changes already reported in the COSMIC v68 database.
Table 1: Characteristics of the whole B-CLL patient study group and of the patient subgroups harboring TP53, ATM or NOTCH1 mutations
Characteristics |
All patients (n=108)* |
TP53 mutated (n=10)* |
ATM mutated (n=18)* |
NOTCH1 mutated (n=10)* |
|||
Age >70 y |
55.5 |
60 |
72.2 |
60 |
|||
Male |
63.0 |
70 |
55.6 |
70 |
|||
Rai Stage 0 |
66.0 |
50 |
72.2 |
60 |
|||
Rai Stage I |
12.6 |
0 |
0 |
10 |
|||
Rai Stage II |
11.7 |
10 |
22.2 |
20 |
|||
Rai Stage III |
1.0 |
0 |
0 |
0 |
|||
Rai Stage IV |
8.7 |
40 |
5.6 |
10 |
|||
IGHV mutated |
69.3 |
60 |
55.6 |
60 |
|||
CD38 high |
19.4 |
10 |
27.8 |
50 |
|||
In therapy |
36.1 |
70 |
38.9 |
50 |
|||
Trisomy 12 |
15.6 |
10 |
11.1 |
40 |
|||
13q deletion |
63.5 |
70 |
100 |
30 |
|||
11q deletion |
6.3 |
10 |
22.2 |
0 |
|||
17p deletion |
9.4 |
70 |
11.1 |
0 |
|||
TP53 mutations |
9.3 |
- |
11.1 |
0 |
|||
ATM mutations |
16.7 |
20 |
- |
10 |
|||
NOTCH1 mutations |
9.3 |
0 |
5.6 |
- |
|||
*: all values are expressed as percentage.
Table 2: List of NGS mutation validated by Sanger sequencing
Exon |
nt change |
aa change |
COSMIC reported |
HLT tumor |
Mutated Patients (n) |
Freq. % |
ATM |
||||||
8 |
c.946T>C |
p.Tyr316His |
No |
- |
1 |
51.2 |
9 |
c.1229T>C |
p.Val410Ala |
Yes |
Yes, and others |
2 |
50.6, 51.1 |
12 |
c.1810C>T |
p.Pro604Ser |
Yes |
Yes |
1 |
48.6 |
20 |
c.2932T>C |
p.Ser978Pro |
Yes |
Yes |
1 |
51.2 |
22 |
c.3161C>G |
p.Pro1054Arg |
Yes |
Yes |
6 |
43.2 - 46.8 |
29 |
c.4388T>G |
p.Phe1463Cys |
No |
- |
1 |
50.0 |
37 |
c.5558A>T |
p.Asp1853Val |
Yes |
Yes |
2 |
36.2, 46.8 |
39 |
c.5818G>T |
Glu1940* |
No |
- |
1 |
87.8 |
40 |
c.5975_5979del |
p.Ser1993Argfs*23 |
No |
- |
1 |
55.0 |
50 |
c.7342G>A |
p.Asp2448Asn |
Yes |
Yes |
1 |
93.6 |
50 |
c.7390T>C |
p.Cys2464Arg |
Yes |
Yes |
1 |
49.7 |
52 |
c.7671_7674del |
p.Phe2558Leufs*5 |
No |
- |
1 |
89.4 |
58 |
c.8492T>C |
p.Phe2831Ser |
No |
- |
1 |
20.9 |
CDKN1A |
||||||
2 |
c.350G>A |
p.Cys117Tyr |
No |
- |
1 |
47.5 |
NOTCH1 |
||||||
17 |
c.2734C>T |
p.Arg912Trp |
No |
- |
1 |
50.9 |
19 |
c.3011C>T |
p.Ser1004Leu |
No |
- |
1 |
48.5 |
23 |
c.3853G>A |
p.Val1285Met |
No |
- |
1 |
54.5 |
31 |
c.5690C>T |
p.Thr1897Met |
No |
- |
1 |
48.4 |
34 |
c.6941T>C |
p.Leu2314Pro |
No |
- |
1 |
53.5 |
34 |
c.7375C>T |
p.Gln2459* |
Yes |
Yes |
1 |
53.2 |
34 |
c.7541_7542del |
p.Pro2514Argfs*4 |
Yes |
Yes, and others |
4 |
36.1 - 52.8 |
NOTCH2 |
||||||
22 |
c.3625T>G |
p.Phe1209Val |
Yes |
Yes |
1 |
46.4 |
34 |
c.7242C>G |
p.Tyr2414* |
No |
- |
1 |
44.7 |
TP53 |
||||||
6 |
c.376-2A>G |
p.Tyr126_Lys132del |
Yes |
Others |
1 |
94.5 |
5 |
c.470T>A |
p.Val157Asp |
Yes |
Others |
1 |
96.2 |
7 |
c.701A>G |
p.Tyr234Cys |
Yes |
Yes, and others |
1 |
26.1 |
7 |
c.733G>A |
p.Gly245Ser |
Yes |
Yes, and others |
1 |
77.0 |
7 |
c.742C>T |
p.Arg248Trp |
Yes |
Yes, and others |
1 |
97.5 |
7 |
c.770T>C |
p.Leu257Pro |
Yes |
Yes, and others |
1 |
75.7 |
8 |
c.817C>T |
p.Arg273Cys |
Yes |
Yes, and others |
2 |
5.4, 13.4 |
8 |
c.818G>A |
p.Arg273His |
Yes |
Yes, and others |
1 |
95.4 |
8 |
c.832C>T |
p.Pro278Ser |
Yes |
Others |
1 |
18.9 |
8 |
c.838A>G |
p.Arg280Gly |
Yes |
Yes, and others |
1 |
82.3 |
nt: nucleotide; aa: aminoacid; HLT: hematopoietic and lymphoid tissue; Others: mutation identified in other tumors; Freq.: mutated allele frequency estimated by NGS (multiple values are referred either to 2 different patients or to the range when the mutation was documented in >2 patients).
