
INTRODUCTION
CML, primarily induced by the expression of an abnormal BCR/ABL fusion tyrosine kinase, is characterized by a gradual progression from a benign chronic phase displaying hyperproliferation of normally maturing myeloid cells to a lethal blast crisis, in many cases resembling AML where myeloid differentiation is impaired. Although treatment of chronic-phase patients with tyrosine kinase inhibitors (TKIs) has been shown to induce disease remission and to prevent progression in most cases, a significant population of patients still developed blast crisis for which treatment options are very limited. Targeting the self-renewal of leukemia-initiating cells (LICs) represents a promising strategy for the treatment of leukemia; however, the self-renewal mechanisms for LICs in CML myeloid blast crisis remain unclear. Different from the chronic phase, where the LICs likely are derived directly from HSCs [1, 2], an origin from the more differentiated granulocyte-macrophage progenitors (GMPs) has been suggested for LICs in CML myeloid blast crisis as the GMP population in this phase is significantly expanded and displays increased self-renewal capability [3]. While abnormal activation of a number of genes/pathways has been implicated in the self-renewal of GMPs in myeloid blast crisis, including WNT/β-CATENIN, RUNX1, RUNX1/EVI1, RUNX1/PRDM16, GATA-2, and Msi2 [3–8], direct evidence in supporting this concept has come from studies on several genes including NUP98/HOXA9, Hes1 and Setbp1, showing that they can cooperate with BCR/ABL to transform GMPs into LICs for development of CML myeloid blast crisis in vivo [2, 9, 10]. Identification and characterization of downstream targets of these genes should lead to a better understanding of the generation and self-renewal mechanisms of LICs of CML myeloid blast crisis.
Homeobox transcription factor genes Hoxa9 and Hoxa10 have been identified as critical activation targets of Setbp1 for its ability to induce self-renewal of myeloid progenitors and development of CML myeloid blast crisis [10]. Both Hoxa9 and Hoxa10 are validated oncogenes capable of inducing AMLs [11, 12]. Past studies also have suggested that overexpression of either gene may stimulate the self-renewal of myeloid progenitors. Immortalized myeloid progenitor lines can be generated by transducing mouse bone marrow progenitors with retrovirus expressing Hoxa9 [13]. Overexpression of either gene in GMPs also has been found to induce serial re-plating activity [14]. In addition, both are also key targets of MLL translocation genes, which have been shown to induce self-renewal and transformation of GMPs in vivo [14]. Therefore, it is possible that overexpression of HOXA9 or HOXA10 alone may be sufficient to cooperate with BCR/ABL to induce transformation of GMPs, leading to CML progression into myeloid blast crisis; however, this hypothesis has not been tested.
Here we show that overexpression of Hoxa9 or Hoxa10 is sufficient to confer unlimited self-renewal capability to myeloid progenitors in vitro and to cooperate with BCR/ABL to induce transformation of GMPs in vivo, leading to the development of myeloid blast crisis. Our study also identifies Myb as a shared critical target of Hoxa9 and Hoxa10 for their self-renewal inducing activity. Our findings on the capability of Myb to drive CML myeloid blast crisis development and the prevalent overexpression of MYB in CML myeloid blast crisis patients further suggest that inhibition of MYB activity could be an effective strategy for inhibiting the self-renewal of LICs in CML myeloid blast crisis.
RESULTS
Both Hoxa9 and Hoxa10 are capable of inducing immortalization of myeloid progenitors
We have found previously that overexpression of Setbp1 can confer self-renewal capability to myeloid progenitors, and that this activity of Setbp1 is critically dependent on its activation of Hoxa9 and Hoxa10. To further explore the mechanism(s) underlying Setbp1-induced self-renewal, we tested whether overexpression of Hoxa9 or Hoxa10 alone also is sufficient to confer unlimited self-renewal capability to myeloid progenitors by first assessing their ability to immortalize myeloid progenitors in culture. We transduced primary murine myeloid progenitors with MSCV retroviruses expressing Hoxa9 cDNA (MSCV-Hoxa9-PGK-Neo) or Hoxa10 cDNA (MSCV-Hoxa10-PGK-Puro), and subsequently passaged the cells in culture in the presence of stem cell factor (SCF) and interleukin-3 (IL-3) as described previously [10]. Infections were carried out using a low viral titer of 1 × 105 cfu, which was shown previously to be insufficient for empty MSCV retroviral vectors to induce immortalization through insertional mutagenesis [15]. As controls, cells infected by same titers of empty MSCV viruses (MSCV-PGK-Neo and MSCV-PGK-Puro) were passaged together. At two weeks after infection, significant reduction in proliferation and increase in macrophage differentiation were observed in the cultures of empty virus-infected cells. In contrast, cultures from Hoxa9 or Hoxa10 virus-transduced cells were still dominated by proliferating myeloid cells. These cells are immortalized as they can be continuously passaged for six months until the experiments are terminated (Figure 1). Consistent with their myeloid lineage, these cells express myeloid markers including Gr-1 and Mac-1, and are negative for markers of lymphoid and erythroid lineages (Figure 1). Similar to progenitors immortalized by Setbp1, these cells are dependent on IL-3 for their proliferation (data not shown). Immortalized myeloid progenitor lines with similar phenotypes also could be established from purified mouse GMPs and 5-FU-treated bone marrow progenitors by transduction with Hoxa9 and Hoxa10 viruses (data not shown). These results suggest that either Hoxa9 or Hoxa10 alone can induce immortalization of myeloid progenitors.
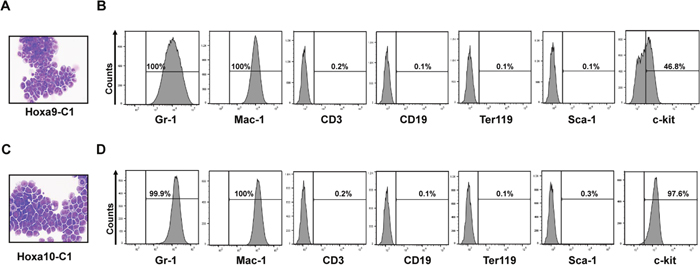
Figure 1: Constitutive expression of Hoxa9 or Hoxa10 alone is capable of inducing immortalization of myeloid progenitor cells. Representative Wright-Giemsa staining and FACS analysis of indicated marker expression of cells immortalized by transduction with retroviruses expressing either Hoxa9 (A and B) or Hoxa10 (C and D) after passaging in liquid media containing SCF and IL-3 for 2 months.
