
INTRODUCTION
Many of the most recent advances in oncology have been related to the field of precision medicine. For example, the introduction of targeted therapies in lung, colon, and breast cancer have contributed to a significant increase in overall survival (OS) related to these diseases [1–3]. Patients with pancreatic ductal adenocarcinoma (PDA) have not seen the same benefit of these precision oncology advances [4, 5]. The 5-year survival rate of patients with locally advanced or metastatic PDA has only seen modest increases over the last several decades, from 5% to more recent estimates of 8–9% [4–6].
Identification and utilization of reliable and informative biomarkers will play a pivotal role in precision oncology, not only for triaging patients to appropriate molecularly guided therapies, but also to inform therapeutic decision making [7]. In PDA, carbohydrate antigen 19-9 (CA19-9) has long been the biomarker of choice, has a sensitivity and specificity of 79–81% and 82–90% respectively, and is recommended for use by the National Comprehensive Cancer Network (NCCN) (v.02.2016) [8–11]. In patients with resectable disease, which accounts for less than 20% of those diagnosed [12], pre-operative serum CA19-9 levels, and serial serum CA19-9 measurements before and after surgery can be prognostic, and post-operative serum CA19-9 levels can help determine adjuvant therapy course [13]. In patients with advanced disease, the ability of serum CA19-9 to help determine treatment benefit during chemotherapy has generated conflicting results [14–20].
Currently, there are no clinically useful biomarkers established for treatment selection in patients with unresectable PDA. Daily clinical decision-making is based on imaging techniques and CA19-9. However, these tools are not always accurate in patient identification or outcome prediction. Serum CA19-9 has its limitations, primarily because 10–15% of patients do not express Lewis Body antigen, meaning that their CA19-9 levels are not informative [21]. In these patients, there is no validated biomarker for determining prognosis at time of diagnosis or for use in monitoring of therapeutic response. Additionally, CA19-9 can be elevated in non-neoplastic, inflammatory conditions, like pancreatitis, as well as due to biologic conditions like stent blockage [21, 22].
Detection of circulating tumor DNA (ctDNA) has rapidly emerged as an alternative to tissue biopsy in identifying the presence or absence of clinically actionable genomic alterations within a tumor [23–25]. Longitudinal monitoring of ctDNA has been shown to serve as a biomarker for predicting patient outcome by quantitating dynamic changes in ctDNA mutation load in response to a therapeutic intervention. Multiple studies have reported correlation between dynamic changes in the levels of a specific ctDNA mutation with radiographic response [26–28]. However, these studies have examined individual patients (i.e., anecdotal data) and not the development and testing of a clinical tool within a patient population that could be used to monitor and predict response. PDA presents a unique opportunity for molecular diagnosis and monitoring given that over 90–95% of patients with PDA harbor somatic KRAS G12/G13 mutations as the primary driver of mutagenesis [29–32]. Given this fact, measurement of ctDNA for detection and quantitative monitoring of KRAS mutations may offer a complementary diagnostic and prognostic biomarker to CA19-9. Researchers have attempted to harness advancing ctDNA technologies to understand the clinical utility of ctDNA KRAS in patients with PDA as a diagnostic and prognostic biomarker, biomarker for drug response, and molecular monitoring tool. Previous studies have reported low ctDNA KRAS detection rates ranging from 27% to 71%, hampering the advancement of ctDNA KRAS as a reliable clinical tool [23, 27, 33–38].
Here we report on the application of an ultrasensitive ctDNA KRAS assay used in patients with unresectable PDA, and its use as a prognostic biomarker independent and complementary to CA19-9. We also introduce novel work utilizing dynamic changes in ctDNA KRAS mutation load from serial measurements as an assessment of therapeutic response.
RESULTS
In this prospective, retrospective analysis of CA19-9 and ctDNA KRAS in patients with newly diagnosed American Joint Committee on Cancer (AJCC) stage III or stage IV PDA, 189 patients met inclusion criteria and had baseline samples that passed preliminary quality control analysis (Table 1). The majority of patients (78.8%) were diagnosed with Stage IV disease. In this cohort, all patients were treated with chemotherapy with the primary first line chemotherapy being gemcitabine (66.7%) which is consistent with primary therapy for this patient population during the recruitment period. Median number of ctDNA KRAS timepoints per patient was 3 (interquartile range (IQR) 2–4) and median number of CA19-9 timepoints per patient was 8 (IQR 4–16).
Table 1: Demographics for 189 patients
Characteristic |
Median (Q1–Q3) |
|---|---|
Age, years |
67 (60–72) |
Number (%) |
|
Gender |
|
Male |
95 (50.3) |
Female |
94 (49.7) |
ECOG Performance Status |
|
0 |
70 (37.4) |
1 |
99 (52.4) |
2 |
20 (10.6) |
Stage |
|
III |
40 (21.2) |
IV |
149 (78.8) |
Chemotherapy type |
|
Gemcitabine |
126 (66.7) |
FOLFIRINOX |
63 (33.3) |
ctDNA KRAS mutation detected at baseline1 |
|
G12A |
4 (2.3) |
G12C |
18 (10.2) |
G12D |
72 (40.7) |
G12R |
22 (12.4) |
G12S |
4 (2.3) |
G12V |
55 (31.1) |
G13D |
2 (1.1) |
1Available for 177 patients.
Q - quartiles.
Baseline statistics and primary variates
Plasma ctDNA KRAS mutation detection rate in this 189 patient cohort was 93.7% (N = 177; 95% confidence interval (CI) 89.2%–96.7%) (Table 2). The most frequently detected KRAS mutations were G12D (40.7%) and G12V (31.1%) (Table 1), which is consistent with the most common mutations identified through somatic testing of pancreatic adenocarcinoma tumors [39]. The median number of ctDNA KRAS copies detected at baseline was 335.4 copies per 105 Genome Equivalents (GEq) (IQR of 54–2,214 copies per 105 GEq). For CA19-9, 167 patients had baseline levels above 37 units per milliliter (U/mL) and were considered elevated (88.4%; 95% CI 82.9%–92.6%) (Table 2). The median U/mL was 1,890 (IQR 412 – 12, 160 U/mL). ctDNA KRAS was detectable at baseline for 19 of 22 patients that had non-elevated CA19-9 levels (86.4%; 95% CI 65.1%–97.1%) (Table 2). The addition of baseline ctDNA KRAS to this cohort significantly increased patient identification (p = 0.0020). For baseline positive samples, there was a significant, positive relationship between CA19-9 and ctDNA KRAS (Figure 1) (Pearson’s Correlation Coefficient r = 0.295; p = 0.0002; Spearman’s rank correlation r = 0.312; p < 0.0001).
Table 2: Detection concordance between ctDNA KRAS and serum CA19-9
CA19-9 |
||||
|---|---|---|---|---|
Detected |
Not Detected |
Total |
||
ctDNA KRAS |
Detected |
158 |
19 |
177 |
Not Detected |
9 |
3 |
12 |
|
Total |
167 |
22 |
189 |
|
ctDNA – circulating tumor DNA.
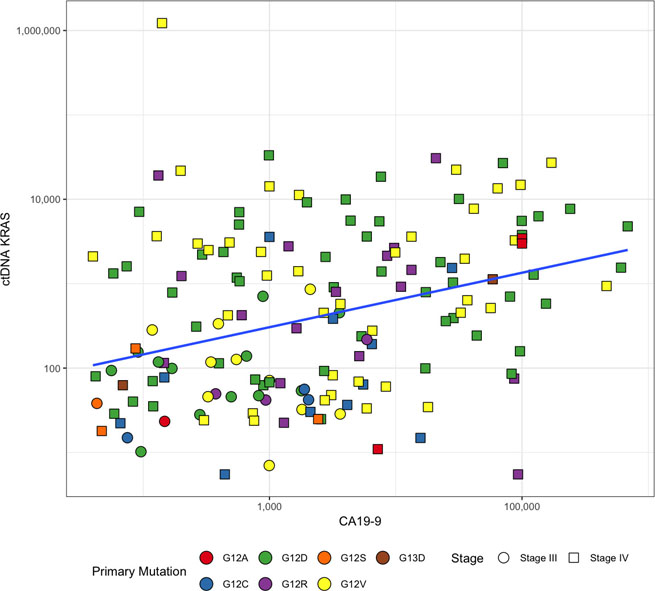
Figure 1: Significant positive relationship between baseline CA19-9 and ctDNA KRAS for positive samples (N = 158). Pearson’s correlation coefficient r = 0.295; p = 0.0002. Spearman’s Rank Correlation r = 0.312; p < 0.0001.