Potentially TP53 pathogenetic mutations were identified in 10 out of 108 B-CLL patients (9.3%), consistently with previously reported studies carried out with the same genetic approach [27]. These mutations included 10 non-synonymous and 1 splicing site mutation leading to in frame protein deletion of 7 aminoacids (Figure 1A), with one patient harboring 2 different mutations. Of interest, all aminoacid changes afflicted the DNA-binding domain (102-292 aa; Figure 1A-B), with a particular high hot spot of mutations on the protein region involved in the DNA interaction (273-280 aa) [28,29].
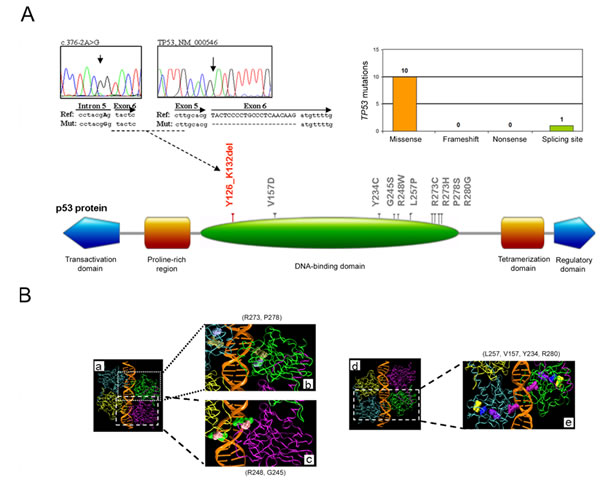
Figure 1: Molecular profile of the TP53 mutations. In A, distribution of the TP53 mutations per type of mutation and schematic representation of the p53 protein domains with indication of the aminoacid alterations: in gray, the non-synonymous mutations; in red, the inframe deletions. Partial electropherograms of the validated splicing site mutation c.376-2A>G on DNA and cDNA, leading to an inframe 7 aminoacid deletion (p.Tyr126_Lys132delYSPALNK) are shown. In B, mutated aminoacids are also evidenced on the structure reconstruction of the p53 tetramer-DNA complex (a and d; PDB accession code 2AC0): (b) tan-R273 and ice blue-P278; (c) green-R248 and pink-G245; (e) yellow-L257, blue-V157, violet-Y234 and purple-R280.
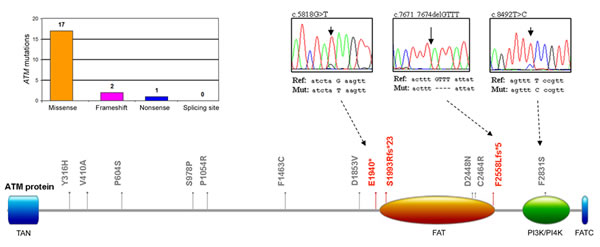
Figure 2: Molecular profile of the ATM mutations. Distribution of the ATM mutations per type of mutation and schematic representation of the ATM protein domains with indication of the aminoacid alterations: in gray, non-synonymous mutations; in red, nonsense and frameshift deletions. Partial electropherograms of one nonsense (c.5818G>T, Glu1940*), one frameshift deletion (c.7471_7674delGTTT, p.Phe2558Leufs*5), and one non-synonymous (c.8492T>C, p.Phe2831Ser) mutations are shown.
