
INTRODUCTION
The High-Mobility Group A (HMGA) family consists of three proteins: HMGA1a, HMGA1b, and HMGA2 [1]. HMGA proteins do not have transcriptional activity per se; however, by interacting with the transcription machinery, they alter the chromatin structure and thereby regulate the transcriptional activity of various genes [2, 3]. The levels of HMGA proteins are low or absent in normal cells and adult tissues [4]. In contrast, their constitutive expression is remarkably high in neoplastically transformed cells and in embryonic cells [5-7]. Their overexpression is associated with a highly malignant phenotype and correlates with the presence of metastasis and reduced survival [8, 9]. Several studies implicate the expression of the HMGA genes in the process of carcinogenesis [10-18]. However, although HMGA overexpression is known to play a critical role in malignant cell transformation, the mechanisms regulating HMGA protein levels remain largely obscure.
Non-coding RNAs, including pseudogenes, have long been viewed as non-functional genomic relicts of evolution, but a large body of evidence now suggests they are important in both physiology and disease. Pseudogenes are usually defined as defunct copies of genes that have lost their potential as DNA templates for functional products [19-26] because they harbour premature or delayed stop codons, deletions/insertions and frameshift mutations that abrogate their translation into functional proteins. There are two types of pseudogenes: processed pseudogenes, which have been retrotransposed back into a genome via an RNA intermediate; and nonprocessed pseudogenes, which are the genomic remnants of duplicated genes or residues of dead genes. Processed pseudogenes contain no introns, and share 5’ and 3’ untranslated region (UTR) sequences with their ancestral genes [27]. Since miRNAs repress target gene expression by binding to complementary sequences in the 3’ UTR of target mRNA, pseudogenes can be targeted by miRNAs that modulate the expression of coding genes. Indeed, several pseudogene transcripts exert regulatory control of their ancestral gene expression levels by competing for the same miRNAs [28], which is in keeping with the notion that miRNA activity is theoretically affected by the availability of target microRNA response elements (MRE) in the cellular milieu [28-30]. Given this scenario, we studied the possible functional relationship between the mRNAs produced by the HMGA1 oncogene and its pseudogenes (HMGA1Ps), and the consequences of this interaction especially in the process of carcinogenesis in which HMGA1 overexpression plays a critical role.
RESULTS
HMGA1P6 and HMGA1P7 are targeted by HMGA1-targeting miRNAs
We first identified seven HMGA1Ps by bioinformatics analysis. Of these, we focused on the HMGA1P6 and HMGA1P7 processed pseudogenes located at 13q12.12 and 6q23.2, respectively, because of their very high sequence homology with HMGA1 in the coding region and in the 5’ and 3’ UTRs (Figure 1A). A missense mutation of the initiator methionine codon prevents translation of HMGA1P7 whereas HMGA1P6 is non-protein coding since it carries a mutation in the stop codon and so generates a non-translatable mRNA. Within the high homology regions, we found perfectly conserved seed matches for miRNAs that have been predicted (miR-103, miR-142-3p, miR-370, and miR-432) or already demonstrated (miR-15 [31], miR-16 [31], miR-26a [32], miR-214 [33], miR-548c-3p [34] and miR-761 [33]) to target the HMGA1 gene (Figure 1B and 1C).
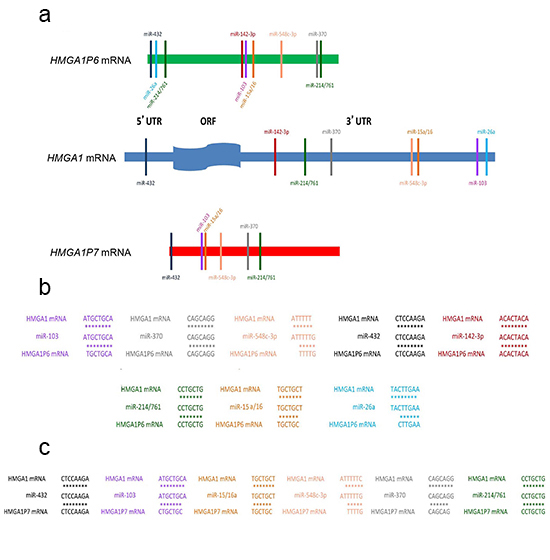
Figure 1: HMGA1P6 and HMGA1P7 show the same seed sequences of HMGA1-targeting miRNAs. (A) HMGA1 (middle), HMGA1P6 (top) and HMGA1P7 (bottom) UTRs contain highly conserved regions. HMGA1-targeting miRNA seed matches within the high homology region are conserved between HMGA1 and HMGA1Ps. (B) and (C) binding of HMGA1-targeting miRNAs to HMGA1P6 (B) and HMGA1P7 (C).
To evaluate the ability of these miRNAs to target HMGA1P6 and HMGA1P7, we transfected miR-15, miR-16, miR-214 and miR-761 into MCF7 cells (human breast adenocarcinoma), and measured HMGA1P6, HMGA1P7, and HMGA1 mRNA levels using Real-time PCR and PCR primer sets (see Methods) that discriminate the three mRNA transcripts. As shown in Figure 2A, the transfection of the HMGA1-targeting miRNAs led to a significant reduction of HMGA1, HMGA1P6 and HMGA1P7 mRNA levels.
To determine whether the HMGA1-targeting miRNAs directly interacted with the HMGA1P mRNAs, we inserted the full-length HMGA1P6 and HMGA1P7 mRNAs downstream of the luciferase open reading frame. These reporter vectors were transfected into human embryonic kidney (HEK293) cells together with miRNA precursors and a control non-targeting scrambled oligonucleotide. The luciferase activity was much lower after miR-15, miR-16, miR-214 and miR-761 transfection compared with the scrambled oligonucleotide (Figure 2B). These results indicate that the HMGA1Ps and HMGA1 undergo the same miRNA-mediated post-transcriptional regulation.
HMGA1P6 and HMGA1P7 act as decoys for HMGA1-targeting miRNAs
Subsequently, we examined the ability of HMGA1P6 and HMGA1P7 to function as a decoy for HMGA1-targeting miRNAs using a vector carrying the 3’ UTR of the HMGA1 mRNA downstream of the luciferase open reading frame. This reporter vector was transfected into HEK293 cells together with HMGA1P6- or HMGA1P7-expressing vectors. As expected, luciferase activity was much higher in HMGA1Ps-transfected cells than in the control vector (Figure 2C). Moreover, overexpression of different amounts of HMGA1P6 or HMGA1P7 drastically and dose-dependently reduced the effects exerted by miRNA on the levels of both the HMGA1 transcript and protein (Figure 3A). Conversely, HMGA1P6 and HMGA1P7 knockdown resulted in decreased HMGA1 mRNA and protein levels (Figure 3B) thereby mirroring the results obtained with HMGA1P6 and HMGA1P7 overexpression (Figure 3A). Therefore, the HMGA1Ps compete for the endogenous miRNA-binding sites.