The median number of baseline plasma ctDNA KRAS copies was higher in patients with stage IV versus stage III PDA (608.2 versus 56.0 copies per 105 GEq; p < 0.0001) (Figure 2A). This was also observed in baseline serum CA19-9 (3,230 versus 540 U/mL; p < 0.0001) (Figure 2B). When the quantitative baseline values of ctDNA KRAS and CA19-9 were plotted versus disease stage, there was a notable bi-modal distribution for ctDNA KRAS that was not observed for CA19-9 (Figure 3). The resulting natural cut-point was approximately 250 ctDNA KRAS copies per 105 GEq (Figure 3A). This natural cut-point was utilized in subsequent calculations to categorize the number of ctDNA KRAS copies into “High” and “Low” bins. For serum CA19-9, the median (1,890 U/mL) was utilized in subsequent calculations for a similar categorization (Figure 3B).
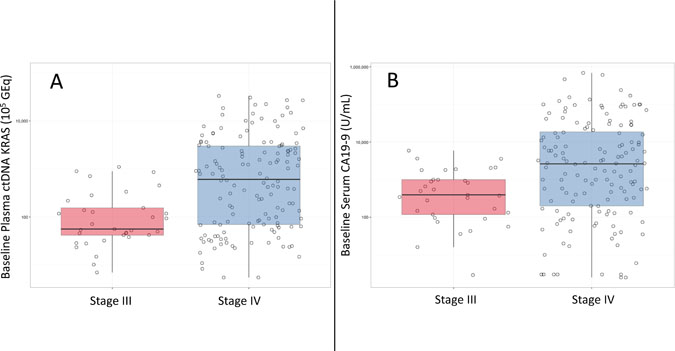
Figure 2: Boxplots of ctDNA KRAS (A) and CA19-9 (B) for baseline detected samples.
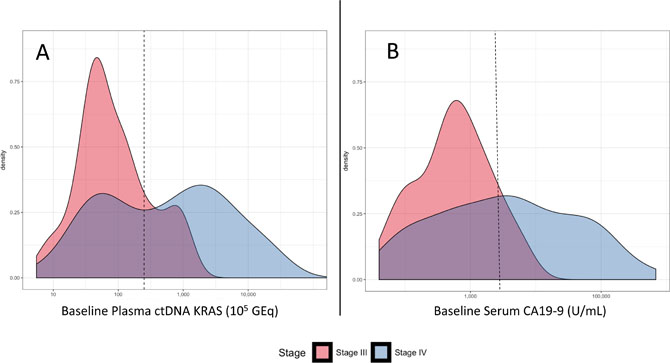
Figure 3: (A) Density plot of ctDNA KRAS copies per 105 GEq in 177 patients with detectable baseline results stratified by stage. Dotted vertical line denotes the bimodal cutpoint of approximately 250 copies per 105 GEq. (B) Density plot of CA19-9 U/mL for 167 patients with detected baseline samples stratified by stage. Dotted vertical line denotes the median cutpoint of approximately 1,890 units per milliliter.
Association with time to disease progression
One-hundred and thirteen patients achieved disease progression (PD) during first line of palliative chemotherapy (59.8%; 95% CI 52.4%–66.8%). Primary reasons for not reaching PD were death and loss to follow-up (missing CT evaluation). Median time to progression (TTP) during first line of chemotherapy was 85 days and did not differ significantly based on disease stage (stage IV: 84 days versus stage III: 151 days; p = 0.1094) (Figure 4A). However, there was a difference in TTP for stage IV versus III disease that trended towards significance (Hazard Ratio (HR) = 1.26, 95% CI 1.00–1.59, p = 0.0504). Irrespective of disease stage, baseline ctDNA KRAS above the natural bimodal cutpoint was associated with PD (HR 1.29; 95% CI 1.06–1.57, p = 0.0105), as was CA19-9 above its respective median (HR 1.25; 95% CI 1.02–1.53, p = 0.0354).
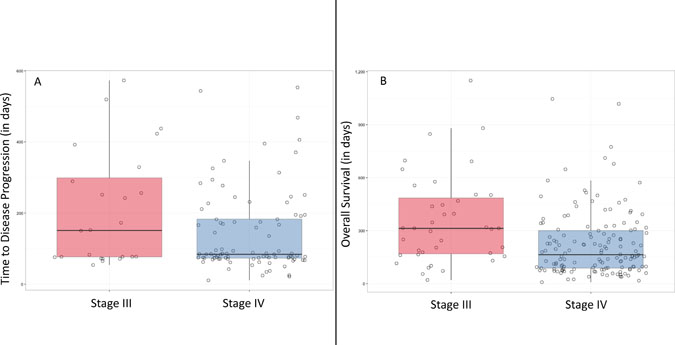
Figure 4: Boxplots of time to disease progression in days (A) and overall survival in days (B) for baseline samples stratified by stage.
In a univariate analysis, baseline ctDNA KRAS was associated with TTP (HR = 1.42; 95% CI 1.12–1.76, p = 0.0013). In a multivariate regression analysis, utilizing baseline CA19-9, and baseline ctDNA KRAS as continuous variables, along with the co-variates of interest, stage, gender (sex), and baseline European Cooperative Oncology Group (ECOG) performance score, ctDNA KRAS remained highly significant for TTP (HR = 1.45; 95% CI 1.15–1.83, p = 0.0017), while baseline CA19-9 was less significant (HR = 1.27; 95% CI 1.03–1.58, p = 0.0284) along with sex (HR = 0.79; 95% CI 0.63–0.99, p = 0.0400) (Table 3).
Table 3: Multivariate regression analysis: prognostic performance of continuous variable ctDNA KRAS and CA19-9
Variate |
HR (95% CI) |
Coefficient |
|
|---|---|---|---|
Overall Survival* |
|||
CA19-9 |
1.30 (1.11–1.53) |
0.0014 |
|
ctDNA KRAS |
1.45 (1.17–1.80) |
0.0007 |
|
Sex |
0.79 (0.67–0.93) |
0.0060 |
|
Age |
1.03 (1.01–1.05) |
0.0068 |
|
Performance Score 1 and 2 |
0.80 (0.67–0.96) |
0.0148 |
|
Time to Progression* |
|||
CA19-9 |
1.27 (1.03–1.58) |
0.0284 |
|
ctDNA KRAS |
1.45 (1.15–1.83) |
0.0017 |
|
Sex |
0.79 (0.63–0.99) |
0.0400 |
*Overall model fit was assessed by Likelihood and Wald scores. Corresponding p-values of the model were < 0.0001 indicating a good fit.
When analyzing the categorical cutpoints and TTP, there was not a significant difference in the median TTP in patients with stage IV disease based on their baseline CA19-9 level (p = 0.2550; Figure 5A). In contrast, those with a diagnosis of stage IV disease had a borderline significant association based on their ctDNA KRAS level (p = 0.0554; Figure 5B). The number of patients with stage III disease and high baseline CA19-9 (N = 4) was too few for calculations, as was the number of patients with stage III disease and high baseline ctDNA KRAS (N = 3).