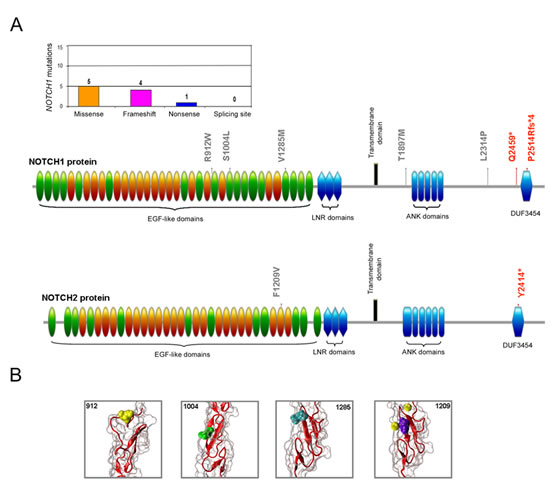
Figure 3: Molecular profile of the NOTCH mutations. In A, distribution of the NOTCH mutations per type of mutation and schematic representation of the NOTCH1 and NOTCH2 protein domains with indication of the aminoacid alterations. In gray, non-synonymous mutations; in red, nonsense and frameshift deletion mutations. In B, mutated aminoacids are also evidenced on the three-dimensional structure reconstruction of the NOTCH EGF-like domain (PDB accession code 1TOZ). In yellow, cyan and green the mutated aminoacids (positions 912, 1004, and 1285 respectively) identified in NOTCH1 protein. In violet the mutated aminoacid (position 1209) identified in NOTCH2 protein and in yellow the calcium ion that is predicted to bind the wild type aminoacid in position 1209.
In parallel, the analysis of the TP53 functional modulators, such as ATM, MDM2, NOTCH1 and NOTCH2, revealed absence of mutations in MDM2, while ATM mutations were identified in 18 (16.7%) patients and NOTCH1 and NOTCH2 mutations were documented in 10 (9.3%) and 2 (1.9%) of the B-CLL patients, respectively (Table 2). In 2 patients ATM mutations were coupled to TP53 mutations (Table 1), and in an additional patient ATM mutation was coupled to NOTCH1 mutation. Studying the potential effect of the 20 identified mutations in the ATM gene at protein level, we observed that the point aminoacid changes and deleterious mutation were distributed across the full length of the serine-protein kinase ATM, with 6 mutations affecting the principal functional domains FAT (1960-2566 aa) and PI3K/PI4K (2712-2962 aa) (Figure 2). With respect to NOTCH proteins, among the aminoacid substitutions some were located in the EGF-like domains (24, 26 and 33 for NOTCH1 and 31 for NOTCH2) (Figure 3). On the other side, mutations affecting the protein length (stop codons and frameshift deletions) were located in the intracellular portion, affecting the DUF3454 domain (Figure 3), as previously described [13]. Interestingly, while 20% of TP53 mutated B-CLL patients also showed ATM mutations, none of the patients showed mutations in both TP53 and NOTCH1 (Table 1).
Analysis of key p53 transcriptional target genes, such as CDKN1A, BAX and TNFRSF10A and TNFRSF10B [21,30,31], revealed mutation of CDKN1A in only 1 B-CLL patient (0.9%) (Tables 1 and 2), while no mutations were detected in BAX, as well as of genes encoding death receptors TNFRSF10A and TNFRSF10B.
Analysis of the transcriptional response of B-CLL to the in vitro treatment with Nutlin-3
In order to investigate the potential impact of the identified mutations on the functionality of p53 pathway, B-CLL patient derived cell cultures were treated in vitro for 24 hours with 10 microM of Nutlin-3, as previously described [19], before assessing the transcriptional activation of canonical target genes. For this purpose, the mRNA levels of CDKN1A/p21, MDM2 and BAX were comparatively measured by quantitative RT-PCR in the untreated and Nutlin-3-treated cultures derived from each B-CLL patient. As shown in Figure 4A, the induction of CDKN1A/p21, MDM2, BAX was comparable among the control group of B-CLL samples, which did not harbor any of the analyzed mutations, and the B-CLL samples characterized by either ATM mutations (n=18) or NOTCH1 mutations (n=10). On the other hand, as expected, the groups of B-CLL samples with TP53 mutations (n=10) showed a significantly (p<0.01) decreased transcriptional activity as compared to all other groups (Figure 4A). However, it is noteworthy that the subgroup of TP53 mutated B-CLL samples characterized by single mutations in the high hot spot region of the DNA binding domain (273-280 aa, n=4) still displayed a residual transcriptional activity in response to Nutlin-3 (Figure 4B). Indeed, in this subgroup of patients we documented induction levels of CDKN1A/p21, BAX and MDM2 significantly higher as compared to all other TP53 mutated samples (n=6), in which the mRNA levels of the target genes were unaffected by Nutlin-3 (mean fold induction <2; Figure 4B).