Hoxa9 cooperates with BCR/ABL to induce transformation of GMPs in vivo
The ability of Hoxa9 and Hoxa10 to immortalize myeloid progenitors in vitro suggested that they may also help confer limitless self-renewal potential to GMP-derived LICs in CML myeloid blast crisis. We first tested this hypothesis with Hoxa9. We co-transduced GMPs (purified from C57BL/6 mice) with MSCV retrovirus expressing BCR/ABL (MSCV-BCR/ABL-IRES-GFP) and Hoxa9-expressing MSCV-Hoxa9-PGK-Neo virus. Co-transduced GMPs were then transplanted into lethally irradiated congenic B6-Ly5.2 recipient mice along with supporting bone marrow. GMPs infected singly by the same titer of either virus were transplanted into mice as controls. Interestingly, all mice receiving co-transduced GMPs became sick within the first four weeks of transplantation with dramatically enlarged spleens (Figure 2A and data not shown). Cytospin analysis of the bone marrow and spleen cells of the moribund mice showed domination of abnormal cells resembling myeloid blasts (Figure 2B), which represent 32 ± 3.2% (Mean ± SD) of all nucleated cells in the bone marrow, suggesting the development of CML myeloid blast crisis. In support of this notion, more than 90% of these cells in the bone marrow were positive for myeloid markers Gr-1, and negative for CD3, CD19 and Ter119, which are markers for T cells, B cells, and erythroid cells, respectively (Figure 2C). Five to 11% of the cells in the bone marrow and spleen also were positive for Sca-1, while less than 1% of the cells were positive for c-kit (Figure 2C). Pathological examination of these mice further showed extensive spleen and liver infiltrations by the abnormal cells (Figure 2D). As expected, these cells expressed high levels of BCR/ABL and Hoxa9 mRNAs (Supplementary Figure 1). Transplantation of 1 × 106 spleen cells from these moribund mice into secondary recipient mice also led to development of same disease in four weeks (Figure 2A). We also found that neomycin-resistant colonies formed by the spleen cells are positive for GFP (data not shown), further suggesting the disease development required both Hoxa9 and BCR/ABL expression within the same cells. In contrast, consistent with previous studies demonstrating the inability of BCR/ABL to induce self-renewal and transformation of GMPs [1, 2, 9, 10], mice transplanted with GMPs that were singly transduced with BCR/ABL virus remained healthy for four months (Figure 2A). Mice receiving cells transduced by Hoxa9 virus alone also did not develop any leukemias, suggesting Hoxa9 by itself is not sufficient to transform GMPs. In combination, these results suggest that Hoxa9 is capable of cooperating with BCR/ABL to induce development of CML myeloid blast crisis.
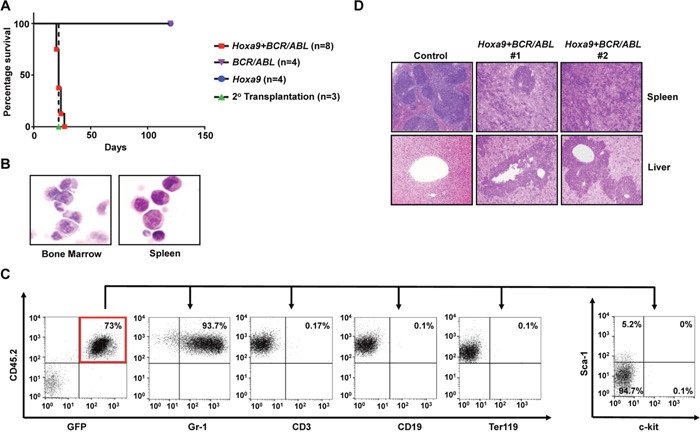
Figure 2: Hoxa9 cooperates with BCR/ABL to induce development of CML myeloid blast crisis. (A). Survival curves of lethally-irradiated B6-Ly5.2 mice receiving GMPs transduced with MSCV-Hoxa9-PGK-Neo virus alone, MSCV-BCR/ABL-IRES-GFP virus alone, the combination of both viruses, or 1 x 106 spleen cells from primary leukemic mice. (B). Representative cytospin analysis of bone marrow and spleen cells from leukemic mice. (C). Representative FACS analysis of GFP and CD45.2 double positive leukemia cells from the bone marrow of leukemic mice using the indicated antibodies. Numbers represent the percentages of gated events. (D). H&E staining of spleen and liver tissue sections showing leukemic infiltration in two Hoxa9+BCR/ABL leukemic mice in comparison to a healthy control mouse.
Hoxa10 is also capable of cooperating with BCR/ABL to induce GMP transformation in vivo
Utilizing the same approach, we also examined the capability of Hoxa10 to cooperate with BCR/ABL to transform normal GMPs in vivo. Interestingly, we found that all mice receiving GMPs co-transduced by BCR/ABL and Hoxa10 viruses also developed myeloid leukemias, although with longer latencies than leukemias induced by Hoxa9+BCR/ABL (Figure 3A). Again, none of the recipient mice for GMPs singly infected with Hoxa10 or BCR/ABL virus developed leukemia (Figure 3A). Similar to leukemias induced by Hoxa9+BCR/ABL, these leukemias also are characterized by expansion and infiltration of myeloid blasts into the bone marrow, spleen, and liver (Figure 3B and 3D). Myeloid blasts represent 25 ± 1.4% (Mean ± SD) of all nucleated cells in the bone marrow, suggesting development of myeloid blast crisis. Consistent with their myeloid origins, the leukemia cells are also mostly positive for Gr-1 expression and negative for the expression of CD3, CD19, and Ter119 (Figure 3C). Additionally, less than 1% and 6 to 10% of the leukemia cells expressed c-kit and Sca-1, respectively (Figure 3C). Consistent with their co-transduction by BCR/ABL and Hoxa10 viruses, these leukemia cells expressed high levels of BCR/ABL and Hoxa10 (Supplementary Figure 1). These leukemias also are transplantable, as secondary transplantation of the leukemia cells led to development of the same disease in recipient mice in 40 days (Figure 3A). All puromycin-resistant colonies formed by the leukemia cells also were GFP positive (data not shown), suggesting that both need to be expressed in the same cells to induce transformation. These data suggest that overexpression of Hoxa10 is also able to cooperate with BCR/ABL to induce CML myeloid blast crisis.