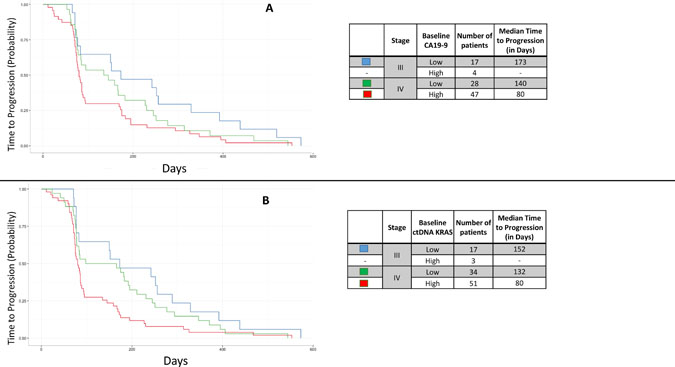
Figure 5: Kaplan Meier curves demonstrating the patient time to progression (TTP) by stage and categorical baseline CA19-9 (A) and categorical baseline ctDNA KRAS status (B). High/low CA19-9 status was based on the median in this cohort (1,890 U/mL) while high/low ctDNA KRAS status was based on the bimodal cutpoint of 250 copies per 105 Genome Equivalents. Patients with high baseline values of ctDNA KRAS had significantly shorter TTP than those with low baseline values (p = 0.0358) which was not appreciated in high/low CA19-9 status (p = 0.1237). The number of patients with Stage III disease and high baseline CA19-9 was too few for calculation, as was the number of patients with Stage III disease and high baseline ctDNA KRAS.
Association with overall survival
OS was calculated for 183 patients for a median OS of 188 days (IQR of 96–326 days). Patients with stage IV disease had a significantly shorter OS than those with stage III disease (stage IV: 164 days versus stage III: 313 days; p = 0.0005) (Figure 4B). In addition to this, a baseline ctDNA KRAS above the natural bimodal cut-point (Hazard ratio (HR) = 1.36; 95% CI 1.16–1.59, p = 0.0001), a baseline CA19-9 value above the median (1,890 U/mL) (HR = 1.34; 95% CI 1.15–1.57, p = 0.0003), and disease stage (stage III versus stage IV) (HR = 1.34; 95% CI 1.12–1.61, p = 0.0014) were each significantly associated with OS.
Univariate analysis demonstrated that baseline plasma ctDNA KRAS as a continuous variable (HR = 1.42; 95% CI 1.19–1.69, p < 0.0001) and baseline serum CA19-9 as a continuous variable (HR = 1.36; 95% CI 1.17–1.59, p < 0.0001) were each directly associated with OS significantly. In a multivariate regression analysis, categorical ctDNA KRAS and CA19-9 along with the other co-variates (stage, gender (sex), baseline ECOG performance status) were utilized to determine their respective associations with OS. Both ctDNA KRAS and CA19-9 when analyzed with the co-variates of interest were associated with OS (ctDNA KRAS: HR = 1.45; 95% CI 1.17–1.80, p = 0.0007; CA19-9: HR = 1.31; 95% CI 1.11–1.53, p = 0.0014). Other significant variates included gender and baseline ECOG performance status (Table 3).
Patients with stage III disease and low baseline CA19-9 (N = 28) had a longer OS than those with stage IV disease, regardless of which categorical CA19-9 status (p < 0.0001 for CA19-9 high, p = 0.0333 for CA19-9 low; Figure 6A). Patients with stage IV disease and a high baseline CA19-9 level (N = 74) had a significantly shorter OS than those with stage IV disease and a low baseline CA19-9 (N = 52; p = 0.0009) (Figure 6A). The number of patients with stage III disease and high baseline CA19-9 was too few for calculations (N = 7). Similarly, for baseline ctDNA KRAS, patients with stage III disease and low baseline ctDNA KRAS levels (N = 26) had a longer OS than patients with stage IV disease, regardless of baseline ctDNA KRAS level (p = 0.0170 and < 0.0001; Figure 6B). Patients with stage IV disease and a high baseline ctDNA KRAS (N = 85) had a significantly shorter OS than those with stage IV disease and low baseline ctDNA KRAS levels (N = 55; p = 0.0425) (Figure 6B). The number of patients with stage III disease and high baseline ctDNA KRAS was too few for calculation (N = 6), as was the number of patients with Stage III disease and high baseline CA19-9 (N = 7).
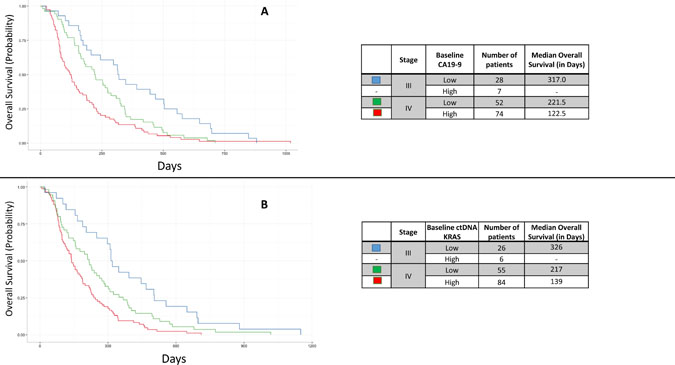
Figure 6: Kaplan Meier curves demonstrating the overall survival (OS) by stage and categorical baseline CA19-9 (A) and categorical baseline ctDNA KRAS status (B). High/low CA19-9 status was based on the median in this cohort (1,890 U/mL) while high/low ctDNA KRAS status was based on the bimodal cutpoint of 250 copies per 105 Genome Equivalents. Patients with high baseline values had significantly shorter OS than those with low baseline values. The number of patients with Stage III disease and high baseline CA19-9 was too few for calculation, as was the number of patients with Stage III disease and high baseline ctDNA KRAS.
Association with performance status
Association of OS with the patient’s pre-treatment ECOG performance status was examined with baseline high and low ctDNA KRAS and CA19-9 levels. In patients with a performance status of 0, there was a statistically significant difference in OS between those patients with high versus low baseline CA19-9 levels (p = 0.0024). This also held true in patients with a performance status of 1 (p = 0.0121) (Figure 7A). The differences in OS were also significant for those with a performance status of 0 and high versus low baseline ctDNA KRAS (p = 0.0053) as well as a performance status of 1 (p = 0.0102) (Figure 7B).
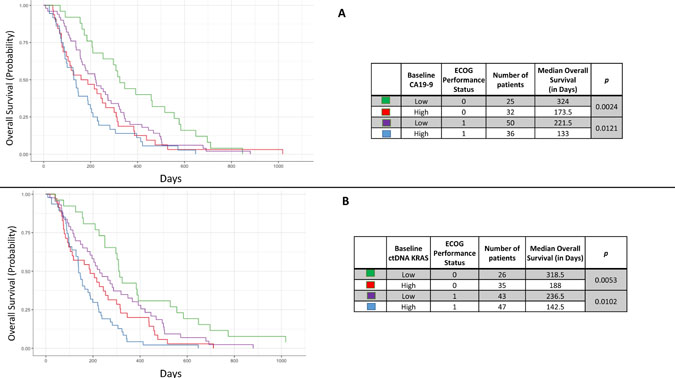
Figure 7: Kaplan Meier curves demonstrating the overall survival based on categorical CA19-9 (high/low) and pretreatment ECOG performance score (A) and similarly, the overall survival based categorical ctDNA KRAS (high/low) and pretreatment ECOG performance score (B).