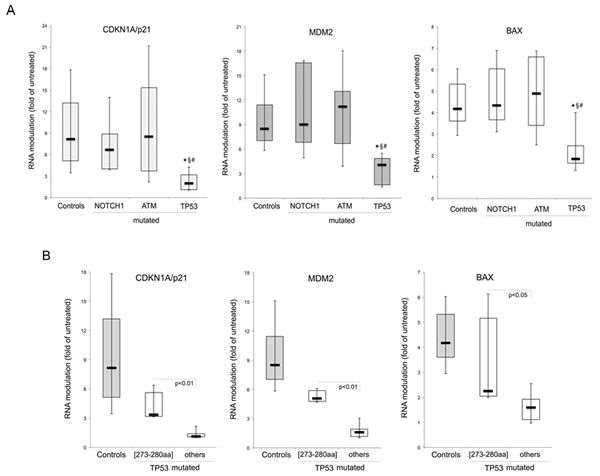
Figure 4: In vitro activation of p53 pathway by Nutlin-3. B-CLL patients’ cell cultures were exposed in vitro to Nultin-3 (10 microM) for 24 hours before RNA extraction. Transcriptional activation of p53 target genes, CDKN1A/p21, MDM2 and BAX, was assessed by quantitative RT-PCR and mRNA levels were expressed as folds of modulation with respect to the control untreated cultures set at 1. In A, the transcriptional response to Nutlin-3 is compared among the B-CLL samples harboring the indicated mutated genes and those without the investigated mutations (Controls). *, p<0.01 compared to Controls for all target genes; §, p<0.05 (for CDK1A/p21) or p<0.01 (for MDM2 and BAX) compared to NOTCH1mutated samples; #, p<0.05 (for CDK1A/p21) or p<0.01 (for MDM2 and BAX) compared to ATMmutated samples. In B, the transcriptional response to Nutlin-3 is compared between the B-CLL samples harboring TP53 mutation in different gene loci, as detailed in the text. In A and B, horizontal bars are median, upper and lower edges of box are 75th and 25th percentiles, lines extending from box are 10th and 90th percentiles.
Discussion
There are numerous reports documenting that TP53 mutations, with or without del17p, are associated with a poor outcome during first-line treatment [1-7]. However, patients with 17p- CLL exhibit marked clinical heterogeneity, with some patients experiencing an indolent course with prolonged survival [32]. A recent study published by Rossi et al demonstrated that ultra-deep-NGS significantly improved the detection of TP53 genetic defects in B-CLL allowing the identification of small TP53 mutated subclones among patients that would be otherwise considered wild type for the TP53 gene according to Sanger sequencing [27]. In agreement with the study of Rossi et al [27], we also found a TP53 mutation frequency around 9-10%, with a prevalence of missense substitutions mapping in the DNA-binding domain of the TP53 protein. We and other groups of investigators have previously demonstrated the potential therapeutic efficacy of Nutlin-3, a small molecule non-genotoxic activator of the p53 pathway, in p53wild-type B-CLL cells [21,22]. In the present study, we documented that the presence of TP53 mutations predicted impairment of the transcriptional activation of TP53 target genes, as assessed by in vitro treatment with Nutlin-3. However, we found that the residual transcriptional activity of TP53 mutations toward CDKN1A/p21 and other TP53 target genes significantly varied among the different TP53mutated B-CLL patient cell samples. In fact, mutations affecting the hot spot of the DNA binding domain [273-280 aa] were accompanied by partial impairment of the p53 transcriptional activity while mutations of other hot spots abrogated p53 transcriptional activity, as also reported by other studies [28,29,33]. In line with our results, it has been established the presence of a significant correlation between the residual transactivation function of individual TP53 alleles and clinical variables in patients with inherited p53 mutations who develop cancer [28]. In addition, it has to be underlined the importance of our experimental approach, based on the use of patient leukemic cell cultures, for the evaluation of the p53 transcriptional activity, which has revealed that the DNA binding itself is not sufficient for activating the p53 target genes in at least some of the TP53 mutants. Therefore, the transcriptional studies performed in yeasts might not always reflect the conditions of primary B-CLL patients cells [33]. Moreover, it is important to specify that the residual p53 transcriptional activity in our primary B-CLL samples harboring mutations affecting the hot spot region [273-280 aa] could not be ascribed to low frequency of the TP53 mutation, since it was observed also in patient samples in which the mutation frequency was >90% (Table 2).