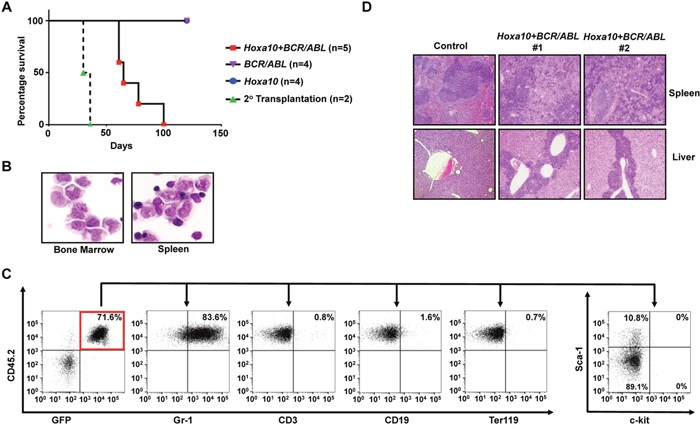
Figure 3: Hoxa10 cooperates with BCR/ABL to induce development of CML myeloid blast crisis. (A). Survival curves of lethally-irradiated B6-Ly5.2 mice receiving GMPs transduced with MSCV-Hoxa10-PGK-Puro virus alone, MSCV-BCR/ABL-IRES-GFP virus alone, the combination of both viruses, or 1 × 106 spleen cells from primary leukemic mice. (B). Representative cytospin analysis of bone marrow and spleen cells from leukemic mice. (C). Representative FACS analysis of GFP and CD45.2 double positive leukemia cells from the bone marrow of leukemic mice using the indicated antibodies. Numbers represent the percentages of gated events. (D). H&E staining of spleen and liver tissue sections showing leukemic infiltration in two Hoxa10+BCR/ABL leukemic mice in comparison to a healthy control mouse.
Myb expression is essential for Hoxa9 and Hoxa10-induced self-renewal
The common capability of Hoxa9 and Hoxa10 to induce the self-renewal of myeloid progenitors suggests that they may activate the same target(s) critical for this process. Interestingly, we found that the expression of Myb, a helix-turn-helix transcription factor important for hematopoietic development and a critical target for Hoxa9-induced myeloid leukemia development [16], was up-regulated to similar levels in Hoxa9+BCR/ABL and Hoxa10+BCR/ABL leukemias (Supplementary Figure 2), suggesting that Myb could be a shared critical target of Hoxa9 and Hoxa10 in their stimulation of self-renewal. Therefore, we examined whether continuous expression of Myb is required for the self-renewal of myeloid progenitors immortalized by Hoxa9 and Hoxa10. As expected, Myb knockdowns using two different Myb-specific lentiviral shRNAs dramatically reduced the colony-forming potential of Hoxa9-immortalized cells (Figure 4A). Similarly, infection with the same lentiviral shRNAs also markedly inhibited the colony formation by Hoxa10-immortalized cells (Figure 4A), suggesting that Myb expression is also critical for Hoxa10-induced self-renewal. Consistent with this idea, cytospin analysis revealed significant differentiation of both Hoxa9 and Hoxa10-immortalized cells into neutrophils after Myb knockdown (Figure 4B), which was confirmed by significantly increased expression of myeloid differentiation markers including Cd11b and Lyz2 (Figure 4C). To further test the possibility that Hoxa10 could directly activate Myb transcription, we transduced 5-FU-treated mouse bone marrow progenitors with Hoxa10-expressing (MSCV-Hoxa10-PGK-puro), Hoxa9-expressing (MSCV-Hoxa9-PGK-puro) or empty virus (MSCV-PGK-puro), and compared the Myb expression levels in the transduced cells at 72hrs after infection following the elimination of non-transduced cells by selection with puromycin at 48hrs. Significant and comparable increases in Myb mRNA levels were observed in both Hoxa9- and Hoxa10-infected cells when compared to control cells infected with empty virus (Figure 4D), suggesting that Myb could be a direct transcriptional target of Hoxa10. In combination, these data suggest that Myb is a downstream target of both Hoxa9 and Hoxa10, and also a critical mediator of their self-renewal promoting activity.