Plasma ctDNA KRAS as a response biomarker
In a subset of patients, details about investigator assessed progression were available in addition to CT scan response by RECIST1.1, and ctDNA KRAS and CA19-9 data (N = 88 of 113 patients who reached PD on first line therapy). Most patients had an investigator assessed response that was confirmed by RECIST 1.1 criteria (N = 62 of 88; 70%). To determine whether dynamic changes in longitudinal ctDNA KRAS levels were consistent with RECIST 1.1 assessment, patients in which there was a plasma ctDNA KRAS within 2 weeks of a RECIST 1.1 assessment were examined. 51.6% of these patients (N = 32) had a ctDNA KRAS collection within 2 weeks of a RECIST 1.1 assessment, and dynamic longitudinal changes in mutation load trended consistent with RECIST 1.1 assessment in 23 patients (71.9%). In the remaining 26 patients where the investigator assessment of response differed from RECIST 1.1, 20 patients had a ctDNA KRAS longitudinal timepoint within 2 weeks of a RECIST 1.1 assessment. We observed that longitudinal monitoring of changes in ctDNA KRAS mutational burden trended consistent with RECIST 1.1 response in nearly half of these patients (N = 9 of 20; Figure 8).
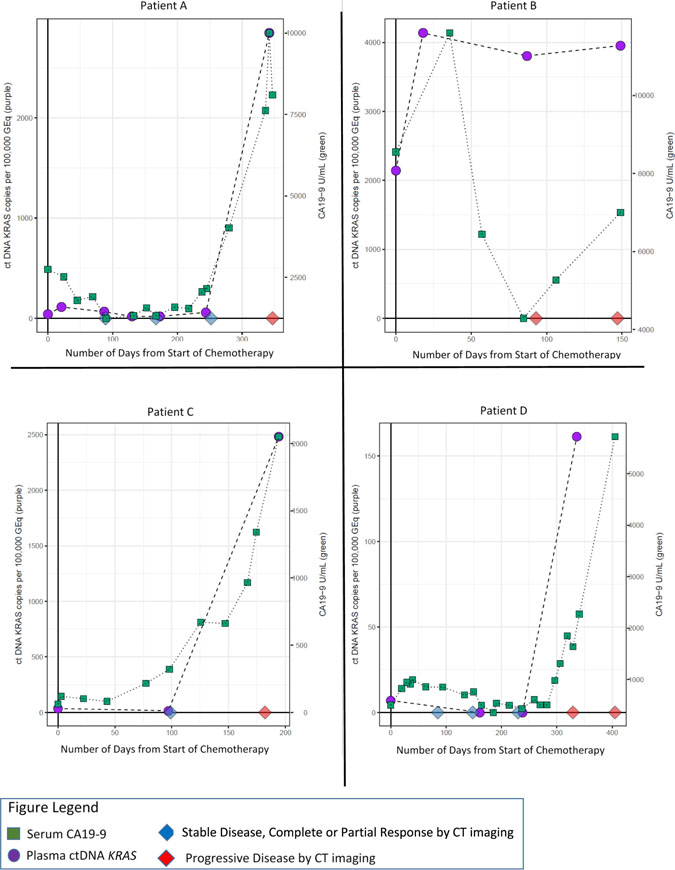
Figure 8: Representative longitudinal plots of patients who had a different investigator assessed response as compared to the confirmatory RECIST 1.1 criteria. Green squares represent longitudinal CA19-9 collection timepoints, while purple circles represent longitudinal ctDNA KRAS collection timepoints. The CT scan RECIST 1.1 assessment is represented by diamonds on the X-axis. CT scans are truncated to include only those that were within 2 weeks of a ctDNA KRAS collection.
These observations were further explored and formalized with predictive modeling (Figure 9). Of the dataset, there were 59 timepoints (matched baseline and longitudinal timepoint) from 46 unique patients that met criteria for predictive modeling (one matched timepoint (N = 37), two matched timepoints (N = 6), three matched timepoints (N = 2) and four matched timepoints (N = 1). Four of the 59 matched timepoints were “undetermined” as they had a baseline ctDNA KRAS GEq that was less than θ. For the remaining 55 samples, utilizing the quantitative baseline ctDNA KRAS level and the longitudinal quantitative ctDNA KRAS level that was within 7 days of a verified RECIST 1.1 timepoint, the model correctly predicted the RECIST 1.1 results (PD versus SD+) in 81.8% of matched timepoints (N = 45 of 55; 95% CI 69.7%–90.9%) (Figure 10). Four of the 10 samples with discrepant calls had an associated longitudinal timepoint that was “not detected” for ctDNA KRAS, therefore the model predicted a response of “SD+” while the RECIST 1.1 response was PD. In the remaining six discrepant matched samples the model predicted a response of SD+, but actual response was PD. In one matched sample ctDNA KRAS value was borderline as the longitudinal timepoint was just below the threshold (Figure 11) and in two matched samples the ctDNA KRAS longitudinal trend that was mirrored by CA19-9 (Figure 11).
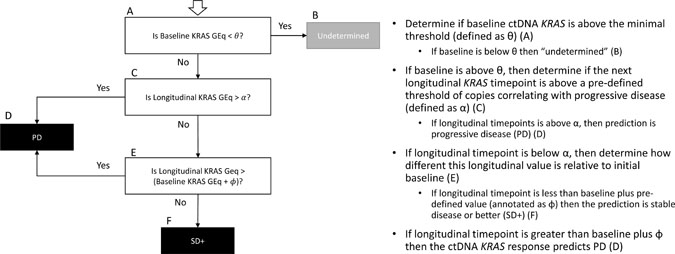
Figure 9: Predictive response model logic.
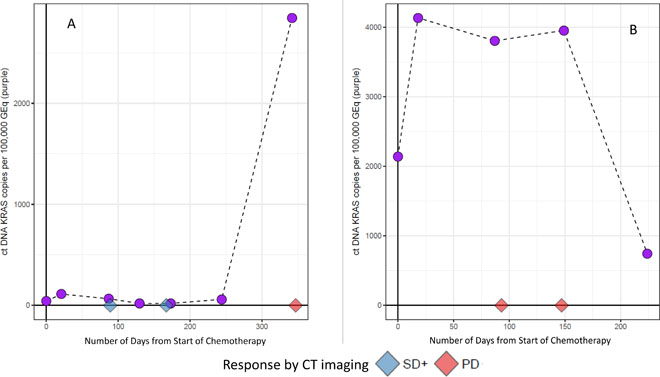
Figure 10: Representative longitudinal plots of patients in whom the ctDNA KRAS predictive model correctly assessed the RECIST 1.1 response. The CT scan RECIST 1.1 assessment is represented by diamonds on the X-axis. CT scans are truncated to include only those that were within 7 days of a ctDNA KRAS collection.
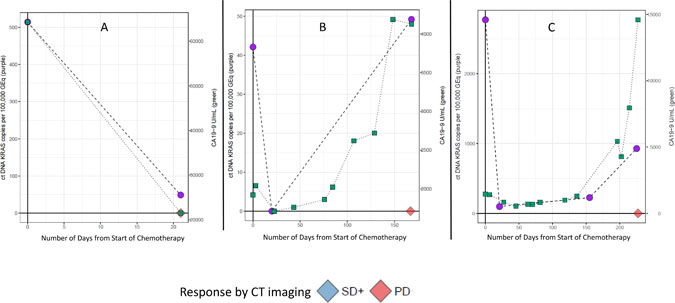
Figure 11: Discrepant predictive modeling plots. Patient A had decreasing CA19-9 and ctDNA KRAS levels as compared to baseline, but a RECIST 1.1 assessment of progressive disease (PD) (A). Patient B had increasing CA19-9 and ctDNA KRAS as compared to baseline, but the difference between the baseline and longitudinal timepoints did not meet threshold for a PD call (B). In Patient C, there was a predicted response of stable disease response, and the longitudinal ctDNA KRAS was just below our threshold, thus this is a borderline case (C).