Another important aim of our study was to assess whether mutations of genes related to the p53 pathway, such as in particular NOTCH1 and ATM might affect the in vitro response to Nutlin-3. Therefore, apart the data on the TP53 mutated B-CLL, it is noteworthy that Nutlin-3 was able to potently induce the transcriptional activity of p53 in ATM mutated B-CLL samples. Although these data were not unexpected since ATM works upstream of p53, it is the confirmation that a therapeutic approach based on non-genotoxic activators of the p53 pathway may be beneficial for B-CLL mutated in ATM, which represented the more frequent mutation in our patient populations (16.5%), according also to the data of other authors [8-11]. Even more importantly, we demonstrated that Nutlin-3 potently activates a p53 target gene signature also in NOTCH1 mutated B-CLL cells. These findings are noteworthy for a number or reasons. First, NOTCH1 mutations are emerging as an independent prognostic factor in B-CLL [12-18]. Second, we have previously demonstrated that Nutlin-3 upregulated NOTCH1 in B-CLL and we have proposed that NOTCH1 activation might represent a negative feed-back loop [19]. Third, it has been shown that NOTCH1 acts by down-modulating the activity of the p53 pathway [20]. Thus, the ability of Nutlin-3 to activate the p53 pathway in NOTCH1 mutated B-CLL cells is noteworthy since suggests that non-genotoxic activators of p53 might be therapeutically active also in NOTCH1 mutated B-CLL patients. This is particularly important since novel therapeutic compounds are under investigation for the therapy of B-CLL [34-37]. An additional finding of our study is that mutations of BAX and MDM2 have not been found in our patient study group. Moreover, at variance to other lymphoid malignancies [38,39], also mutations in the death receptors of TRAIL (TRAIL-R1 and TRAIL-R2), which also represent transcriptional targets of Nutlin-3 [40], have not been found in our study, and CDKN1A/p21 mutation represents a rare event, being observed in only 1 out of 108 patient samples.
Thus, the major conclusion of our study is that the p53 transcriptional pathway is fully preserved in ATM and NOTCH1 mutated B-CLL and partially preserved also in a specific subset of TP53 mutated patients. In consideration of the low number of TP53 mutated B-CLL patient samples examined, further studies on a larger cohort are necessary to ascertain whether Nutlin-3, or other non-genotoxic activator of the p53 pathway, might exhibit therapeutic benefits in a subset of TP53 mutated BCLL patients. Anyhow, these data strength the interest for potential clinical applications of Nutlin-3 also in light of previous reports suggesting that small-molecule p53 activators could have clinical benefits as chemoprotectants for cancer patients bearing p53-mutant tumors, by protecting normal cells from cytotoxicity and nuclear aberrations caused by conventional cancer therapeutics [41,42].
Methods
B-CLL patients
The study population consisted of 108 B-CLL patients. Peripheral blood samples were collected in heparin-coated tubes from all B-CLL patients following informed consent, in accordance with the Declaration of Helsinki and in agreement with institutional guidelines (University-Hospital of Ferrara). The main demographic and clinical parameters of the patients were abstracted from clinical records. B-CLL samples were also characterized by CD38 surface expression, interphase FISH and IgVH status (Table 1). All patients had been without prior therapy at least for three weeks before blood collection. Peripheral blood mononuclear cells (PBMC) were isolated by gradient centrifugation with lymphocyte cell separation medium (Cedarlane Laboratories, Hornby, ON). T lymphocytes, NK lymphocytes, granulocytes and monocytes were negatively depleted from peripheral blood leucocytes (PBL) with immunomagnetic microbeads (MACS microbeads, Miltenyi Biotech, Auburn, CA) and purity (> 93%) of resulting CD19+ B-CLL population was assessed by flow cytometry as previously described [24]. Viability of the cells was analyzed at the end of the purification procedure by Trypan blue dye exclusion as previously described [43].
Targeted deep sequencing and sequencing data analysis
Next generation sequencing of 9 genes related to the p53 pathway, ATM, BAX, CDKN1A, MDM2, NOTCH1, NOTCH2, TNFRSF10A, TNFRSF10B, and TP53 itself (Supplemental Table 1) was performed using Ion Torrent chemistry (Life Technologies, Foster City, CA). For each gene, the coding regions (CCDS), untranslated regions (UTR) 5’ and 3’, and also 50bp exons/introns boundaries were targeted by an Ion AmpliSeq custom panel. Two DNA libraries per sample were prepared and as multi-samples pools were subjected to targeted deep sequencing by an Ion Torrent Personal Genome Machine (IT-PGM) platform. A minimum average coverage per sample was fixed at 500-fold.
Sequencing data were aligned to the hg19 human reference genome and variant calling was performed in accord to the Ion Torrent Suite v4 analysis pipeline (Life Technologies). Single Nucleotides Variations (SNVs) and small insertions and deletions (INDELs) were annotated using ANNOVAR software [44], supplied by the COSMIC v68 database (Catalogue Of Somatic Mutations In Cancer, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) and several web-tools for the prediction of the pathogenicity of the variants. Alignment of all selected variants was visually verified with the Integrative Genome Viewer v2.2 [45]. Further details are available in the Supplementary Material.
Variant prioritization and validation
Nucleotide change were prioritized and assigned in two groups: (1) frameshift deletions and insertions, stop codon gain, stop codon loss and splicing site variants; (2) non-synonymous mutations reported in the COSMIC database or predicted pathogenetic by the web-tools (Supplemental Table 2). All selected variants were confirmed either on genomic DNA or, in case of splicing site mutations, on cDNA by Sanger sequencing. Further details and primer sequencers are available in the Supplementary Material.