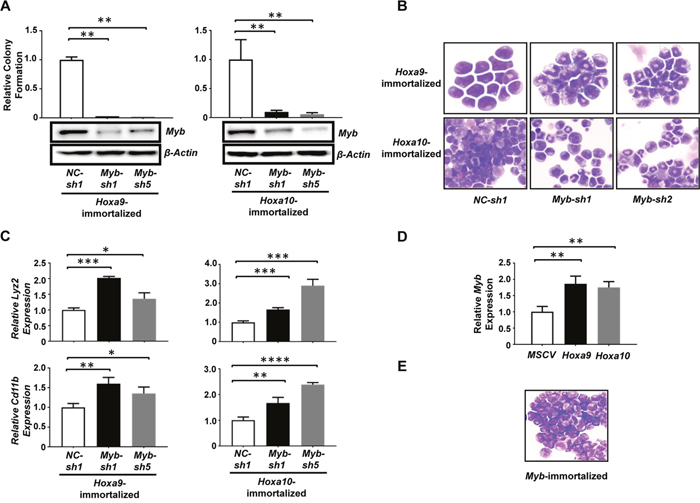
Figure 4: Myb is a critical mediator of Hoxa9/Hoxa10-induced self-renewal of myeloid progenitors. (A). Upper panels, colony-forming potential of myeloid progenitors immortalized by Hoxa9 (left) and Hoxa10 (right) plated 48hrs after infection with lentiviral shRNA targeting Myb (Myb-sh1, Myb-sh5) or control shRNA (NC-sh1). Lower panel, Western blotting analysis of Myb protein at 72hrs after infection in the same cells corresponding to the upper panel. (B). Cytospin analyses of Hoxa9 (top) and Hoxa10-immortalized cells (lower panels) 72hrs after infection with the indicated lentiviral shRNAs. (C). Real-time PCR analysis of Lyz2 and Cd11b mRNA levels in either Hoxa9 or Hoxa10-immortalized myeloid progenitor cells 72hrs after infection with the indicated lentiviral shRNAs. Relative expression levels were calculated by normalizing to β-Actin mRNA levels in the same sample and also in cells infected by NC-sh1. (D). Real-time PCR analysis of Myb mRNA levels in mouse 5-FU treated BM progenitor cells 72hrs after infection with the indicated retrovirus. Non-transduced cells were eliminated by selection with 2 ug/ml of puromycin added at 48 hrs after infection. Relative expression levels were calculated by normalizing to β-Actin mRNA levels in the same sample and also in cells infected by empty virus. The mean and SD of each relative expression level is shown. (E). Representative cytospin analysis of Myb-immortalized cells. *, P <0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (two-tailed Student’s t test).
Myb is capable of inducing the self-renewal of myeloid progenitors in vitro and in vivo
Myb has been indicated as an important self-renewal regulator for normal hematopoietic stem cells [17]. A role of Myb overexpression in promoting self-renewal was also suggested by the generation of an immortalized cell line from mouse fetal liver cells transduced by a mutant retrovirus expressing wild-type Myb [18]. To further understand the role of Myb activation in Hoxa9- and Hoxa10-induced myeloid progenitor self-renewal, we next tested if ectopic expression of Myb also could induce self-renewal of primary myeloid progenitors. Alternative splicing of MYB transcript results in the production of two isoforms: a predominantly expressed 75KDa form and a less expressed 89KDa form [19]. The 75KDa form also is predominantly expressed in Hoxa9- and Hoxa10-immortalized myeloid progenitors and Hoxa9/Hoxa10+BCR/ABL leukemia cells (data not shown). Therefore, we cloned the cDNA for the 75KDa Myb isoform into MSCV and tested the ability of the resulting virus (MSCV-Myb-IRES-GFP) to induce self-renewal of myeloid progenitors in culture under the same condition tested for Hoxa9 and Hoxa10 viruses. We found that expression of this Myb isoform is able to efficiently induce immortalization of myeloid progenitors (Figure 4E and Supplementary Figure 3), suggesting that it could confer unlimited self-renewal capability to myeloid progenitors. Cooperation between Myb and Abl activation in AML development has been suggested previously [20]. We next examined if Myb could cooperate with BCR/ABL in vivo to cause leukemic transformation of GMPs. Similar to our earlier studies, purified GMPs from C57BL/6 mice were co-transduced, with BCR/ABL and Myb virus, and subsequently transplanted into lethally irradiated B6-Ly5.2 mice. GMPs transduced by BCR/ABL or Myb virus alone were transplanted as controls. Interestingly, six out of eight mice receiving GMPs co-transduced with BCR/ABL- and Myb-expressing viruses developed myeloid leukemia in six months after transplantation while mice transplanted with GMP cells that were singly transduced with either BCR/ABL or Myb expressing retrovirus remained healthy during the same period (Figure 5A). Resembling mice with Hoxa9+BCR/ABL and Hoxa10+BCR/ABL leukemias, the bone marrow, spleen and liver of the moribund mice were infiltrated by immature leukemia blasts (Figure 5B and 5D), representing 31 ± 5.7% (Mean ± SD) of all nucleated cells in the bone marrow. These leukemia cells also displayed a similar lineage marker expression profile to that of Hoxa9+BCR/ABL and Hoxa10+BCR/ABL leukemia cells and were transplantable (Figure 5C and 5A). As expected from the co-transduction, these cells expressed significantly elevated levels of Myb and BCR/ABL mRNA (Supplementary Figure 4). Both Myb and BCR/ABL proviruses were detected in single methylcellulose colonies formed by the leukemia cells by PCR (data not shown), further suggesting that the transformation requires their expression within the same cell. In addition, these leukemia cells did not express detectable levels of Hoxa9 or Hoxa10 protein (Supplementary Figure 5), suggesting that their transformation is independent of Hoxa9 or Hoxa10 activation. Taken together, these results indicate that overexpression of Myb is also capable of inducing CML progression by inducing self-renewal of GMPs, although with a lower potency than Hoxa9 and Hoxa10.
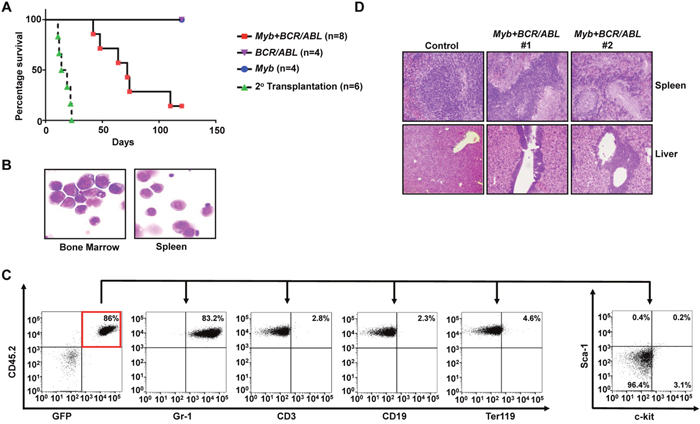
Figure 5: Myb is capable of cooperating with BCR/ABL to induce development of CML myeloid blast crisis. (A). Survival curves of lethally-irradiated C57BL6-Ly5.2 mice receiving GMPs transduced with MSCV-Myb-IRES-GFP virus alone, MSCV-BCR/ABL-IRES-GFP virus alone, the combination of both viruses, or 1 × 106 spleen cells from primary leukemic mice. (B). Representative cytospin analysis of bone marrow and spleen cells from leukemic mice. (C). Representative FACS analysis of GFP and CD45.2 double positive leukemia cells from the bone marrow of leukemic mice using the indicated antibodies. Numbers represent the percentages of gated events. (D). H&E staining of spleen and liver tissue sections showing leukemic infiltration in two Myb+BCR/ABL leukemic mice in comparison to a healthy control mouse.