DISCUSSION
We report on the utilization of a novel mutant allele enrichment ctDNA assay in patients with advanced PDA yielding a circulating tumor DNA (ctDNA) KRAS baseline detection rate of 93.7% (95% CI 89.2%–96.7%), outperforming the detection rate of prior reported assays [23, 27, 33–38]. The percentage of individuals without an informative biomarker was reduced from 11.6% when utilizing CA19-9 alone (22 of 189 patients), to 1.6% when utilizing both serum CA19-9 and plasma ctDNA KRAS (3 of 189 patients). Utilization of CA19-9 and ctDNA KRAS in this cohort increased the yield of positively identified patients by 10.1% and resulted in a positive biomarker result in 186 of 189 patients. ctDNA KRAS and CA19-9 were significantly positively correlated, demonstrating good performance of ctDNA KRAS alongside the currently accepted standard of care biomarker for PDA.
In univariate analysis, both baseline CA19-9 and ctDNA KRAS were associated with overall survival (OS) which was expected given the known positive correlation between the two variates. In multivariate analysis both CA19-9 and ctDNA KRAS were significantly associated with OS, indicating that these two biomarkers capture unique, but complementary prognostic information. The multivariate analysis suggests that assessment of ctDNA KRAS and CA19-9 levels could be used jointly to achieve greater prognostic certainty within the clinical setting.
Baseline ctDNA KRAS was associated with time to progression (TTP). In a multivariate analysis with CA19-9 and the additional co-variates (stage, gender, baseline ECOG performance status), ctDNA KRAS remains highly significant while CA19-9 was less significant.
We observed a natural bimodal distribution of baseline quantitative plasma ctDNA KRAS copy number when analyzed versus PDA disease stage. Upon exploration of this bimodal distribution and natural cutpoint, it was confirmed that there are significant differences in outcome (OS, TTP) between these two patient populations, irrespective of stage of disease. However, when ctDNA KRAS was examined within predefined stage III or stage IV disease, individuals with high versus low baseline ctDNA KRAS copies also had a significantly different OS and TTP. These two observations indicate that assessment of plasma ctDNA KRAS levels for an individual patient may give greater clarity of prognosis than disease stage alone and underscores the potential clinical impact of ctDNA KRAS, a molecular biomarker, to further sub-classify prognosis beyond the current methodologies. Patients with PDA and poorer performance status and low baseline ctDNA KRAS levels had a longer OS than those patients with a better performance status and high baseline ctDNA KRAS levels.
Assessment of response to chemotherapy is challenging in patients with advanced PDA, as evidenced by the cohort of patients where in 30% of cases the investigator assessed response differed from response as confirmed by RECIST 1.1 criteria. In PDA, specifically, given the lethality of the cancer, there is the strong desire for the ability to rapidly ascertain response and inform the decision of whether to continue an effective therapeutic regimen or discontinue an ineffective one [40].
The utilization of serum CA19-9 has been explored as a potential biomarker of response, but with conflicting results [14, 41, 42]. A study of more than 200 patients with metastatic and locally advanced PDA concluded that changes in CA19-9 levels should not be used to make decisions about discontinuing or continuing therapies given the lack of significant positive predictive value. The authors suggest that a more rigorous analysis, limiting the time collection timepoints to within the first two cycles of chemotherapy could be clinically meaningful in treatment decision making [20].
Since the first report in 1948 by Mandel and Metais, describing the presence of cell-free nucleic acid in human blood, ctDNA and its clinical utility have been of clinical interest [43–45]. Assessment of ctDNA, termed as “liquid biopsy”, can provide complementary dynamic information, which can aid in clinical decision making [24]. Somatic KRAS mutations have been reported to be present in greater than 90–95% of PDA cases assessed by tumor tissue analysis suggesting a potential biomarker in detecting and monitoring disease progression [29–32]. Several studies have been published demonstrating the presence of ctDNA in plasma in patients with PDA with varying sensitivity [23, 27, 33–37, 46–51]. Yamada et al. reported that high plasma ctDNA KRAS was positively correlated with stage and that persistent post-treatment detectable ctDNA KRAS mutations was associated with poorer prognosis [46]. Recently, Kinugasa et al. utilized a droplet digital PCR ctDNA technology and identified ctDNA KRAS mutations in 62.6% of patients with PDA (stages II, III and IV). Patients with detectable ctDNA KRAS mutations had significantly shorter survival than patients without detectable mutations [52]. Pietrasz et al. reported that 41.3% of patients with advanced PDA had detectable ctDNA KRAS mutation by NGS and droplet-based dPCR in microfluidics with a high concordance between the two technics [38].
The high prevalence of somatic KRAS mutations in PDA provides an opportunity to have an informative, specific biomarker of response that may aid in assessment of response, identifying patients who are not likely to benefit from additional cytotoxic chemotherapy. In a novel predictive modeling algorithm, examining the quantitative change between ctDNA KRAS mutation load at baseline (pre-treatment) and at subsequent CT scan correctly predicted the RECIST 1.1 assessment in approximately 82% of matched timepoints. This analysis was restricted to timepoints collected while on first line therapy, and those timepoints collected within 7 days of a RECIST 1.1 assessment. The ability of longitudinal monitoring to correctly assess the current disease state would provide clinicians with a rapid, non-invasive assessment of response that could be repeated frequently. Future analysis will expand this modeling in a larger cohort of patients with PDA and with more structured collection timepoints and intervals. We observed that longitudinal monitoring of changes in ctDNA KRAS trended consistent with RECIST 1.1 response in nearly half of the patients. It would also be interesting in future studies to evaluate the correlation between ctDNA KRAS and tumor load or shrinkage, as well as whether the depth of decrease of KRAS ctDNA from baseline could be predictive for survival.
The current study has several strengths. First, the study population was treated and followed within a single hospital system, allowing accurate clinical metric follow-up. Second, the Danish universal health care system allowed for detailed tracking of death dates for the patients. Third, this is one of the largest, if not the largest study of the application of ctDNA in patients with unresectable PDA.
However, the study is not without its limitations. First, matched tumor tissue KRAS analysis was not available for this cohort. It is widely accepted that more than 90–95%, of PDAs harbor somatic KRAS G12/G13 mutations. We relied on this background knowledge, along with the mutation distribution within the cohort that was consistent with what was expected, to estimate that the ultrasensitive ctDNA KRAS assay detected almost all samples correctly, however “tissue truth” was not available. Second, this analysis was retrospectively applied to prospectively collected samples. While the BIOPAC study design outlined the sample collection timepoints, these were not uniformly applied. This impacted the number of plasma ctDNA timepoints available within a RECIST 1.1-assessed CT scans. Future prospectively designed studies will have more rigorous enforcement of study sample collection timepoints and intervals.
In conclusion, plasma ctDNA KRAS was detected in 93.7% of patients with stage III/IV PDA. The addition of plasma ctDNA KRAS to serum CA19-9 at baseline significantly increased patient identification. High baseline ctDNA KRAS mutation levels were significantly associated with OS and TTP in both univariate and multivariate models and were more significantly associated with prognosis than CA19-9. Longitudinal monitoring with ctDNA KRAS as a response biomarker successfully predicted response in over 80% of patients with PDA. The utilization of this ultrasensitive ctDNA KRAS assay not only provides an informative prognostic biomarker, but may also aid in clinical decision making at diagnosis and during treatment.
MATERIALS AND METHODS
Patients
Patients diagnosed and histologically verified with AJCC Stage III or Stage IV unresectable PDA between July 2008 and August 2015 at Herlev and Gentofte Hospital were eligible for the Danish BIOPAC (BIOmarkers in Pancreatic Cancer) Study and were prospectively enrolled [53]. The study is ongoing and approved by the Regional Ethics Committee (VEK ref. KA-20060113) and the Danish Data Protection Agency (j.nr. 2006-41-6848 and HGH-2015-027, I-Suite nr: 03960). Informed consent for research use was obtained from all patients at the enrolling institution (Herlev and Gentofte Hospital, Copenhagen University Hospital, Denmark) before prospective plasma banking.