Structural bioinformatics analysis
The potential effect of the validated mutations on the protein products was analyzed in terms of sequence comparisons and structural analysis. Protein sequences and structures were retrieved from different databases: NCBI (http://www.ncbi.nlm.nih.gov/protein/), PDB (http://pdb.org), UniProt (http://www.uniprot.org), and PFAM (http://pfam.sanger.ac.uk). Protein structures were visualized and graphics represented with VMD (http://www.ks.uiuc.edu/Research/vmd/) and Prosite (http://prosite.expasy.org/cgi-bin/prosite/mydomains/).
B-CLL cultures
In order to evaluate the transcriptional functionality of p53, CD19+ B-CLL patient cells were seeded at a density of 1x106 cells/ml in RPMI-1640 medium containing 10% FBS, L-glutamine and Penicillin/streptomycin (all from Gibco, Grand Island, NY). Cultures were then either left untreated or exposed to Nutlin-3 (used at 10 microM; Cayman Chemical, Ann Arbor, MI) for 24 hours, before total RNA extraction (QIAGEN RNeasy Plus mini kit; QIAGEN) according to the supplier’s instructions. For the integrity measurement, total RNA were analyzed on an Agilent Cary 60 UV-Vis Spectrophotometer (Agilent Technologies Inc., Santa Clara, CA, USA). The expression of relevant p53 target genes (MDM2, CDKN1A/p21, BAX) was quantitatively assessed by reverse transcription-polymerase chain reaction (RT-PCR). For this purpose total RNA was transcribed into cDNA, using the QuantiTect® Reverse Transcription kit (QIAGEN). MDM2, CDKN1A/p21, BAX gene expression was analyzed using the SYBR Green-based real-time quantitative polymerase chain reaction (RT qPCR) detection method with SABiosciences RT2 Real-TimeTM Gene expression assays, which include specific validated primer sets and PCR master mix (SABiosciences). All samples were run in triplicate using the real time thermal analyzer Rotor-GeneTM 6000 (Corbett, Cambridge, UK), as previously described [23,24]. Expression values were normalized to the housekeeping gene POLR2A amplified in the same sample.
Acknowledgements
This study was supported by grants from MIUR-FIRB (RBAP11Z4Z9_002 to GZ; RBAP10447J_002 to PS), and from the Italian Association for Cancer Research (AIRC IG 11465 to GZ). CA has been supported by a “Consorzio Spinner PhD Program”.
Authorship contributions
EA and EM performed the research, collected, analyzed and interpreted the data, and wrote the manuscript; GMR and AC provided experimental material and contributed data; CA, RV, EP and LS performed the molecular and cell biology experiments; DV and SDM contributed to the NGS data analyses; PS and GZ, designed and coordinated the research, interpreted data, wrote and revised the manuscript; all authors assisted in the analysis and/or interpretation of the data, critically revised the manuscript and gave final approval of the manuscript.
Conflict of interest disclosure
All authors declare no conflict of interest.
References
1. Zenz T, Kröber A, Scherer K, Häbe S, Bühler A, Benner A, Denzel T, Winkler D, Edelmann J, Schwänen C, Döhner H, Stilgenbauer S. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008; 112: 3322-3329.
2. Dicker F, Herholz H, Schnittger S, Nakao A, Patten N, Wu L, Kern W, Haferlach T, Haferlach C. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia. 2009; 23: 117-124.
3. Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S, De Paoli L, Spina V, Gattei V, Capello D, Forconi F, Lauria F, Gaidano G. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res. 2009; 15: 995-1004.
4. Malcikova J, Smardova J, Rocnova L, Tichy B, Kuglik P, Vranova V, Cejkova S, Svitakova M, Skuhrova Francova H, Brychtova Y, Doubek M, Brejcha M, Klabusay M et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood. 2009; 114: 5307-5314.
5. Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, Bühler A, Edelmann J, Bergmann M, Hopfinger G, Hensel M, Hallek M, Döhner H et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010; 28: 4473-4479.
6. Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, Dearden CE, Richards SM, Catovsky D, Morgan GJ. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011 Jun 1; 29: 2223-2229.
7. Dufour A, Palermo G, Zellmeier E, Mellert G, Duchateau-Nguyen G, Schneider S, Benthaus T, Kakadia PM, Spiekermann K, Hiddemann W, Braess J, Truong S, Patten N et al. Inactivation of TP53 correlates with disease progression and low miR-34a expression in previously treated chronic lymphocytic leukemia patients. Blood. 2013; 121: 3650-3657.