The upregulation of HMGA1 induced by overexpression of HMGA1P6 and HMGA1P7 was blunted in DICER-silenced cells (Figure 3C). In fact, silencing of DICER, the enzyme that catalyses the last step of miRNA maturation, leads to reduced levels of mature miRNAs compared to control cells. These results support the notion that HMGA1P6 and HMGA1P7 require mature miRNAs to regulate HMGA1 levels. Finally, as expected from our observation that HMGA1Ps increase HMGA1 levels, we found that the HMGA1 3’ UTR upregulates HMGA1P levels (Figure 3D).
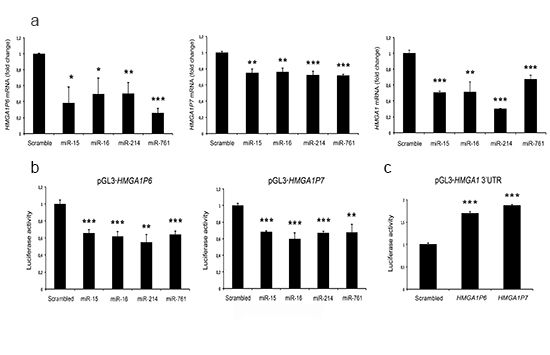
Figure 2: HMGA1P6 and HMGA1P7 are targeted by HMGA1-targeting miRNAs. (A) qRT-PCR analysis of HMGA1P6 (left), HMGA1P7 (middle) and HMGA1 (right) mRNA from the MCF7 cells transfected with scrambled-oligonucleotide, miR-15, miR-16, miR-214 and miR-761.(B) HMGA1Ps were cloned into the pGL3 control vector. Relative luciferase activity in HEK293 cells transiently transfected with miR-15, miR-16, miR-214, miR-761 and a control scrambled oligonucleotide. (C) The 3’UTR of HMGA1 was cloned into the pGL3 control vector. Relative luciferase activity in HEK293 cells transiently transfected with the empty vector, HMGA1P6 and HMGA1P7.. The results are reported as the mean of values. Error bars represent mean ± SD; n=3. *, P < 0.05 **, P < 0.01 ***, P < 0.001 (t test).
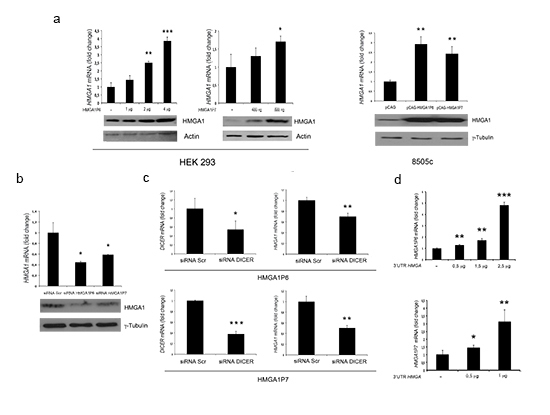
Figure 3: HMGA1 is positively regulated by HMGA1Ps. (A) (upper panels) qRT-PCR analysis of HMGA1 mRNA levels in HEK293 and 8505c cells transfected with the empty vector, HMGA1P6 and HMGA1P7. (Lower panels) Western blot analysis of HMGA1 protein levels from the same samples shown in the upper panels. (B) (Upper panel) qRT-PCR analysis of HMGA1 mRNA levels in 8505c cells transfected with the scrambled oligonucleotide, siRNA-HMGA1P6 and siRNA-HMGA1P7s. (Lower panel) Western blot analysis of HMGA1 protein levels from the same samples shown in the upper panel. (C) HMGA1 mRNA levels 24 h after the transfection of HMGA1P6 and HMGA1P7 in scrambled oligonucleotide or siRNA-DICER 8505c transfected cells. (D) HMGA1P6 and HMGA1P7 mRNA levels after the transfection of the 3’UTR of the HMGA1 plasmids in MCF7 cells. The results are reported as the mean of values; Error bars represent means ± SD; n =3. *, P < 0.05 **, P < 0.01 ***, P < 0.001 (t test).
HMGA1P6 and HMGA1P7 exert oncogenic activity
The HMGA1 pseudogenes can be transcribed but they cannot code for protein. However, the above-reported results suggest that they derepress HMGA1 transcript and protein levels (Figure 3A). To evaluate the functional consequences of HMGA1P6 and HMGA1P7 overexpression, we investigated their role in cellular proliferation, apoptosis, migration and invasion in cells expressing HMGA1.
As shown in Figure 4A and B, HEK293 cells and 8505c cells (derived from a human anaplastic thyroid carcinoma) transfected with HMGA1P6- or HMGA1P7-expressing vectors grew significantly faster than the empty vector-transfected cells. Cell cycle analysis of the cells overexpressing HMGA1P6 and HMGA1P7 revealed an increased number of cells in the S phase and a reduced number of cells in G1 compared with control cells (Figure 4C). This was not unexpected given the increased HMGA1 levels induced by HMGA1P6 and HMGA1P7 expression. Moreover, in 8505c cells knocked down for the HMGA1Ps, we found that 8505c-siRNA-HMGA1P6 and 8505c-siRNA-HMGA1P7 cells grew at a significantly slower rate than the 8505c-siRNA negative control (Figure 4D). Interestingly, cell cycle analysis of the 8505C-siRNA-HMGA1P6 and 8505C-siRNA-HMGA1P7 cells revealed an increase in the number of cells in the sub-G1 phase, which corresponds to apoptotic cells, compared with control cells (data not shown). This result is in agreement with the finding that HMGA silencing induces apoptosis in cancer cells [12].
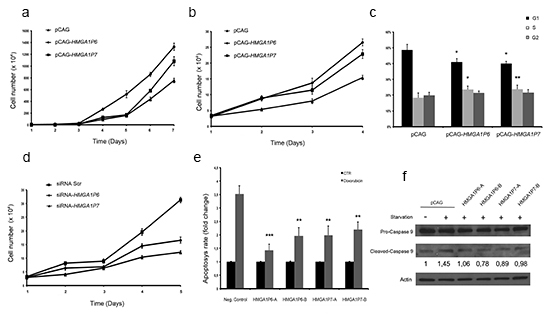
Figure 4: HMGA1P6 and HMGA1P7 expression increases cell proliferation and reduces apoptosis. (A) and (B) HEK293 and 8505c cell proliferation in HMGA1P6- and HMGA1P7-transfected cells. (C) HEK293 cells were transfected with the control, HMGA1P6 or HMGA1P7 vectors. The DNA content of the transfected HEK293 cells was analyzed by flow cytometry after propidium iodine staining. (D) 8505c cell proliferation in siRNA-HMGA1P6- and siRNA-HMGA1P7-transfected cells. (E) HMGA1P6- and HMGA1P7-transfected cells were treated with doxorubicin, and apoptosis was assessed by FACS. (F) HEK293 cells were starved, and apoptosis was assessed by Western blot analysis of Caspase 9 cleavage. The results are reported as the mean of values; Error bars represent means ± SD; n =3. *, P < 0.05 **, P < 0.01 ***, P < 0.001 (t test).