Patients were qualified for inclusion if they had a diagnosis of PDA, confirmed with CT scan and histology/cytology and were of age 18 years or older at the time of diagnosis. Patients were treatment naïve at the time of enrollment and were treated with first line gemcitabine or fluorouracil (5-FU), leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX). During the study period, 241 patients were recruited to the study, 52 of whom were excluded for the following reasons: no baseline (pre-treatment) sample received (N = 10), no baseline (pre-treatment) CT scan (N = 11), data was from second line of chemotherapy (N = 2), patient had resectable disease (N = 2), patient was diagnosed with non-pancreas cancer (N = 2), patient was treated with radiation (N = 1), and ctDNA assay failed (N = 24) leaving 189 patients for analysis (Table 1). During treatment CT evaluation was missed in 76 patients due to cancer related death or rapid clinical deterioration. In some cases the data collection intervals were irregular due to different schedules of chemotherapy (Gemcitabine 3 weeks cycle vs. FOLFIRINOX 2 weeks cycle), accelerated sample collection because of accelerated CT scan or missing samples. Last follow-up was August 2016.
Sample collection and processing
Baseline and longitudinal plasma samples were collected on all enrolled patients when possible. Study design outlined plasma collection timepoints as baseline (pre-treatment, prior to initiation of first line of chemotherapy), before administration of the second cycle of first line of chemotherapy, and thereafter with every CT scan. Actual collection time-points varied. Biobanking strategy in all enrolled patients was designed with the same standard operating procedures for the acquisition, handling and storage of blood samples. Plasma samples were collected in sodium citrate tubes and centrifuged within ½–2 hours (2330 g for 10 min at 4°C), and stored at –80°C with temperature alarm. 1.2–2 mL of plasma ctDNA was isolated using the QIAamp Circulating Nucleic Acid kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions.
Clinical data collection
Medical records and outcome data of all included patients were collected and reviewed retrospectively. Changes in tumor burden on the consecutive scans were assessed according to the RECIST 1.1 by an independent radiologist, blinded to all clinical data and ctDNA KRAS information. In 30% of cases the investigator assessed response differed from response as confirmed by RECIST 1.1 criteria. One-hundred and thirteen patients achieved disease progression as assessed by RECIST criteria. Thus, appearance of undoubtedly new lesions was classified as progressive disease (PD). We followed RECIST guidelines in case of a new lesion was equivocal. If repeated scans confirmed definitely a new lesion, then progression date was stated using the initial scan.
Plasma ctDNA KRAS analysis
Quantitative analysis of the seven most common mutations in KRAS exon 2, codons 12 and 13 (G12A, G12C, G12D, G12R, G12S, G12V, G13D) was performed using a mutation-enrichment polymerase chain reaction (PCR) coupled with next generation sequencing (NGS) utilizing an ultra-short footprint PCR assay (gene specific footprint 31 bp, with overall amplicon length of 75 bp) to amplify highly degraded ctDNA KRAS fragments [54].
A proprietary pipeline processed the FASTQ files from the Illumina’s MiSeq Sequencing Platform with algorithms that were validated with a sensitivity of 96.0% (95% CI of 86.3%–99.5%) and specificity of 97.9% (95% CI of 88.7%–99.9%) with samples obtained from healthy volunteers, patients with wild-type KRAS G12/G13 cancer and patients with mutant KRAS G12/G13 cancer. Given that detectable KRAS G12/G13 mutation levels were identified, the pipeline then accurately quantifies the mutational load using a corresponding reference set of standard samples that are processed on the same run. Final mutation copy numbers were scaled to 105 Genome Equivalents (GEq, refers to amount of DNA in a single cell) [54]. The concordance was 94% between KRAS G12/G13 mutations in plasma cell-free DNA and FFPE tumor tissue [54].
Serum CA19-9 analysis
Pretreatment and longitudinal serum CA 19-9 concentration was analyzed using the Immulite 2000 GI-MA (Siemens, Catalogue Number L2KG12) assay, a solid-phase, two-site sequential chemiluminescent immunometric assay. The upper normal limit for serum CA 19.9 was 37 U/mL, as this is the reference range for CA19-9.
Statistical analysis
The results of this project are reported in accordance with the REMARK (Reporting recommendations for tumor marker prognostic study) guidelines [55]. Results were compiled for the 189 patients with available baseline, pre-treatment ctDNA specimens successfully processed through the proprietary pipeline along with the demographic information collected at baseline, CT imaging scans assessed with RECIST 1.1 criteria and serum CA19-9 results taken throughout treatment. At each timepoint, specimens with ctDNA KRAS G12/G13 quantitative copy numbers greater than the established lower limit of detection were labeled as “detected”. At each timepoint, specimens with CA19-9 levels with greater than or equal to 37 U/mL were considered “elevated” according to the CA19-9 reference range. Analyzed longitudinal plasma and serum samples were collected after baseline, prior to the start of the second line of chemotherapy (when occurred) and at each RECIST 1.1 assessed CT imaging within two weeks of collection.
The response end points examined were overall survival (OS) and Time to Progression (TTP). OS is defined as the number of days from the start of the first line of chemotherapy to death. TTP is defined here as the number of days from the start of the first line of chemotherapy to PD given that progression is achieved prior to the start of any subsequent lines of chemotherapy. PD is defined using the RECIST 1.1 criteria as have a greater than 20% increase in sum of the diameters of target lesions with an absolute increase of ≥ 5 mm [56].
DNA analyses were performed retrospectively and independently from the clinical data review. Predictive modeling was implemented in 46 patients, in which ctDNA KRAS level was within 7 days of a verified RECIST 1.1 timepoint. Statistical analysis examined both primary variates of interest, ctDNA KRAS copies per 105 GEqand CA19-9 U/mL, against each other and with the response end points. Relationships for both response end points and primary variates along with co-variates of age, gender (sex), chemotherapy type, stage, and pre-treatment ECOG performance status were evaluated. Non-significant variates were removed from consideration using backwards selection.
Cox proportional hazards regression models were used to assess the significance of the variates of interest. Confidence intervals were assessed and reported at 95% unless otherwise stated, and significance testing was two-sided with p < 0.05 considered statistically significant. Due to the high skewness of the data, a log10 transformation was applied to both plasma ctDNA KRAS copy number and serum CA19-9 prior to analysis in order to preserve the normality assumption. The model was tested for fit utilizing Wald and Likelihood scores.
Predictive model for response
Additional analyses were performed for those samples with two or more plasma collection timepoints and RECIST 1.1 CT scan data available. Analysis was restricted to patients with a detectable baseline ctDNA KRAS result and at least one subsequent longitudinal ctDNA timepoint within 7 days of a CT scan with RECIST 1.1 assessment. RECIST 1.1 response was simplified and reported as PD or SD+. SD+ includes a RECIST 1.1 assessment of stable disease, partial response, or complete response [56].
First, we assessed the longitudinal trends in quantitative ctDNA KRAS mutation load and correlated with RECIST 1.1 data and investigator assessed response. The trends observed between longitudinal measurements of ctDNA KRAS and RECIST 1.1 status were strong enough to be explicitly quantified and demonstrated the ability to identify RECIST 1.1 response by measuring quantitative changes in ctDNA KRAS mutation load as compared to baseline. Given a minimal threshold of ctDNA KRAS copies at baseline (annotated as θ), trends were analyzed over large search grids to determine the minimum ctDNA KRAS copies correlating with ctDNA KRAS response of PD (annotated as α). If a longitudinal measurement was less than the α minimum PD threshold and less than the baseline measurement plus a corresponding shift threshold (annotated as ϕ), the ctDNA KRAS response was assessed as SD+. Otherwise, the ctDNA KRAS response was called to be PD (Figure 5).
All statistical analyses were carried out using R version 3.2.3 statistical software.