Increased Myb expression is detected in CML blast crisis patients
The ability of Myb to cooperate with BCR/ABL to induce transformation of GMPs into LICs in vivo suggests that its activation may play an important role in the progression of human CML. To test this hypothesis, we examined MYB mRNA levels in bone marrow aspirates of normal and CML patients at different stages of CML progression. Relatively low levels of MYB mRNA were observed in the BM of normal human volunteers, chronic phase CML patients, and accelerated phase patients. In contrast, significantly higher levels of MYB mRNA were detected in all myeloid blast crisis CML patients (Figure 6). This result suggests a possibility that MYB overexpression could be a major contributor to LIC self-renewal in blast crisis progression of CML patients.
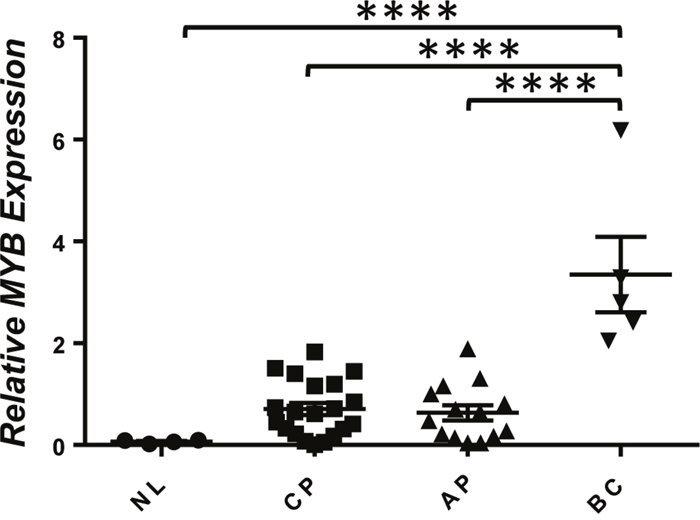
Figure 6: Increased expression of MYB in CML blast crisis patients. Real-time RT-PCR analysis of MYB mRNA levels in total RNA isolated from whole bone marrow of healthy volunteers (NL) and CML chronic phase (CP), advanced phase (AP), and blast crisis phase (BC) patients. Relative expression levels were calculated by normalizing to BCR mRNA levels in the same sample. ****, P < 0.0001 (two-tailed Student’s t test).
DISCUSSION
The molecular mechanisms underlying CML myeloid blast crisis development are poorly understood. It has been suggested that LICs in these leukemias are derived from GMPs with unlimited self-renewal capability [3]. BCR/ABL alone is not capable of stimulating the self-renewal of GMPs [1], suggesting that additional mutations must be acquired to confer self-renewal capability to chronic phase GMPs and to drive disease progression into myeloid blast crisis. We have shown previously that overexpression of Setbp1 can cooperate with BCR/ABL to induce development of CML myeloid blast crisis by stimulating the self-renewal of GMPs [10]. To gain further insights into the mechanisms inducing GMP self-renewal in CML blast crisis, we tested whether overexpression of Hoxa9 or Hoxa10, both critical targets of Setbp1, is sufficient to confer unlimited self-renewal capability to myeloid progenitors and to induce the development of CML blast crisis. Previous studies have shown that purified mouse GMPs transduced with retrovirus expressing either Hoxa9 or Hoxa10 could be serially plated multiple times in colony assays [14]; however, the full potential of their expression in inducing long-term self-renewal of myeloid progenitors in vitro and in vivo remained to be determined. Our results strongly suggest that overexpression of either homeobox gene alone is adequate to confer unlimited self-renewal capability to myeloid progenitors in vitro as transduction of these cells with even low titers of retroviral vectors expressing either gene efficiently induced their immortalization in culture. In addition, as normal mouse GMPs differentiate quickly and do not engraft in recipient mice after transplantation [21], the leukemic transformation of these cells induced by co-expression of Hoxa9/Hoxa10 and BCR/ABL in our transplantation studies further suggests that both Hoxa9 and Hoxa10 activation also are able to confer unlimited self-renewal capability to chronic phase GMPs in vivo. Although both HOXA9 and HOXA10 were previously found to be overexpressed in CML blast crisis patients [8], their roles in CML progression have been unclear. Our in vivo results strongly identify HOXA9 and HOXA10 as potent drivers of human CML myeloid blast crisis development.
Different from GMP-derived leukemias induced by Setbp1 and BCR/ABL [10], Hoxa9+BCR/ABL and Hoxa10+BCR/ABL leukemias are mostly monoclonal in origin (Supplementary Figure 6), suggesting that additional mutation(s) is likely required for the transformation in both cases. These mutations may be required for further stimulation of proliferation and/or inhibition of apoptosis of GMPs. Our results also suggest that Hoxa9 is more potent than Hoxa10 in inducing CML myeloid blast crisis, as the Hoxa9+BCR/ABL mice developed leukemia with a significantly shorter latency than Hoxa10+BCR/ABL mice. This could be due to less mutations required for GMPs to become transformed after transduction by Hoxa9 and BCR/ABL viruses or the potential inhibitory effect of high levels of Hoxa10 expression on cell proliferation described previously [22].