To probe further the role of HMGA1 pseudogenes in apoptotic cell death, we incubated HEK293 cells with doxorubicin in the presence or absence of the HMGA1Ps. As shown in Figure 4E, HMGA1P6 and HMGA1P7 overexpression significantly reduced the programmed cell death induced by doxorubicin. The same result was obtained with HEK293 cells in which apoptosis was induced by serum-starvation. Indeed, the overexpression of the HMGA1Ps counteracted caspase 9 cleavage (Figure 4F).
Since HMGA1 promotes cell migration and invasion [8] we carried out cell migration and invasion assays in cells transfected with the HMGA1Ps. As expected, cell migration was significantly higher in HEK293 and 8505c cells overexpressing HMGA1P6 or HMGA1P7 than in control cells (Figure 5A). Moreover, 8505c-siRNA-HMGA1P6 and 8505c-siRNA-HMGA1P7 cells migrated more slowly than the 8505c-siRNA negative control (Figure 5B). Accordingly, the invasion matrigel assay revealed invasion activity in HEK293 cells transfected with HMGA1P6 or HMGA1P6 (Figure 5C). Similar results were obtained with the HMGA1Ps-8505c cells (data not shown). These results indicate that cell proliferation, motility and invasion is driven by regulation HMGA1Ps-mediated of HMGA1.
Bioinformatic analysis revealed that HMGA1P6 and HMGA1P7 contain sequences that can be targeted by miRNAs that target High Mobility Group A2 (HMGA2), Vascular Endothelial Growth Factor (VEGF) and Enhancer of Zeste Homolog 2 (EZH2), all of which are known to be involved in carcinogenesis [34-36]. Accordingly, we found that HMGA1P6 or HMGA1P7 overexpression increased the level of the proteins coded for by these genes (Figure 5D). Consequently, it appears that HMGA1P6 and HMGA1P7 expression may contribute to cancer progression by acting as decoys for cancer-related genes other than HMGA1.
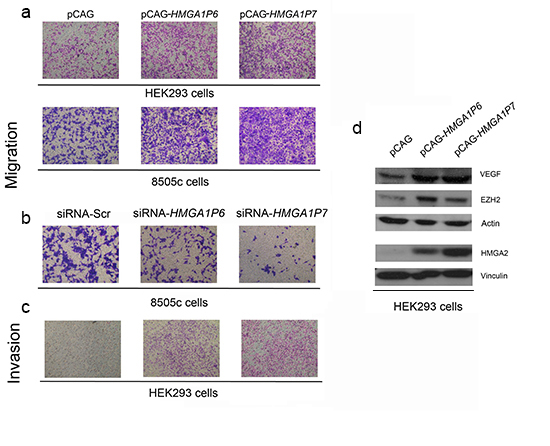
Figure 5: The expression of the HMGA1Ps affects cell migration and invasion. (A) Cell migration assays of HEK293 and 8505c cells transfected with HMGA1P6 or HMGA1P7 or with a control vector. One representative experiment is reported. (B) Cell migration assays of 8505c cells transfected with siRNA-HMGA1P6 or siRNA-HMGA1P7 or with a empty vector. One representative experiment is reported. (C) Cell invasion assays of HEK293 cells transfected with HMGA1Ps or with the backbone vector. One representative experiment is reported. (D) Extracts from HEK293 transfected with HMGA1P6 or HMGA1P7 or with a control vector were analyzed by Western blotting.
Correlation of HMGA1 and the overexpression of the HMGA1Ps in human cancer
To verify whether the two HMGA1Ps function as decoys in the regulation of HMGA1 protein levels also in human cancer, we analyzed the expression of HMGA1 and of the HMGA1Ps in a panel of differentiated and undifferentiated thyroid carcinomas by Western Blotting and Real-time PCR. As shown in Figure 6, papillary (PTC) thyroid carcinomas, which are well differentiated and poorly aggressive, expressed low levels of HMGA1P6 and HMGA1P7 (Figure 6A). Conversely, anaplastic thyroid carcinoma (ATC), which is one of the most aggressive human tumours, expressed very high HMGA1P levels that, moreover, correlated with HMGA1 protein levels (Figure 6B). Accordingly, HMGA1 expression, which was undetectable in normal thyroid tissue, was much higher in ATC than in PTC. Similar results were obtained in human ovarian carcinomas (see Figure 6C and 6D). The direct correlation between HMGA1 and HMGA1P6 expression (r=0.6553, P<0.0001) and between HMGA1 and HMGA1P7 expression (r=0.7001, P<0.0001) suggests that these genes are co-regulated (Figure 6C and 6D). Taken together, these results indicate that HMGA1P6, HMGA1P7 and HMGA1 expression is correlated with cancer aggressiveness.
HMGA1P7 overexpressing mouse embryonic fibroblasts grow faster and senesce later
To establish the role of the HMGA1Ps in vivo, we generated transgenic mice overexpressing HMGA1P7. The expression of HMGA1P7 in transgenic mice was verified by RT-PCR using RNAs extracted from liver, spleen, lung and mouse embryonic fibroblasts (MEFs). HMGA1P7 mRNA levels were high in the tissues of HMGA1P7 transgenic mice and absent in the WT counterpart (Figure 7A). Consistently, HMGA1 transcript and protein levels were significantly higher in HMGA1P7-MEFs than in the WT control (Figure 7B). Notably, HMGA1P7-MEFs also expressed increased levels of the Hmga2, Ezh2 and Vegf proteins, which are coded for by genes that share miRNAs with HMGA1 (Figure 7C), and increased levels of the HMGA1-regulated genes Ccna, Ccnb, Ccnd2 and E2f-1 (Figure 7D), which play a critical role in cell cycle regulation [8].