Abbreviations
BIOPAC, Biomarkers in Pancreatic Cancer; CA19-9, Carbohydrate Antigen 19-9; ctDNA, circulating tumor DNA; CI, confidence interval; GEq, Genome Equivalents; HR, Hazard ratio; IQR, interquartile range; NGS, next generation sequencing; OS, overall survival; p, p value; PCR, polymerase chain reaction; PD, disease progression; PDA, Pancreatic Ductal Adenocarcinoma; RECIST, Response Evaluation Criteria In Solid Tumors; SD, Stable disease; TTP, time to progression; U/mL, units per milliliter.
Author contributions
All authors contributed to study design and editing and review of the manuscript. JAG was responsible for statistical analysis. IC, VMR, JAG, MGE, and JSJ were responsible for compilation of data and manuscript writing, KLH was responsible for radiology review.
ACKNOWLEDGMENTS
The authors would like to acknowledge the patients and families for their participation in this research. We would also like to acknowledge Latifa Hassaine, Saege Hancock, Errin Samuelsz, Cecile Rose Vibat and Vlada Melnikova for their work in sequencing analysis and manuscript review. Many thanks to the biomedical laboratory scientists Charlotte Falk, Vibeke Hintze Holm, Dorthe Baunbjerg Nielsen and Helle Bekker Sørensen, Department of Oncology, Herlev & Gentofte Hospital for excellent technical assistance. Thanks to Dr. Stig E. Bojesen, Department of Clinical Biochemistry, Herlev & Gentofte Hospital for measurement of serum CA 19-9. We also thank Astrid Z. Johansen for data collection in the BIOPAC database.
CONFLICTS OF INTEREST
VMR, JAG and MGE are full time employees of Trovagene, Inc.
EAC has received consulting fees from Qiagen and Guardant and research support from Ignyta, Daiichi-Sankyo.
REFERENCES
1. Chan BA, Hughes BGM. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015; 4:36–54. https://doi.org/10.3978/j.issn.2218-6751.2014.05.01.
2. Mendes D, Alves C, Afonso N, Cardoso F, Passos-Coelho JL, Costa L, Andrade S, Batel-Marques F. The benefit of HER2-targeted therapies on overall survival of patients with metastatic HER2-positive breast cancer – a systematic review. Breast Cancer Res. 2015; 17:140. https://doi.org/10.1186/s13058-015-0648-2.
3. El Zouhairi M, Charabaty A, Pishvaian MJ. Molecularly targeted therapy for metastatic colon cancer: proven treatments and promising new agents. Gastrointest Cancer Res. 2011; 4:15–21.
4. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013; 369:1691–703. https://doi.org/10.1056/NEJMoa1304369.
5. Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364:1817–25. https://doi.org/10.1056/NEJMoa1011923.
6. Howlader N, Noone A, Krapcho M, Miller D, Bishop K, Altekruse S, Kosary C, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis D, Chen H, et al. SEER Cancer Statistics Review, 1975-2013. http://seer.cancer.gov/csr/1975_2013.
7. Carethers JM, Braun J, Sands BE. Genetics, genetic testing, and biomarkers of digestive diseases. Gastroenterology. 2015; 149:1131–3. https://doi.org/10.1053/j.gastro.2015.08.049.
8. Cwik G, Wallner G, Skoczylas T, Ciechanski A, Zinkiewicz K. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch Surg. 2006; 141:968–973. https://doi.org/10.1001/archsurg.141.10.968.
9. Malesci A, Tommasini MA, Bonato C, Bocchia P, Bersani M, Zerbi A, Beretta E, Di Carlo V. Determination of CA 19-9 antigen in serum and pancreatic juice for differential diagnosis of pancreatic adenocarcinoma from chronic pancreatitis. Gastroenterology. 1987; 92:60–7.
10. Paganuzzi M, Onetto M, Marroni P, Barone D, Conio M, Aste H, Pugliese V. CA 19-9 and CA 50 in benign and malignant pancreatic and biliary diseases. Cancer. 1988; 61:2100–8.
11. Pleskow DK, Berger HJ, Gyves J, Allen E, McLean A, Podolsky DK. Evaluation of a serologic marker, CA19-9, in the diagnosis of pancreatic cancer. Ann Intern Med. 1989; 110:704–9.
12. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. The Lancet. 2016; 388:73–85. https://doi.org/10.1016/S0140-6736(16)00141-0.
13. Ballehaninna UK, Chamberlain RS. The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: an evidence based appraisal. J Gastrointest Oncol. 2011; 3:105–119.
14. Hess V, Glimelius B, Grawe P, Dietrich D, Bodoky G, Ruhstaller T, Bajetta E, Saletti P, Figer A, Scheithauer W, Herrmann R. CA 19-9 tumour-marker response to chemotherapy in patients with advanced pancreatic cancer enrolled in a randomised controlled trial. Lancet Oncol. 2008; 9:132–8. https://doi.org/10.1016/S1470-2045(08)70001-9.
15. Pelzer U, Hilbig A, Sinn M, Stieler J, Bahra M, Dörken B, Riess H. Value of carbohydrate antigen 19-9 in predicting response and therapy control in patients with metastatic pancreatic cancer undergoing first-line therapy. Front Oncol. 2013. https://doi.org/10.3389/fonc.2013.00155.
16. Halm U, Schumann T, Schiefke I, Witzigmann H, Mössner J, Keim V. Decrease of CA 19–9 during chemotherapy with gemcitabine predicts survival time in patients with advanced pancreatic cancer. Br J Cancer. 2000; 82:1013.
17. Kanazu M, Maruyama K, Ando M, Asami K, Ishii M, Uehira K, Minomo S, Matsuda Y, Kawaguchi T, Atagi S, Ogawa Y, Kusunoki Y, Takada M, et al. Early pharmacodynamic assessment using 18F-fluorodeoxyglucose positron-emission tomography on molecular targeted therapy and cytotoxic chemotherapy for clinical outcome prediction. Clin Lung Cancer. 2014; 15:182–7. https://doi.org/10.1016/j.cllc.2014.01.001.
18. Ko AH, Hwang J, Venook AP, Abbruzzese JL, Bergsland EK, Tempero MA. Serum CA19-9 response as a surrogate for clinical outcome in patients receiving fixed-dose rate gemcitabine for advanced pancreatic cancer. Br J Cancer. 2005; 93:195–9. https://doi.org/10.1038/sj.bjc.6602687.
19. Wong D, Ko AH, Hwang J, Venook AP, Bergsland EK, Tempero MA. Serum CA19-9 decline compared to radiographic response as a surrogate for clinical outcomes in patients with metastatic pancreatic cancer receiving chemotherapy. Pancreas. 2008; 37:269–74.
20. Bauer TM, El-Rayes BF, Li X, Hammad N, Philip PA, Shields AF, Zalupski MM, Bekaii-Saab T. Carbohydrate antigen 19-9 is a prognostic and predictive biomarker in patients with advanced pancreatic cancer who receive gemcitabine-containing chemotherapy: a pooled analysis of 6 prospective trials. Cancer. 2013; 119:285–92. https://doi.org/10.1002/cncr.27734.
21. Tempero MA, Uchida E, Takasaki H, Burnett DA, Steplewski Z, Pour PM. Relationship of carbohydrate antigen 19-9 and Lewis antigens in pancreatic cancer. Cancer Res. 1987; 47:5501–3.
22. Galli C, Basso D, Plebani M. CA 19-9: handle with care. Clin Chem Lab Med. 2013. https://doi.org/10.1515/cclm-2012-0744.
23. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014; 6:224ra24. https://doi.org/10.1126/scitranslmed.3007094.
24. Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014; 32:579–86. https://doi.org/10.1200/JCO.2012.45.2011.
25. Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014; 4:650–61. https://doi.org/10.1158/2159-8290.CD-13-1014.