Myb is a master regulator of normal hematopoietic development [17, 23]. In addition, Myb is a significant contributor to leukemogenesis as it has been shown to be a critical target of multiple known oncogenes in the hematopoietic system, including HOXA9 [16], MLL fusions [24], and recently Setbp1 [25]. Our study additionally identifies Myb as a critical target of Hoxa10 for its self-renewal stimulating activity, as Myb knockdown caused differentiation of Hoxa10-immortalized myeloid progenitors. It remains unclear how Hoxa10 activates Myb transcription. Studies have suggested that Hoxa9 can directly activate Myb transcription through binding to regions of Myb promoter and first intron, and also a recently identified distal regulatory element [26, 27]. It is possible that the same sites could be responsible for mediating the activation by Hoxa10, as Hox proteins are known to recognize similar binding sites [28].
Previous studies have suggested a critical requirement for MYB in the maintenance of CML cells. Inhibition of MYB expression by anti-sense oligonucleotides previously has been shown to reduce the expansion of human CML blast crisis cells in vitro [29]. Genetic reduction in Myb levels also has been found to suppress BCR/ABL-induced CML development [30]. It was unclear, however, whether MYB plays any causal role in driving CML progression. Our bone marrow transplantation studies show for the first time that overexpression of Myb is capable of inducing the development of CML myeloid blast crisis in vivo, when expressed together with BCR/ABL in GMPs. Our in vitro immortalization studies further indicate that this capability of Myb is likely due to its ability to induce self-renewal of myeloid progenitors, which is consistent with its potential function in maintaining the self-renewal of hematopoietic stem cells during normal hematopoietic development [17]. The incomplete penetrance of the leukemia development induced by Myb and BCR/ABL and the monoclonal origins of the resulting leukemias (Supplementary Figure 6) also suggest that additional mutations, potentially with effects of increasing proliferation and/or inhibiting apoptosis, are required for the transformation of GMPs. Significantly higher levels of MYB expression also were detected in all CML myeloid blast crisis patients than chronic and accelerated phase cases examined in our study, further suggesting a possibility that abnormal activation of MYB expression could be a major “driver” for CML progression into myeloid blast crisis by conferring self-renewal capability to GMPs. Therefore, our study also suggest that strategies to block MYB activity may prove effective for inhibiting LIC self-renewal in CML blast crisis, for which effective treatments are lacking.
MATERIALS AND METHODS
Mice
7-12 week-old C57BL/6 and B6-Ly5.2 mice were purchased from Charles River, Frederick, MD. These mice were maintained in the animal facility of Laboratory of Animal Medicine at Uniformed Services University of the Health Sciences (USUHS). All mouse experiments were carried out according to protocols approved by the USUHS Institutional Animal Care and Use Committee.
Patient samples
Primary human cells were collected after signing the informed consent, according to the protocols approved by the Institutional Review Board of City of Hope (COH) or the University of Alabama at Birmingham (UAB), in accordance with assurances filed with the Department of Health and Human Services, and met all requirements of the Declaration of Helsinki.
Retrovirus generation
MSCV-BCR/ABL-IRES-GFP and MSCV-Hoxa9-PGK-Neo viruses have been described previously [11, 31]. For the generation of MSCV-Hoxa10-PGK-Puro and MSCV-Myb-IRES-GFP retroviral constructs, cDNAs for Hoxa10 and the 75 KDa form of Myb were amplified by RT-PCR, confirmed by sequencing, and cloned into MSCV-PGK-Puro and MSCV-IRES-GFP vectors, respectively. The primers utilized were: Hoxa10 S, 5’-CGC GGG ATC Ccc cac aac aat gtc atg ctc -3’; Hoxa10 AS, 5’-CGC GGA ATT CGC GAA AAG ACG TTG TCT GGA AG -3’; Myb S, 5’-CGC GCT CGA GCC TCG CCA TGG CCC GGA GAC -3’; Myb AS, 5’-CGC GGC GGC CGC GGA AAT GTC TCA CAT GAC CA-3’. To generate retroviral construct MSCV-Hoxa9-PGK-Puro, Hoxa9 cDNA was excised from MSCV-Hoxa9-PGK-Neo by XhoI/EcoRI double digestion and subsequently ligated to the same restriction sites in MSCV-PGK-Puro vector. Viruses were produced by the transfection of the Hoxa9, Hoxa10, BCR/ABL, Myb retroviral plasmids into Plat-E cells using Fugene 6 (Promega, Madison, WI). Viral titers were determined by infecting NIH3T3 cells.
Immortalization of myeloid progenitors
Immortalization of myeloid progenitors was carried out as described [10]. Briefly, bone marrow cells harvested from C57BL/6 mice were cultured first in Stemspan medium (Stemcell Technologies Inc., Cambridge, MA) containing mouse SCF, TPO, IGF-2, and human FGF-1 for six days to expand hematopoietic stem cells, and then in IMDM plus 20% heat-inactivated horse serum with mouse SCF and IL-3 (Biolegend, San Diego, CA) for four days to induce production of myeloid progenitors. 5 × 105 resulting cells were then infected with retrovirus (1 × 105 cfu) on plates coated with Retronectin (Takara Bio USA, Mountain View, CA) for 48 hours. Infected cells were then continuously passaged at 1:10 ratio every three days for at least four weeks to assess immortalization of myeloid progenitors.
Retroviral transduction and transplantation of GMPs
GMPs purified from C57BL/6 mice (8-12 weeks-old females) were transduced and subsequently transplanted as described previously. Briefly, freshly sorted GMPs were infected immediately with retrovirus at 4:1 ratio of viral titer to cell number on Retronectin-coated plates in the presence of mouse SCF (100ng/ml) and IL-11(10ng/ml). After 24 hours, a second infection was carried out under the same condition to increase transduction efficiency. At 48 hours after the first transduction, transduced cells were transplanted into B6-Ly5.2 recipient mice (7-12 weeks-old females) at 1.5 × 105 cells per recipient along with supporting bone marrow cells (7.5 × 105 cells/recipient) via tail vein injection. Before injection, recipient mice were irradiated twice at a total dose of 1100 rads from a 137Cs source. For secondary transplantation, 1 × 106 spleen cells from primary recipients with leukemia were injected into lethally irradiated secondary recipients along with 7.5 × 105 supporting bone marrow cells.