8. Skowronska A, Parker A, Ahmed G, Oldreive C, Davis Z, Richards S, Dyer M, Matutes E, Gonzalez D, Taylor AM, Moss P, Thomas P, Oscier D et al. Biallelic ATM inactivation significantly reduces survival in patients treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 trial. J Clin Oncol. 2012; 30: 4524-4532.
9. Rose-Zerilli MJ, Forster J, Parker H, Parker A, Rodríguez AE, Chaplin T, Gardiner A, Steele AJ, Collins A, Young BD, Skowronska A, Catovsky D, Stankovic T et al. ATM mutation rather than BIRC3 deletion and/or mutation predicts reduced survival in 11q-deleted chronic lymphocytic leukemia: data from the UK LRF CLL4 trial. Haematologica. 2014; 99: 736-742.
10. Ouillette P, Li J, Shaknovich R, Li Y, Melnick A, Shedden K, Malek SN. Incidence and clinical implications of ATM aberrations in chronic lymphocytic leukemia. Genes Chromosomes Cancer. 2012; 51: 1125-1132.
11. Navrkalova V, Sebejova L, Zemanova J, Kminkova J, Kubesova B, Malcikova J, Mraz M, Smardova J, Pavlova S, Doubek M, Brychtova Y, Potesil D, Nemethova V et al. ATM mutations uniformly lead to ATM dysfunction in chronic lymphocytic leukemia: application of functional test using doxorubicin. Haematologica. 2013; 98: 1124-1131.
12. Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz M, Bassaganyas L, Baumann T, Juan M et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011; 475: 101-105.
13. Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, Grunn A, Fangazio M, Capello D, Monti S, Cresta S, Gargiulo E, Forconi F et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011; 208: 1389-1401.
14. Del Giudice I, Rossi D, Chiaretti S, Marinelli M, Tavolaro S, Gabrielli S, Laurenti L, Marasca R, Rasi S, Fangazio M, Guarini A, Gaidano G, Foà R. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012; 97: 437-441.
15. Villamor N, Conde L, Martínez-Trillos A, Cazorla M, Navarro A, Beà S, López C, Colomer D, Pinyol M, Aymerich M, Rozman M, Abrisqueta P, Baumann T et al. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia. 2013; 27: 1100-1106.
16. Lionetti M, Fabris S, Cutrona G, Agnelli L, Ciardullo C, Matis S, Ciceri G, Colombo M, Maura F, Mosca L, Gentile M, Recchia AG, Ilariucci F et al. High-throughput sequencing for the identification of NOTCH1 mutations in early stage chronic lymphocytic leukaemia: biological and clinical implications. Br J Haematol. 2014; 165: 629-639.
17. Arruga F, Gizdic B, Serra S, Vaisitti T, Ciardullo C, Coscia M, Laurenti L, D’Arena G, Jaksic O, Inghirami G, Rossi D, Gaidano G, Deaglio S. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia. 2014; 28: 1060-1070.
18. López-Guerra M, Xargay-Torrent S, Rosich L, Montraveta A, Roldán J, Matas-Céspedes A, Villamor N, Aymerich M, López-Otín C, Pérez-Galán P, Roué G, Campo E, Colomer D. The γ-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia. 2014. doi: 10.1038/leu.2014.143. [Epub ahead of print].
19. Secchiero P, Melloni E, di Iasio MG, Tiribelli M, Rimondi E, Corallini F, Gattei V, Zauli G. Nutlin-3 up-regulates the expression of Notch1 in both myeloid and lymphoid leukemic cells, as part of a negative feedback antiapoptotic mechanism. Blood. 2009; 113: 4300-4308.
20. Licciulli S, Avila JL, Hanlon L, Troutman S, Cesaroni M, Kota S, Keith B, Simon MC, Puré E, Radtke F, Capobianco AJ, Kissil JL. Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer Res. 2013; 73: 5974-5984.
21. Secchiero P, di Iasio MG, Gonelli A, Zauli G. The MDM2 inhibitors Nutlins as an innovative therapeutic tool for the treatment of hematological malignancies. Current Pharmaceutical Design. 2008; 14: 2100-2110.
22. Zauli G, Voltan R, Bosco R, Melloni E, Marmiroli S, Rigolin GM, Cuneo A, Secchiero P. Dasatinib plus Nutlin-3 shows synergistic anti-leukemic activity in both p53wild-type and p53mutated B chronic lymphocytic leukemias by inhibiting the Akt pathway. Clinical Cancer Research. 2011; 17: 762-770.
23. Zauli G, Voltan R, di Iasio MG, Bosco R, Melloni E, Sana ME, Secchiero P. miR-34a induces the down-regulation of both E2F1 and B-Myb oncogenes in leukemic cells. Clinical Cancer Research. 2011; 17: 2712-2724.