26. Reckamp KL, Melnikova VO, Karlovich C, Sequist LV, Camidge DR, Wakelee H, Perol M, Oxnard GR, Kosco K, Croucher P, Samuelsz E, Vibat CR, Guerrero S, et al. A highly sensitive and quantitative test platform for detection of NSCLC EGFR mutations in urine and plasma. J Thorac Oncol. 2016. https://doi.org/10.1016/j.jtho.2016.05.035.
27. Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B, Søreide JA, Javle M, Nordgård O, Smaaland R. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol. 2016; 10:635–43. https://doi.org/10.1016/j.molonc.2015.11.012.
28. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, Lamba S, Hobor S, Avallone A, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015; 21:795–801. https://doi.org/10.1038/nm.3870.
29. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988; 53: 549–54. https://doi.org/10.1016/0092-8674(88)90571-5.
30. Hruban RH, van Mansfeld AD, Offerhaus GJ, van Weering DH, Allison DC, Goodman SN, Kensler TW, Bose KK, Cameron JL, Bos JL. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol. 1993; 143:545–54.
31. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MCJ, Robertson AJ, Fadlullah MZH, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015; 518:495–501. https://doi.org/10.1038/nature14169.
32. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, Choti MA, Yeo CJ, McCue P, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015. https://doi.org/10.1038/ncomms7744.
33. Castells A, Puig P, Móra J, Boadas J, Boix L, Urgell E, Solé M, Capellà G, Lluís F, Fernández-Cruz L, Navarro S, Farré A. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. J Clin Oncol. 1999; 17:578–84.
34. Chen H, Tu H, Meng ZQ, Chen Z, Wang P, Liu LM. K-ras mutational status predicts poor prognosis in unresectable pancreatic cancer. Eur J Surg Oncol. 2010; 36:657–62. https://doi.org/10.1016/j.ejso.2010.05.014.
35. Maire F, Micard S, Hammel P, Voitot H, Lévy P, Cugnenc PH, Ruszniewski P, Puig PL. Differential diagnosis between chronic pancreatitis and pancreatic cancer: value of the detection of KRAS2 mutations in circulating DNA. Br J Cancer. 2002; 87:551–4. https://doi.org/10.1038/sj.bjc.6600475.
36. Pishvaian MJ, Bender RJ, Matrisian LM, Rahib L, Hendifar A, Hoos WA, Mikhail S, Chung V, Picozzi V, Heartwell C, Mason K, Varieur K, Aberra M, et al. A pilot study evaluating concordance between blood-based and patient-matched tumor molecular testing within pancreatic cancer patients participating in the Know Your Tumor (KYT) initiative. Oncotarget. 2017; 8:83446–56. https://doi.org/10.18632/oncotarget.13225.
37. Brychta N, Krahn T, von Ahsen O. Detection of KRAS mutations in circulating tumor DNA by digital PCR in early stages of pancreatic cancer. Clin Chem. 2016. https://doi.org/10.1373/clinchem.2016.257469.
38. Pietrasz D, Pécuchet N, Garlan F. Plasma circulating tumor DNA in pancreatic cancer patients is a prognostic marker. Clin Cancer Res. 2017. https://doi.org/10.1158/1078-0432.CCR-16-0806.
39. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012; 72:2457–67. https://doi.org/10.1158/0008-5472.CAN-11-2612.
40. Rahib L, Fleshman JM, Matrisian LM, Berlin JD. Evaluation of pancreatic cancer clinical trials and benchmarks for clinically meaningful future trials: a systematic review. JAMA Oncol. 2016; 2:1209. https://doi.org/10.1001/jamaoncol.2016.0585.
41. Hammad N, Heilbrun LK, Philip PA, Shields AF, Zalupski MM, Venkatramanamoorthy R, El-Rayes BF. CA19-9 as a predictor of tumor response and survival in patients with advanced pancreatic cancer treated with gemcitabine based chemotherapy. Asia Pac J Clin Oncol. 2010; 6:98–105. https://doi.org/10.1111/j.1743-7563.2010.01290.x.
42. Ziske C, Schlie C, Gorschlüter M, Glasmacher A, Mey U, Strehl J, Sauerbruch T, Schmidt-Wolf IGH. Prognostic value of CA 19-9 levels in patients with inoperable adenocarcinoma of the pancreas treated with gemcitabine. Br J Cancer. 2003; 89:1413–7. https://doi.org/10.1038/sj.bjc.6601263.
43. Mandel P, Metais P. Les acides nucléiques du plasma sanguin chez l’homme. C R Seances Soc Biol Fil. 1948; 142:241–3.
44. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977; 37:646–50.
45. Shapiro B, Chakrabarty M, Cohn EM, Leon SA. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer. 1983; 51:2116–20. https://doi.org/10.1002/1097-0142(19830601)51:11<2116::AID-CNCR2820511127>3.0.CO;2-S.
46. Yamada T, Nakamori S, Ohzato H, Oshima S, Aoki T, Higaki N, Sugimoto K, Akagi K, Fujiwara Y, Nishisho I, Sakon M, Gotoh M, Monden M. Detection of K-ras gene mutations in plasma DNA of patients with pancreatic adenocarcinoma: correlation with clinicopathological features. Clin Cancer Res. 1998; 4:1527–32.
47. Mulcahy HE, Lyautey J, Lederrey C, qi Chen X, Anker P, Alstead EM, Ballinger A, Farthing MJ, Stroun M. A prospective study of K-ras mutations in the plasma of pancreatic cancer patients. Clin Cancer Res. 1998; 4:271–5.
48. Dianxu F, Shengdao Z, Tianquan H, Yu J, Ruoqing L, Zurong Y, Xuezhi W. A prospective study of detection of pancreatic carcinoma by combined plasma K-ras mutations and serum CA19-9 analysis. Pancreas. 2002; 25:336–41.
49. Uemura T, Hibi K, Kaneko T, Takeda S, Inoue S, Okochi O, Nagasaka T, Nakao A. Detection of K-ras mutations in the plasma DNA of pancreatic cancer patients. J Gastroenterol. 2004; 39:56–60. https://doi.org/10.1007/s00535-003-1245-1.
50. Takai E, Totoki Y, Nakamura H, Kato M, Shibata T, Yachida S. Clinical utility of circulating tumor DNA for molecular assessment and precision medicine in pancreatic cancer. Adv Exp Med Biol. 2016; 924:13–7. https://doi.org/10.1007/978-3-319-42044-8_3.
51. Däbritz J, Hänfler J, Preston R, Stieler J, Oettle H. Detection of Ki-ras mutations in tissue and plasma samples of patients with pancreatic cancer using PNA-mediated PCR clamping and hybridisation probes. Br J Cancer. 2005. https://doi.org/10.1038/sj.bjc.6602319.
52. Kinugasa H, Nouso K, Miyahara K, Morimoto Y, Dohi C, Tsutsumi K, Kato H, Matsubara T, Okada H, Yamamoto K. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer: K-ras in blood with pancreatic cancer. Cancer. 2015; 121:2271–80. https://doi.org/10.1002/cncr.29364.
53. Schultz NA, Dehlendorff C, Jensen BV, Bjerregaard JK, Nielsen KR, Bojesen SE, Calatayud D, Nielsen SE, Yilmaz M, Holländer NH, Andersen KK, Johansen JS. MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA. 2014; 311:392–404. https://doi.org/10.1001/jama.2013.284664.
54. Fujii T, Barzi A, Sartore-Bianchi A, Cassingena A, Siravegna G, Karp DD, Piha-Paul SA, Subbiah V, Tsimberridou AM, Huang HJ, Veronese S, Di Nicolantonio FD, Pingle S, et al. Mutation-enrichment next-generation sequencing for quantitative detection of KRAS mutations in urine cell-free DNA from patients with advanced cancers. Clin Cancer Res. 2017.
55. McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM, Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics. REporting recommendations for tumor MARKer prognostic studies (REMARK). Nat Clin Pract Urol. 2005; 2:416–22.
56. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45:228–47. https://doi.org/10.1016/j.ejca.2008.10.026.