Flow cytometry
Purification of mouse GMPs (IL-7RαˉSca-1ˉc-Kit+FcγR-II/IIIHiCD34+) and flow cytometry analyses of immortalized myeloid progenitors and leukemic bone marrow and spleen cells were performed as described previously [10].
Lentiviral production, infection, and analysis
Myb shRNAs (Myb-sh1 and -sh5) were described previously [25] and cloned into the pLKO.1-blast lentiviral vector [32]. pLKO.1-blast-SCRAMBLE [32] was used as the negative control (NC-sh1). Both pLKO.1-blast and pLKO.1-blast-SCRAMBLE were gifts from Dr. Keith Mostov (Addgene plasmid #26655 and #26701). To generate infectious lentivirus, the constructs were co-transfected using Fugene 6 (Promega, Madison, WI) into 293T cells along with packaging plasmid Δ8.9 and a plasmid expressing VSV-G, and virus was harvested at 72 hours after transfection. Viral titers were calculated by infecting NIH-3T3 cells with serial dilutions of viral stocks and isolating blasticidin resistant colonies. Lentiviral infections were performed by spinoculation in which a mixture of lentivirus and target cells at 4:1 ratio in 48-well or 24-well plates were centrifuged at 2000 x g for 90 minutes at 37°C. Blasticidin (12 μg/ml) was added to the infected cells 24 hours after infection. Colony formation assays were performed at 48 hours after infection using 1 x104 blasticidin-resistant cells on IMDM methylcellulose medium supplemented with 15% fetal bovine serum, mouse SCF (50ng/ml), IL-3 (6ng/ml), and blasticidin (12 μg/ml). Colony numbers were counted after seven days.
Western blotting analysis
Western blotting analyses were carried out as previously described [10]. Immunoblotting was carried outusing anti-Hoxa9 (07-178, Merck Millipore, Billerica, MA), anti-Hoxa10 (SC-17159, Santa Cruz Biotechnology, Santa Cruz, CA), anti-Myb (05-175 Millipore), β-Actin (MAB1501R, Millipore) primary antibodies, and goat anti-rabbit IgG-HRP (sc-2004, Santa Cruz Biotechnology) and rabbit anti-mouse IgG-HRP (a-9044, Sigma Aldrich, St. Louis, MO) secondary antibodies. Protein bands were visualized by incubation with SuperSignal West chemi-luminescent substrate (Pierce, Thermo Fisher Scientific, Waltham, MA).
Real-time RT-PCR
For mouse cells or tissues, total RNA was extracted using RNAeasy Plus mini kit (Qiagen, Germantown, MD). Oligo-dT-primed cDNA samples were prepared from total RNA using Superscript III (Life Technologies, Carlsbad, CA), and real-time PCR analysis was performed in triplicates using SYBR green detection reagents (Life Technologies) according to the manufacturer’s instructions in 20-μL final volume on a 7500 real time PCR system (Applied Biosystems, Foster City, CA). Relative changes in expression of BCR/ABL, Hoxa9, Hoxa10, and Myb were calculated according to the ΔΔCt method. The cycling conditions were 50°C for 2 minutes, followed by 95°C for 2 minutes, and then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. The following gene-specific primer sequences were used: BCR/ABL S, 5’-TCC GCT GAC CAT CAA YAA GGA-3’; BCR/ABL AS, 5’- CAC TCA GAC CCT GAG GCT CAA-3’; Hoxa9 S, 5’-TGT CTC CTC TCC CCC AAA CC-3’; Hoxa9 AS, 5’-GAG ATG AGG CCT GGG ATTTAG A-3’; Hoxa10 S, 5’-CCA CAG GCC ACT TCG TGT T-3’; Hoxa10 AS, 5’-TCG TAG AGG CAG TAG GAG CTC TCT-3’; Myb S, 5’-CCA TGA AAG CTC GGG CTT AG-3’; Myb AS, 5’-CTC GAC ATG GTG TCA GTT GTG-3’; Rpl4 S, 5’-ATG ATG AAC ACC GAC CTT AGC A-3’; Rpl4 AS, 5’-CGG AGG GCT CTT TGG ATT TC-3’.
For human bone marrow cells, total RNA was extracted with TRizol (Life Technologies) according to manufacturer’s instruction. First-strand cDNA was synthesized using the Superscript III first strand kit (Life Technologies). All reactions were performed in a total volume of 20 μl. Quantitative detection of MYB and BCR transcripts was performed by q-PCR analysis using a TaqMan universal PCR master mix kit and the ABI Prism 7900 sequence detector (Applied Biosystems). TaqMan® Gene Expression Assays probes were purchased from Applied Biosystems. The housekeeping gene BCR was measured as an internal control. The levels of MYB were calculated based on standard curves and expressed as relative ratio to BCR.
ACKNOWLEDGMENTS
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Uniformed Services University of the Health Sciences, Department of Defense, or the U.S. Government. Some authors are U.S. Government employees. This work was prepared as part of their official duties. Title 17 U.S.C. 101 defines a United States Government work as a work prepared by a military service member or employee of the United States Government as part of that person’s official duties.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
GRANT SUPPORT
This work was supported by National Institutes of Health (NIH) grants RO1CA143193 (Y.D.) and USUHS Pediatrics Grant QP86GI (Y.D).
REFERENCES
1. Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, Akashi K, Gilliland DG. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004; 6: 587-96.
2. Neering SJ, Bushnell T, Sozer S, Ashton J, Rossi RM, Wang PY, Bell DR, Heinrich D, Bottaro A, Jordan CT. Leukemia stem cells in a genetically defined murine model of blast-crisis CML. Blood. 2007; 110: 2578-85.
3. Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004; 351: 657-67.
4. Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG, Giles FJ, Durocher J, Creusot RS, Karimi M, Jones C, Zehnder JL, Keating A, et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci U S A. 2009; 106: 3925-9.
5. Cuenco GM, Ren R. Cooperation of BCR-ABL and AML1/MDS1/EVI1 in blocking myeloid differentiation and rapid induction of an acute myelogenous leukemia. Oncogene. 2001; 20: 8236-48.