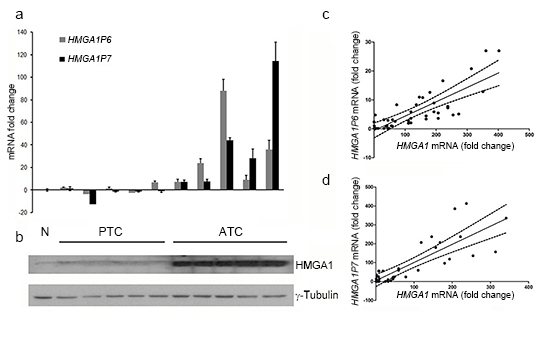
Figure 6: HMGA1 protein expression positively correlates with the expression of the HMGA1Ps in ATC. (A) HMGA1P6 and HMGA1P7 qRT-PCR analysis in normal thyroid tissue (NT), papillary thyroid carcinoma (PTC) and anaplastic thyroid carcinoma (ATC). The results are reported as the mean of expression values. The error bars represent mean ± SD; n = 3. (B) Western blot analysis of HMGA1 protein expression in the same samples as in A. (C) and (D) Ovarian sample expression values derived from commercial sources were combined for correlation analysis. Linear regressions of HMGA1 versus HMGA1P6 (C) and HMGA1 versus HMGA1P7 (D) are shown.
As expected from the high HMGA1 expression in the HMGA1P7-MEFs, the growth rate of these MEFs was significantly higher than that of the WT controls (Figure 7E). To determine whether the higher growth rate of HMGA1P7-MEFs was caused by altered progression through the cell cycle, we examined asynchronously growing MEFs by flow cytometry. The number of HMGA1P7-MEFs was lower in G1 and higher in the S phase of the cell cycle compared with WT MEFs (Figure 7F).
We, next, examined the susceptibility of MEFs to senescence by measuring senescence-associated β-gal (SA-β-gal) activity. At culture passage 6, SA-β-gal activity was present in WT MEFs, but not in the HMGA1P7 transgenic counterparts (Figure 7G). These findings indicate that HMGA1P7 overexpression reduced susceptibility to cellular senescence.
Identification of the genes modulated by HMGA1P7 expression
To identify the genes regulated by HMGA1P7 expression, we analyzed the expression profile of WT and HMGA1P7 transgenic MEFs in microarray analyses. To this aim, RNAs extracted from WT and HMGA1P7-MEFs were hybridized to the Affymetrix GeneChip Mouse Gene 2.0 ST oligonucleotide arrays. Seventy transcripts that had a significant fold change variation (p <0.05) were examined as candidate genes involved in HMGA1P7 tumour-promoting activity. Interestingly, we found five upregulated cancer-related genes (Epha3, Hjurp, Kif26, S1pr3 and Pde3B) that shared miRNAs with the HMGA1P7 transcript. These genes are involved in various human cancers (glioblastoma, breast and hematological cancers), and are candidate therapeutic targets [37-41]. Real-time PCR experiments confirmed upregulation of all these genes in HMGA1P7-MEFs (Supplementary Figure 1). These results support the concept that HMGA1P7 modulates the expression of several cancer-related genes by acting as a ceRNA.
DISCUSSION
The HMGA proteins play a critical role in carcinogenesis. Recently, several miRNAs have been demonstrated to target these genes [31-34], and their dysregulation may contribute to HMGA1 protein overexpression in human neoplasias [31, 32, 34]. Moreover, an important role in the regulation of protein synthesis has recently been ascribed to pseudogenes [27, 28]: the presence of the same miRNA-targeted seed sequences in the HMGA1 and in the HMGA1Ps UTR regions could block the access of miRNAs to their protein-coding target genes. Finally, it has been outlined a novel gene-expression pathway in which an HMGA protein-coding gene, Hmga2, operates largely independently of its protein-coding function to promote cancer progression as a competing endogenous RNA [42].

Figure 7: HMGA1P7 overexpressing MEFs show a higher growth rate, and lower susceptibility to senescence. (A) qRT-PCR analysis of total RNA from livers, spleens, lungs, and MEFs of WT and HMGA1P7 transgenic mice. (B) (upper panel) qRT-PCR analysis of HMGA1 mRNA levels in WT and HMGA1P7 transgenic MEFs. (Lower panel) Western blot analysis of HMGA1 protein expression in the same samples. (C) and (D) qRT-PCR and Western blot analysis of genes that share common miRNAs with HMGA1 (Left panel) and HMGA1-regulated genes (Right panel). (E) MEFs were prepared from WT and HMGA1P7 overexpressing embryos at 12.5 dpc. At culture passage 3, they were plated and counted daily for 8 days. (F) Propidium iodide flow cytometry of asynchronous growing WT and HMGA1P7 overexpressing MEFs. (G) Light microscopy of representative WT and HMGA1P7 overexpressing MEFs stained for β-galactosidase activity at culture passages 6. The results are reported as the mean of values with error bars indicating SD (mean ± SD); n =3. *, P < 0.05 **, P < 0.01 ***, P < 0.001 (t test).
We asked whether HMGA1 pseudogenes affect HMGA1 protein levels, and, consequently, whether they play a critical role in cancer progression. We focused on HMGA1P6 and HMGA1P7, which have conserved seed matches for miRNAs targeting the HMGA1 gene in the high homology regions. We demonstrate that overexpression of these HMGA1 pseudogenes increases HMGA1 protein levels, and inhibits the suppression of HMGA1 protein synthesis by miRNAs that target the HMGA1 gene, namely, miR-15, miR-16, miR-214, and miR-761 [31-34]. Consistent with these results, our functional studies demonstrate that HMGA1P6 and HMGA1P7 overexpression increases the cell growth rate by decreasing the number of G1-phase cells and increasing the number of S-phase cells, compared with the backbone vector-transfected cells. Therefore, HMGA1P6 and HMGA1P7 affect cell cycle progression, as expected, given their ability to increase the protein levels of HMGA1, which is involved in the regulation of the G1-S transition phase of the cell cycle [34]. Moreover, HMGA1P6 and HMGA1P7 overexpression increased cell migration and invasiveness, and decreased the apoptotic rate.
These results prompted us to verify whether HMGA1P6 and HMGA1P7 overexpression is involved also in human carcinogenesis. Interestingly, HMGA1P6 and HMGA1P7 were abundantly expressed in ATC, which are very aggressive and express very high HMGA1 protein levels [43]. Conversely, HMGA1P6 and HMGA1P7 expression was low in PTC, which are well differentiated and poorly aggressive, and express moderate HMGA1 protein levels. We obtained similar results in human ovarian carcinomas suggesting that HMGA1Ps can regulate HMGA1 protein levels also in vivo.
Interestingly, HMGA1P6 and HMGA1P7 seem to affect cancer progression also by binding to the same miRNAs that target proteins involved in cancer progression. Indeed, the overexpression of the HMGA1Ps increased also the levels of HMGA2, VEGF and EZH2 that are coded for by genes targeted by HMGA1-targeting miRNAs. Notably EZH2, which is involved in carcinogenesis, is overexpressed in ATC but not in PTC [44].