24. Zauli G, Celeghini C, Melloni E, Voltan R, Ongari M, Tiribelli M, di Iasio MG, Lanza F, Secchiero P. The Sorafenib plus Nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of the FLT3 and p53 status. Haematologica-The Hematology Journal. 2012; 97: 1722-1730.
25. Secchiero P, Bosco R, Celeghini C, Zauli G. Recent advances in the therapeutic perspectives in nutlin-3. Current Pharmaceutical Design. 2011; 17: 569-577.
26. Lin K, Adamson J, Johson GG, Carter A, Oates M, Wade R, Richards S, Gonzalez D, Matutes E, Dearden C, Oscier DG, Catovsky D, Pettitt AR. Functional analysis of the ATM-p53-p21 pathway in the LRF CLL4 trial: blockade at the level of p21 is associated with short response duration. Clin Cancer Res. 2012; 18: 4191-4200.
27. Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Famà R, Rasi S, Monti S, Deambrogi C, De Paoli L, Wang J, Gattei V, Guarini A et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014; 123: 2139-2147.
28. Monti P, Ciribilli Y, Jordan J, Menichini P, Umbach DM, Resnick MA, Luzzatto L, Inga A, Fronza G. Transcriptional functionality of germ line p53 mutants influences cancer phenotype. Clin Cancer Res. 2007; 13: 3789-3795.
29. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, Wan Y, Zhang W, Shukla SA et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013; 152: 714-726.
30. Secchiero P, Zauli G. TNF-related apoptosis-inducing ligand and the regulation of hematopoiesis. Current Opinion in Hematology. 2008; 15: 42-48.
31. Zauli G, Melloni E, Capitani S, Secchiero P. Role of full-length osteoprotegerin in tumor cell biology. Cellular and molecular life sciences. 2009; 66: 841-851.
32. Tam CS, Shanafelt TD, Wierda WG, Abruzzo LV, Van Dyke DL, O’Brien S, Ferrajoli A, Lerner SA, Lynn A, Kay NE, Keating MJ. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009; 114: 957-964.
33. Malcikova J1, Tichy B, Damborsky J, Kabathova J, Trbusek M, Mayer J, Pospisilova S. Analysis of the DNA-binding activity of p53 mutants using functional protein microarrays and its relationship to transcriptional activation. J Biol Chem. 2010; 391: 197-205.
34. Burke RT, Meadows S, Loriaux MM, Currie KS, Mitchell SA, Maciejewski P, Clarke AS, Dipaolo JA, Druker BJ, Lannutti BJ, Spurgeon SE. A potential therapeutic strategy for chronic lymphocytic leukemia by combining Idelalisib and GS-9973, a novel spleen tyrosine kinase (Syk) inhibitor. Oncotarget. 2014; 5: 908-915.
35. Billard C. Apoptosis inducers in chronic lymphocytic leukemia. Oncotarget. 2014 30; 5: 309-325.
36. Walsby E, Pratt G, Shao H, Abbas AY, Fischer PM, Bradshaw TD, Brennan P, Fegan C, Wang S, Pepper C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. Oncotarget. 2014; 5: 375-385.
37. Martins LR, Perera Y, Lúcio P, Silva MG, Perea SE, Barata JT. Targeting chronic lymphocytic leukemia using CIGB-300, a clinical-stage CK2-specific cell-permeable peptide inhibitor. Oncotarget. 2014; 5: 258-263.
38. MacFarlane M, Kohlhaas SL, Sutcliffe MJ, Dyer MJ, Cohen GM. TRAIL receptor-selective mutants signal to apoptosis via TRAIL-R1 in primary lymphoid malignancies. Cancer Res. 2005; 65: 11265-11270.
39. Rubio-Moscardo F, Blesa D, Mestre C, Siebert R, Balasas T, Benito A, Rosenwald A, Climent J, Martinez JI, Schilhabel M, Karran EL, Gesk S, Esteller M et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood. 2005; 106: 3214-3222.
40. Secchiero P, Zerbinati C, di Iasio MG, Melloni E, Tiribelli M, Grill V, Zauli G. Synergistic cytotoxic activity of recombinant TRAIL plus the non-genotoxic activator of the p53 pathway nutlin-3 in acute myeloid leukemia cells. Current Drug Metabolism. 2007; 8: 395-403.
41. Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011; 2:222-233.
42. van Leeuwen IM, Rao B, Sachweh MC, Laín S. An evaluation of small-molecule p53 activators as chemoprotectants ameliorating adverse effects of anticancer drugs in normal cells. Cell Cycle. 2012; 11:1851-1856.
43. Re MC, Zauli G, Gibellini D, Furlini G, Ramazzotti E, Monari P, Ranieri S, Capitani S, La Placa M. Uninfected haematopoietic progenitor (CD34+) cells purified from the bone marrow of AIDS patients are committed to apoptotic cell death in culture. AIDS. 1993; 7: 1049-1055.
44. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38: e164.
45. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29: 24-26.