6. Roche-Lestienne C, Deluche L, Corm S, Tigaud I, Joha S, Philippe N, Geffroy S, Lai JL, Nicolini FE, Preudhomme C, Fi-LMC Group. RUNX1 DNA-binding mutations and RUNX1-PRDM16 cryptic fusions in BCR-ABL+ leukemias are frequently associated with secondary trisomy 21 and may contribute to clonal evolution and imatinib resistance. Blood. 2008; 111: 3735-41.
7. Zhang SJ, Ma LY, Huang QH, Li G, Gu BW, Gao XD, Shi JY, Wang YY, Gao L, Cai X, Ren RB, Zhu J, Chen Z, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2008; 105: 2076-81.
8. Ito T, Kwon HY, Zimdahl B, Congdon KL, Blum J, Lento WE, Zhao C, Lagoo A, Gerrard G, Foroni L, Goldman J, Goh H, Kim SH, et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature. 2010; 466: 765-8.
9. Nakahara F, Sakata-Yanagimoto M, Komeno Y, Kato N, Uchida T, Haraguchi K, Kumano K, Harada Y, Harada H, Kitaura J, Ogawa S, Kurokawa M, Kitamura T, et al. Hes1 immortalizes committed progenitors and plays a role in blast crisis transition in chronic myelogenous leukemia. Blood. 2010; 115: 2872-81.
10. Oakley K, Han Y, Vishwakarma BA, Chu S, Bhatia R, Gudmundsson KO, Keller J, Chen X, Vasko V, Jenkins NA, Copeland NG, Du Y. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood. 2012; 119: 6099-108.
11. Thorsteinsdottir U, Kroon E, Jerome L, Blasi F, Sauvageau G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol Cell Biol. 2001; 21: 224-34.
12. Thorsteinsdottir U, Sauvageau G, Hough MR, Dragowska W, Lansdorp PM, Lawrence HJ, Largman C, Humphries RK. Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol Cell Biol. 1997; 17: 495-505.
13. Calvo KR, Sykes DB, Pasillas M, Kamps MP. Hoxa9 immortalizes a granulocyte-macrophage colony-stimulating factor-dependent promyelocyte capable of biphenotypic differentiation to neutrophils or macrophages, independent of enforced meis expression. Mol Cell Biol. 2000; 20: 3274-85.
14. Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, Golub TR, Armstrong SA. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006; 442: 818-22.
15. Du Y, Jenkins NA, Copeland NG. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood. 2005; 106: 3932-9.
16. Hess JL, Bittner CB, Zeisig DT, Bach C, Fuchs U, Borkhardt A, Frampton J, Slany RK. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood. 2006; 108: 297-304.
17. Lieu YK, Reddy EP. Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proc Natl Acad Sci U S A. 2009; 106: 21689-94.
18. Gonda TJ, Ramsay RG, Johnson GR. Murine myeloid cell lines derived by in vitro infection with recombinant c-myb retroviruses express myb from rearranged vector proviruses. EMBO J. 1989; 8: 1767-75.
19. Manzotti G, Mariani SA, Corradini F, Bussolari R, Cesi V, Vergalli J, Ferrari-Amorotti G, Fragliasso V, Soliera AR, Cattelani S, Raschella G, Holyoake TL, Calabretta B. Expression of p89(c-Mybex9b), an alternatively spliced form of c-Myb, is required for proliferation and survival of p210BCR/ABL-expressing cells. Blood Cancer J. 2012; 2: e71.
20. Gonda TJ, Buckmaster C, Ramsay RG. Activation of c-myb by carboxy-terminal truncation: relationship to transformation of murine haemopoietic cells in vitro. EMBO J. 1989; 8: 1777-83.
21. Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000; 404: 193-7.
22. Magnusson M, Brun AC, Miyake N, Larsson J, Ehinger M, Bjornsson JM, Wutz A, Sigvardsson M, Karlsson S. HOXA10 is a critical regulator for hematopoietic stem cells and erythroid/megakaryocyte development. Blood. 2007; 109: 3687-96.
23. Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer. 2008; 8: 523-34.
24. Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, Shi J, Weissmueller S, Fellmann C, Taylor MJ, Weissenboeck M, Graeber TG, Kogan SC, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011; 25: 1628-40.
25. Nguyen N, Vishwakarma BA, Oakley K, Han Y, Przychodzen B, Maciejewski JP, Du Y. Myb expression is critical for myeloid leukemia development induced by Setbp1 activation. Oncotarget. 2016; 7: 86300-12. https://doi.org/10.18632/oncotarget.13383.
26. Dasse E, Volpe G, Walton DS, Wilson N, Del Pozzo W, O’Neill LP, Slany RK, Frampton J, Dumon S. Distinct regulation of c-myb gene expression by HoxA9, Meis1 and Pbx proteins in normal hematopoietic progenitors and transformed myeloid cells. Blood Cancer J. 2012; 2: e76.
27. Zhang J, Han B, Li X, Bies J, Jiang P, Koller RP, Wolff L. Distal regulation of c-myb expression during IL-6-induced differentiation in murine myeloid progenitor M1 cells. Cell Death Dis. 2016; 7: e2364.
28. Pellerin I, Schnabel C, Catron KM, Abate C. Hox proteins have different affinities for a consensus DNA site that correlate with the positions of their genes on the hox cluster. Mol Cell Biol. 1994; 14: 4532-45.
29. Calabretta B, Sims RB, Valtieri M, Caracciolo D, Szczylik C, Venturelli D, Ratajczak M, Beran M, Gewirtz AM. Normal and leukemic hematopoietic cells manifest differential sensitivity to inhibitory effects of c-myb antisense oligodeoxynucleotides: an in vitro study relevant to bone marrow purging. Proc Natl Acad Sci U S A. 1991; 88: 2351-5.
30. Lidonnici MR, Corradini F, Waldron T, Bender TP, Calabretta B. Requirement of c-Myb for p210(BCR/ABL)-dependent transformation of hematopoietic progenitors and leukemogenesis. Blood. 2008; 111: 4771-9.
31. Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998; 92: 3780-92.
32. Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol. 2010; 12: 1035-45.