Data obtained with transgenic mice overexpressing HMGA1P7 and with the relative MEFs support the concept that HMGA1P7 plays an oncogenic role. Indeed, MEFs derived from transgenic mice overexpressing HMGA1P7 show a higher growth rate, and lower susceptibility to senescence with respect to the WT counterpart. Moreover, flow cytometry showed an increase of cells in S phase as expected given the ability of HMGA1 to increase the E2F1 activity [45].
In contrast to a report that ectopic overexpression of HMGA1 reduces the lifespan of IMR90 cells [46], HMGA1P7-MEFs that have more abundant HMGA1 protein levels, senesce later with respect to WT MEFs. In agreement with our findings, we obtained the opposite result in Hmga1-null MEFs [47]. It is likely that the cellular context influences the effect exerted by HMGA proteins on cell growth. Moreover, the different experimental approach, one in vivo and one in vitro, may account for these contradictory results. In fact, discrepancies between in vitro and transfection approaches were reported in a study of the p53 pathway [48]. The behaviour of the Hmga1-null and HMGA1P7-MEFs described here supports the oncogenic role of HMGA overexpression, which is a feature of malignant neoplasias. In conclusion, our finding that HMGA1P7-overexpressing MEFs grow faster and senesce later than their WT counterpart sustains our model in which HMGA1Ps act as ceRNAs that regulate HMGA1 and other genes by competing for shared miRNAs thus contributing to cancer progression.
MATERIALS AND METHODS
Cell culture and transfections
HEK293, MCF7, 8505c, and MEF (from 12.5-day-old embryos) cells were maintained in DMEM supplemented with 10% foetal calf serum (GIBCO; Invitrogen), glutamine and antibiotics. Cells were regularly tested with MycoAlert (Lonza) to ascertain that cells were not infected with mycoplasma. Cells were transfected using Lipofectamine plus reagent (Invitrogen) according to the manufacturer’s instructions. The transfected cells were selected in a medium containing geneticin (Sigma). For each transfection, several geneticin-resistant mass cell populations were isolated and expanded for further analysis. Transfection efficiency was verified for each experiment by evaluating GFP expression. To inhibit HMGA1P6 and HMGA1P7 expression, small interfering RNAs and corresponding scramble small interfering RNAs were designed and used as suggested by the manufacturer (RIBOXX).
Human thyroid and ovary tissue samples
Neoplastic and normal human thyroid tissues were obtained from surgical specimens and immediately frozen in liquid nitrogen. Thyroid tumours were collected at the Service d’Anatomo-Pathologie, Centre Hospitalier Lyon Sud, Pierre Bénite, France. The tumour samples were frozen until required for RNA or protein extraction. We declare that informed consent for the scientific use of biological material was obtained from all patients. TissueScan Ovarian Cancer Tissue Real-time PCR Panel were purchased from Origene (HORT302).
RNA extraction and quantitative reverse transcription PCR
Total RNA was extracted from tissues and cell cultures with Trizol (Gibco) according to the manufacturer’s instructions. For mRNA detection, we reverse transcribed total RNA from cell lines by using the QuantiTect Reverse Transcription Kit (Qiagen), and then Real-time PCR was performed by using Power SYBR Green PCR Master Mix (Applied Biosystems) and the following primers:
HMGA1-Fw 5’-aaggggcagacccaaaaa-3’ HMGA1-Rev 5’-tccagtcccagaaggaagc-3’
HMGA1P6-Fw 5’-gcagacccacaaaactgga-3’ HMGA1P6-Rev 5’-gagcaaagctgtcccatcc-3’
HMGA1P7-Fw 5’-gctccttctcggctcctc-3’ HMGA1P7-Rev 5’-gcttgggcctcttttatgg-3’
G6PD-Fw 5’-acagagtgagcccttcttcaa-3’ G6PD-Rev 5’-ataggagttgcgggcaaag-3’
Hmga1-Fw 5’-ggcagacccaagaaactgg-3’ Hmga1-Rev 5’-ggcactgcgagtggtgat-3’
Ccna-Fw 5’-cttggctgcaccaacagtaa-3’ Ccna-Rev 5’-caaactcagttctcccaaaaaca-3’
Ccnb-Fw 5’-gcgctgaaaattcttgacaac-3’ Ccnb-Rev 5’-ttcttagccaggtgctgcat-3’
G6pd-Fw 5’-cagcggcaactaaactcaga-3’ G6pd-Rev 5’-ttccctcaggatcccacac-3’
Epha3-Fw 5’-tggctccttggacagtttct-3’ Epha3-Rev 5’-ttcccacaagctccatgact-3’
Hjurp-Fw 5’-gagaactggccatcttgcag-3’ Hjurp-Rev 5’-aaggtgtttccgggcact-3’
Kif26b-Fw 5’-aagaggcaggctctcaagc-3’ Kif26b-Rev 5’-gcagagaaagcaagggtcctt-3’
S1pr3-Fw 5’-agatgcgccttgcagaac-3’ S1pr3-Rev 5’-agagtggtggtgggttcct-3’
Pde3B-Fw 5’-ccttgtatttcccgagaacagat-3’ Pde3B-Rev 5’-ggtaatgaggtttacaccactgc-3’
Hmga2-Fw 5’-aaggcagcaaaaacaagagc-3’ Hmga2-Rev 5’-ttgtggccatttcctaggtc-3’
Ezh2-Fw 5’-tggaagcagcggaggata-3’ Ezh2-Rev 5’-gtcactggtgactgaacactcc-3’
Vegf-Fw 5’-aaaaacgaaagcgcaagaaa-3’ Vegf-Rev 5’-tttctccgctctgaacaagg-3’
The 2−ΔΔCt formula was used to calculate the differential gene expression.
Plasmids
For transfection of miRNA oligonucleotides, cells were transfected with 50 nmol/ml of miRNA precursors or with a control no-targeting scrambled oligonucleotides (Ambion, Austin, TX) using siPORT neoFX Transfection Agent (Ambion). For the HMGA1P6 expression construct (pCAG-HMGA1P6) and the HMGA1P6 luciferase reporter construct (pGL3-HMGA1P6), the entire sequence of HMGA1P6 gene (ENST00000418454.1) was amplified by using the primers Fw HMGA1P6 5’-tcctctaattgggactccga-3’ and Rev HMGA1P6 5’-ttactcagatcccaggcaga-3’. The amplified fragment was cloned into pCAG vector kindly given by Dr. S. Soddu, and into pGL3-Control firefly luciferase reporter vector (Promega), respectively. For the HMGA1P7 construct (pCAG-HMGA1P7) and the HMGA1P7 luciferase reporter construct (pGL3-HMGA1P6), the entire sequence of the HMGA1P7 gene (ENST00000406908.1) was amplified by using the primers Fw HMGA1P7 5’-agccagtcgagctggaggtc-3’ and Rev HMGA1P7 5’-ctgcaatgtgtactcagagc-3’. The amplified fragment was cloned as described for the HMGA1P6 constructs. All the generated vectors were confirmed by sequencing. The Renilla luciferase vector (pRL-CMV), for transient transfection efficiency, was purchased from Promega. The 3′ UTR region of the HMGA1 gene has been previously described [34].
Protein extraction, western blotting and antibodies
Protein extraction and Western blotting were performed as previously described [49]. The primary antibodies used were anti-EZH2 (AC22) and anti-Cyclin D2 (2924) from Cell Signaling; anti-Actin (sc-1615), anti-Vinculin (sc-7649), anti-γ-Tubulin (sc-17787), and anti-E2f1 (sc-193) from Santa Cruz Biotechnology; anti-VEGF (ab46154) from Abcam. Antibodies versus the HMGA1 and HMGA2 proteins are described elsewhere [50, 51]. Blots were visualized by using the Western blotting detection reagents (GE Healthcare).
Cell migration and invasion assay
Cell migration and invasion experiments were performed as previously described [44].
Dual-luciferase reporter assay
For dual-luciferase reporter assay, 3 × 105 HEK293 cells were co-transfected in 6-well plates with the pGL3-HMGA1P6 or the pGL3-HMGA1P7 luciferase reporter vectors, together with the Renilla luciferase plasmid and miRNA precursors or a control no-targeting scrambled oligonucleotides (Ambion), using siPORT neoFX Transfection Agent (Ambion). The pRL-TK control vector expressing Renilla luciferase (Promega) was used for normalization of cell number and transfection efficiency. Luciferase activity was measured 48 hours after transfection using the Dual-Luciferase Reporter Assay System (Promega) with a Lumat LB 9507 apparatus (Berthold Technologies).
Flow cytometric analysis
HEK293 cells were transfected with HMGA1P6, HMGA1P7 and the empty vector, and analysed by flow cytometry after 48 hours of growth under normal culture conditions. Primary MEFs were obtained from 12.5-day-old embryos. The MEFs were minced and used to establish single cell suspensions and then analysed by flow cytometry after 48 hours of growth under normal culture conditions. Briefly, cells were harvested in PBS containing 2 mmol/l EDTA, washed once with PBS, and fixed for 2 hours in cold ethanol (70%). Fixed cells were washed once in PBS and treated with 40 μg/ml RNase A in PBS for 30 minutes. They were then washed once in PBS and stained with 50 μg/ml propidium iodide (Roche). Stained cells were analysed with a fluorescence activated cell sorter (FACS) Calibur (Becton-Dickinson), and the data were analysed using a mod-fit cell cycle analysis programme.
Generation and genotyping of mutant mice
The 3.5 kb HMGA1P7 of the pCAG-HMGA1P7 expression plasmid was excised with SalI & HindIII restriction endonucleases by cleaving 10 μg of the plasmid. The fragment was purified from SeaKem GTG agarose (avoiding exposure to UV light) using the Qbiogene Geneclean Spin kit, then dialysed 24 h against 2 l microinjection buffer (10 mM Tris.HCl pH 7.2, 0.1 mM EDTA), and diluted to a concentration of 4 ng/μl. The DNA was injected in three sessions into C57BL/6N-derived zygotes. For this purpose, C57BL/6N female mice (bred at PolyGene from parents obtained from Charles River) were superovulated at 28-34 days of age and mated in the PolyGene mouse facility to C57BL/6N breeder males, originally also obtained from Charles River. Injected zygotes were cultivated overnight and transferred into pseudopregnant B6CBAF1 females, also from Charles River. The animals were kept in individually ventilated cages. Injections were performed at the PolyGene labs in Rümlang, Switzerland. Pups were biopsied at weaning and analysed for transgene integration by PCR, using the PCR primer combination: Fw 5’-ggcatgtcccactctatt-3’; Rev 5’-caattcctgcaatgtgtactc-3’. All mice were maintained under standardized nonbarrier conditions in the Laboratory Animal Facility of the Istituto dei Tumori di Napoli (Naples, Italy), and all studies were conducted in accordance with Italian regulations for experimentations on animals.
SA-β-gal assay
4 × 104 MEFs, plated 24 hours before the assay, were washed twice with PBS and immersed in fixation buffer (2% [w/v] formaldehyde, 0.2% [w/v] glutaraldehyde in PBS) for 7 minutes. After 3 additional PBS washes, the cells were allowed to stain overnight in staining solution (40 mM citric acid/sodium phosphate, pH 6.0; 150 mM NaCl; 2.0 mM MgCl2; 1 mg/ml X-gal) at 37°C without CO2 to avoid changes in pH. The next day, the staining solution was replaced with PBS, and the stained and unstained cells were counted by light microscopy (at least 24 fields).
Microarray analyses
RNAs extracted from HMGA1P7 transgenic and WT MEFs (two biological replicates for each sample) were hybridized to the Affymetrix GeneChip Mouse Gene 2.0 ST oligonucleotide arrays. Hybridization, washing, staining, scanning, and data analysis were performed by the Affymetrix Microarray Unit at the IFOM-IEO campus, Milan, Italy, according to the manufacturer’s instructions. Data were analyzed using Partek Genomics Suite version 6.6. Transcripts showing a significant fold change variation (p<0.05) were examined.
Statistical analysis
Data were analyzed using a two-sided unpaired Student’s t test (GraphPad Prism, GraphPad Software, Inc.). Values of P<0.05 were considered statistically significant. The mean +/− s.d. of three or more independent experiments is reported. Regression analyses and correlation coefficients were generated using GraphPad Prism, GraphPad Software, Inc.
ACKNOWLEDGMENTS
We thank Mario Berardone for the art-work and Jean Ann Gilder (Scientific Communication srl) for substantive text editing.
GRANT AND FUND SUPPORT
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC-IG 5346), the Project “Invecchiamento” of the National Research Council (CNR).
AUTHOR CONTRIBUTIONS
F.E. and A.F. conceived and designed the study. F.E., with help from M.D.M., M.G.P., F.F., M.T., A.F., G.M.P. and A.F., performed all experiments, and F.E., M.D.M and A.F. analyzed most data. F.E. and A.F. wrote the manuscript with contributions from all authors.
AUTHOR INFORMATION
The authors declare no competing financial interests.
REFERENCES
1. Johnson KR, Lehn DA and Reeves R. Alternative processing of mRNAs encoding mammalian chromosomal high-mobility-group proteins HMG-I and HMG-Y. Mol Cell Biol. 1989; 9:2114–2123. doi:10.1128/MCB.9.5.2114.
2. Grosschedl R, Giese K and Pagel J. HMG domain proteins: architectural elements in the assembly of nucleoprotein structures. Trends Genet. 1994; 10:94–100. doi:10.1016/0168-9525(94)90232-1.
3. Thanos D and Maniatis T. The high mobility group protein HMG I(Y) is required for NF-κB-dependent virus induction of the human IFN-β gene. Cell. 1992; 71:777–789. doi:10.1016/0092-8674(92)90554-P.
4. Zhou X, Benson KF, Ashar HR and Chada K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature. 1995; 376:771–774. doi:10.1038/376771a0.
5. Chiappetta G, Avantaggiato V, Visconti R, Fedele M, Battista S, Trapasso F, Merciai BM, Fidanza V, Giancotti V, Santoro M, Simeone and Fusco A. High level expression of the HMGA1 gene during embryonic development. Oncogene. 1996; 13:2439–2446.
6. Pierantoni GM, Agosti V, Fedele M, Bond H, Caliendo I, Chiappetta G, Lo Coco F, Pane F, Turco MC, Morrone G, Venuta S and Fusco A. High-mobility group A1 proteins are overexpressed in human leukaemias. Biochem J. 2003; 372:145–150.
7. Chieffi P, Battista S, Barchi M, Di Agostino S, Pierantoni GM, Fedele M, Chiariotti L, Tramontano D and Fusco A. HMGA1 and HMGA2 protein expression in mouse spermatogenesis. Oncogene. 2002; 21:3644–3650.
8. Fusco A and Fedele M. Roles of the HMGA proteins in cancer. Nat Rev Cancer. 2007; 7:899–910. doi:10.1038/nrc2271.
9. Pegoraro S, Ros G, Piazza S, Sommaggio R, Ciani Y, Rosato A, Sgarra R, Del Sal G and Manfioletti G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget. 2013; 4:1293–1308.
10. Berlingieri MT, Manfioletti G, Santoro M, Bandiera A, Visconti R, Giancotti V and Fusco A. Inhibition of HMGI-C protein synthesis suppresses retrovirally induced neoplastic transformation of rat thyroid cells. Mol Cell Biol. 1995; 15:1545–1553.
11. Berlingieri MT, Pierantoni GM, Giancotti V, Santoro M and Fusco A. Thyroid cell transformation requires the expression of the HMG1 proteins. Oncogene. 2002; 21:2971–2980. doi:10.1038/sj/onc/1205368.
12. Scala S, Portella G, Fedele M, Chiappetta G and Fusco A. Adenovirus mediated suppression of the HMGI(Y) protein synthesis as a potential therapy of human malignant neoplasias. Proc Natl Acad Sci USA. 2000; 97:4256–4261. doi:10.1073/pnas.070029997.
13. Wood LJ, Maher JF, Bunton TE and Resar LM. The oncogenic properties of the HMG-I gene family. Cancer Res. 2000; 60:4256–4261.
14. Reeves R, Edberg DD and Li Y. Architectural transcription factor HMGI (Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol Cell Biol. 2001; 21:575–594. doi:10.1128/MCB.21.2.575-594.2001.
15. Xu Y, Sumter TF, Bhattacharya R, Tesfaye A, Fuchs EJ, Wood LJ, Huso DL and Resar LM. The HMG-I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res. 2004; 64:3371–3375. doi:10.1158/0008-5472.CAN-04-0044.
16. Hillion J, Dhara S, Sumter TF, Mukherjee M, Di Cello F, Belton A, Turkson J, Jaganathan S, Cheng L, Ye Z, Jove R, Aplan P and Lin YW, et al. The high-mobility group A1a/signal transducer and activator of transcription-3 axis: an achilles heel for hematopoietic malignancies?. Cancer Res. 2008; 68:10121–10127. doi:10.1158/0008-5472.CAN-08-2121.
17. Tesfaye A, Di Cello F, Hillion J, Ronnett BM, Elbahloul O, Ashfaq F, Dhara S, Prochownik E, Tworkoski K, Reeves R, Elbahlouh O, Kowalski J and Bhattacharya R, et al. The high-mobility group A1 gene up-regulates cyclooxygenase 2 expression in uterine tumorigenesis. Cancer Res. 2007; 67:3998–4004. doi:10.1158/0008-5472.CAN-05-1684.
18. Puca F, Colamaio M, Federico A, Gemei M, Tosti N, Bastos AU, Del Vecchio L, Pece S, Battista S and Fusco A. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget. 2014; 5:3234–3245.
19. Vanin EF. Processed pseudogenes: Characteristics and evolution. Annu Rev Genet. 1985; 19:253–272. doi:10.1146/annurev.ge.19.120185.001345.
20. Mighell AJ, Smith NR, Robinson PA and Markham AF. Vertebrate pseudogenes. FEBS Lett. 2000; 468:109–114. doi:10.1016/S0014-5793(00)01199-6.
21. Harrison PM, Hegyi H, Balasubramanian S, Luscombe NM, Bertone P, Echols N, Johnson T and Gerstein M. Molecular fossils in the human genome: Identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res. 2002; 12:272–280. doi:10.1101/gr.207102.
22. Balakirev ES and Ayala FJ. Pseudogenes: Are they “junk” or functional DNA?. Annu Rev Genet. 2003; 37:123–151. doi:10.1146/annurev.genet.37.040103.103949.
23. Zhang Z, Harrison PM, Liu Y and Gerstein M. Millions of years of evolution preserved: A comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 2003; 13:2541–2558. doi:10.1101/gr.1429003.
24. Zhang Z and Gerstein M. Large-scale analysis of pseudogenes in the human genome. Curr Opin Genet Dev. 2004; 14:328–335. doi:10.1016/j.gde.2004.06.003.
25. Zheng D, Zhang Z, Harrison PM, Karro J, Carriero N and Gerstein M. Integrated pseudogene annotation for human chromosome 22: Evidence for transcription. J Mol Biol. 2005; 349:27–45. doi:10.1016/j.jmb.2005.02.072.
26. D’Errico I, Gadaleta G and Saccone C. Pseudogenes in metazoa: origin and features. Brief Funct Genomic Proteomic. 2004; 3:157–167. doi:10.1093/bfgp/3.2.157.
27. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ and Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010; 465:1033–1038. doi:10.1038/nature09144.
28. Arvey A, Larsson E, Sander C, Leslie CS and Marks DS. Target mRNA abundance dilutes microRNA and siRNA activity. Mol Syst Biol. 2010; 6:363 doi:10.1038/msb.2010.24.
29. Salmena L, Poliseno L, Tay Y, Kats L and Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell. 2011; 146:353–358. doi: 10.1016/j.cell.2011.07.014.
30. Tay Y, Katz L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, Lieberman J, Rigoutsos I and Pandolfi PP. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011; 147:344–357. doi: 10.1016/j.cell.2011.09.029.
31. Palmieri D, D’Angelo D, Valentino T, De Martino I, Ferraro A, Wierinckx A, Fedele M, Trouillas J and Fusco A. Downregulation of HMGA-targeting microRNAs has a critical role in human pituitary tumorigenesis. Oncogene. 2012; 31:3857–3865. doi: 10.1038/onc.2011.557.
32. Li W, Yuan Y, Huang L, Qiao M and Zhang Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Res Clin Pract. 2012; 96:187–195. doi: 10.1016/j.diabres.2011.12.028.
33. Sehic A, Risnes S, Khuu C, Khan QE and Osmundsen H. Effects of in vivo transfection with anti-miR-214 on gene expression in murine molar tooth germ. Physiol Genomics. 2011; 43:488–498. doi:10.1152/physiolgenomics.00248.
34. D’Angelo D, Palmieri D, Mussnich P, Roche M, Wierinckx A, Raverot G, Fedele M, Croce CM, Trouillas J and Fusco A. Altered MicroRNA Expression Profile in Human Pituitary GH Adenomas: Down-Regulation of miRNA Targeting HMGA1, HMGA2, and E2F1. J Clin Endocrinol Metab. 2012; 97:E1128–38. 10.1210/jc.2011-3482.
35. Hua Z, Lv Q, Ye W, Wong CKA, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB and Zhang Y. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006; 1:e116 doi:10.1371/journal.pone.0000116.
36. Huang SD, Yuan Y, Zhuang CW, Li BL, Gong DJ, Wang SG, Zeng ZY and Cheng HZ. MicroRNA-98 and microRNA-214 post-transcriptionally regulate enhancer of zeste homolog 2 and inhibit migration and invasion in human esophageal squamous cell carcinoma. Mol Cancer. 2012; 11:51 doi:10.1186/1476-4598-11-51.
37. Day BW, Stringer BW, Al-Ejeh F, Ting MJ, Wilson J, Ensbey KS, Jamieson PR, Bruce ZC, Lim YC, Offenhauser C, Charmsaz S, Cooper LT and Ellacott JK, et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell. 2013; 23:238–248. doi:10.1016/j.ccr.2013.01.007.
38. Valente V, Serafim RB, de Oliveira LC, Adorni FS, Torrieri R, Tirapelli DP, Espreafico EM, Oba-Shinjo SM, Marie SK, Paçò-Larson ML and Carlotti CG. Modulation of HJURP (Holliday Junction-Recognizing Protein) levels is correlated with glioblastoma cells survival. PLoS One. 2013; 8:e62200 doi:10.1371/journal.pone.0062200.
39. Wang Q, Zhao ZB, Wang G, Hui Z, Wang MH, Pan JF and Zheng H. High expression of KIF26B in breast cancer associates with poor prognosis. PLoS One. 2013; 8:e61640 doi: 10.1371/journal.pone.0061640.
40. Moon E, Lee R, Near R, Weintraub L, Wolda S and Lerner A. Inhibition of PDE3B augments PDE4 inhibitor-induced apoptosis in a subset of patients with chronic lymphocytic leukemia. Clin Cancer Res. 2002; 8:589–595.
41. Hsu A, Zhang W, Lee JF, An j, Ekambaram P, Liu J, Honn KV, Klinge CM and Lee MJ. Sphingosine-1-phosphate receptor-3 signaling up-regulates epidermal growth factor receptor and enhances epidermal growth factor receptor-mediated carcinogenic activities in cultured lung adenocarcinoma cells. Int J Oncol. 2012; 40:1619–1626. doi:10.3892/ijo.2012.1379.
42. Kumar MS, Armenteros-Monterroso E, East P, Chakravorty P, Matthews N and Winslow MM. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature. 2014; 505:212–217. doi:10.1038/nature12785.
43. Chiappetta G, Tallini G, De Biasio MC, Manfioletti G, Martinez-Tello FJ, Pentimalli F, de Nigris F, Mastro A, Botti G, Fedele M, Berger N, Santoro M, Giancotti V and Fusco A. Detection of high mobility group I HMGI(Y) protein in the diagnosis of thyroid tumors: HMGI(Y) expression represents a potential diagnostic indicator of carcinoma. Cancer Res. 1998; 58:4193–4198.
44. Borbone E, Troncone G, Ferraro A, Jasencakova Z, Stojic L, Esposito F, Horning N, Fusco A and Orlando V. Enhancer of zeste homolog 2 overexpression has a role in the development of anaplastic thyroid carcinomas. J Clin Endocrinol Metab. 2011; 96:1029–1038. doi:10.1210/jc.2010-1784.
45. Palmieri D, Valentino T, De Martino I, Esposito F, Cappabianca P, Wierinckx A, Vitiello M, Lombardi G, Colao A, Trouillas J, Pierantoni GM, Fusco A and Fedele M. PIT1 upregulation by HMGA proteins has a role in pituitary tumorigenesis. Endocr Relat Cancer. 2012; 19:123–135. doi: 10.1530/ERC-11-0135.
46. Narita M, Narita M, Krizhanovsky V, Nuñez S, Chicas A, Hearn SA, Myers MP and Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006; 126:503–514. doi:10.1016/j.cell.2006.05.052.
47. Federico A, Forzati F, Esposito F, Arra C, Palma G, Barbieri A, Palmieri D, Fedele M, Pierantoni GM, De Martino I and Fusco A. Hmga1/Hmga2 double knock-out mice display a “superpygmy” phenotype. Biol Open. 2014; doi: 10.1242/bio.20146759.
48. Toledo F and Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006; 6:909–923. doi:10.1038/nrc2012.
49. Esposito F, Boscia F, Gigantino V, Tornincasa M, Fusco A, Franco R and Chieffi P. The high-mobility group A1-estrogen receptor β nuclear interaction is impaired in human testicular seminomas. J Cell Physiol. 2012; 227:3749–3755. doi:10.1002/jcp.24087.
50. Esposito F, Tornincasa M, Chieffi P, De Martino I, Pierantoni GM and Fusco A. High-mobility group A1 proteins regulate p53-mediated transcription of Bcl-2 gene. Cancer Res. 2010; 70:5379–5388. doi:10.1158/0008-5472.CAN-09-4199.
51. Piscuoglio S, Zlobec I, Pallante P, Sepe R, Esposito F, Zimmermann A, Diamantis I, Terracciano L, Fusco A and Karamitopoulou E. HMGA1 and HMGA2 protein expression correlates with advanced tumour grade and lymph node metastasis in pancreatic adenocarcinoma. Histopathology. 2012; 60:397–404. doi: 10.1111/j.1365-2559.2011.04121.x.