
Introduction
Common fragile sites (CFS) encompass vast chromosomal regions often containing genomically large, active genes. Although discovered by applying artificial chemical stresses to cell cultures, they can be hotspots of natural rearrangement and deletion in cancer [1]. Point mutations are rarely found in the coding region of genes within CFS [2,3]. Conversely, large intragenic homozygous deletions are frequently observed at some CFS genes, proposed to be the result of selection for disrupted tumor-suppressor genes (TSGs) such as FHIT and WWOX [4].
The analysis of the CFS DNA sequences has not clearly identified the causes of their fragility, but it was observed that they share characteristic features such as AT base richness, high degrees of DNA flexibility, and late DNA replication [5]. Despite their instability, CFSs are evolutionary stable regions as proved by their conserved features across species [6].
FRA4F is the region of common fragility at 4q22.1. Deletion at this locus was reported as a frequent event in many tumor types, suggesting 4q22.1 as the site of a not-yet-identified TSG [6,7,8,9,10]. A more recent study of esophageal cancer proposed FAM190A (family with sequence similarity 190, member A; KIAA1680; MGC48628), mapping to 4q22.1, as the TSG [8]. FAM190A is a large (1.5 Mb), functionally uncharacterized gene with no recognizable protein domain and no sequence similarity to other proteins. At least 15 different DNA sequence variants are known for FAM190A (http://www.ncbi.nlm.nih.gov/projects/SNP/, October 2010) none of which are associated with disease (http://www.ncbi.nlm.nih.gov/omim, October 2010).
FAM190A has known transcript variants (http://insdc.org/, October 2010), which share 100% identity of the 5’ coding sequences to exon 6, but have different 5’and 3’ UTRs and 3’ exons. Variant 1 is the longest variant, containing 11 exons (NM_001145065.1 ); variant 2 contains 7 exons (NM_207491.2). Although the function of the protein is still unknown, the high degree of conservation across vertebrate species suggests that it has a conserved, important function.
In this study we found that the FAM190A transcript was very often rearranged in cancer samples. A predominance of in-frame deletions speculatively suggested a frequent activating mutation such as seen in the EGFRvIII rearrangement [11]. We suggest that the joined sequences of these in-frame deletions may form cancer-specific peptides (neo-antigens) with potential diagnostic and therapeutic relevance.
Results
Structure of the FAM190A coding sequence
We found partially overlapping homozygous genomic deletions (HDs) of 4q22.1 in a pancreatic cancer cell line, BxPc3 (reported in ref [9]), and in two of 60 xenografted pancreatic cancers, PX19 and PX188. A fourth homozygous deletion of the same region was reported in a lung cancer line, H2126 [12], and a somatic out-of-frame deletion of two exons is reported in multiple metastases of a single pancreatic cancer [13]. In all five cases, the overlapping deleted region included the FAM190A gene.
In order to analyze the transcriptional pattern of FAM190A, overlapping primers for the human FAM190A transcript variant 1 were designed (see Materials and Methods and Table S1). We then performed a PCR-based screen of exons 2 to 11 on cDNAs synthesized from 72 cell lines and xenografted cancer of different types. This sample set comprised two panels: 48 unselected samples and 24 samples selected for having a known HD or small heterozygous deletions at 4q22.1 (Table S2) [7]. The gene was expressed in most samples (92%, 66/72 cases). DNA fragments of unexpected size were sequenced. In the former panel we found eight types of rearranged transcripts and internal rearrangements in nearly 40% of the cases (39.6%, 19/48 cases). Among these affected samples, 84% had in-frame structures, 94% of which involved deletion of exon 9.
In the latter panel, we found nine types of rearrangements and 18 aberrant cases (75%) producing exclusively in-frame structures. In 89% of the cases the deletion involved exon 9. For each case having a deleted FAM190A transcript, we found a co-existing spliced form of expected length (wild-type) and/or only a rearranged transcript. Among combined selected and unselected cases (Figure 1), the changes appeared homozygous in 24 samples and heterozygous in 13. Overall, we identified 13 aberrant structural transcript types between exons 2 and 11: 11 represented different in-frame deletions (Figures 1 and 2); the remaining two caused shifting of the reading frame. Two cell lines and one xenograft (H1975, SW780, and MX7) had multiple rearranged spliced forms. Sequencing of the cDNA of AsPc1 revealed no subtle (point) mutation in the coding sequence. The sequencing of the cDNA of BxPc3 revealed (beside the known deletion of exon 9 and 10) the presence of a heterozygous nonsynonymous SNP at nucleotide 1144 resulting in a missense mutation at aminoacid 382. All rearrangements could be replicated in a PCR assay using different primers. In one patient, four independent parallel xenografts (PX19-1, -2, -3, and -4) had been derived from four locations in the resected primary tumor. All four had the identical exon deletion, indicating that the deletion had pre-existed within the patient’s tumor prior to expansion as xenografts.
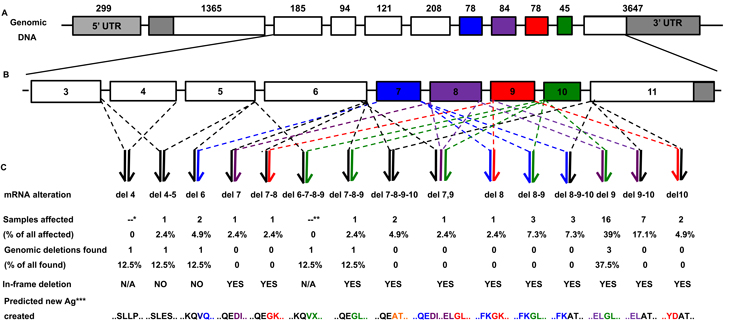
Figure 1. (A) The genomic structure of FAM190A, NM_001145065. Each box represents an exon, numbers above indicate their nucleotide lengths. Grey indicates the UTR; white and colors, the exons having nucleotide lengths not divisible and divisible by 3, respectively. (B) A magnification of exons from 3 to 11. (C) Top to bottom: structural types of the intragenic deletions, percentage of samples (combined panels) affected, percentage of homozygous deletions at the genomic level, type of rearranged transcripts, and predicted aminoacidic sequences at the transcript rearrangement joints. *cDNA not examined. **Lack of a PCR product in cDNA from exons 6 to 11. ***Predicted new antigen at the joint between normally non-contiguous exons
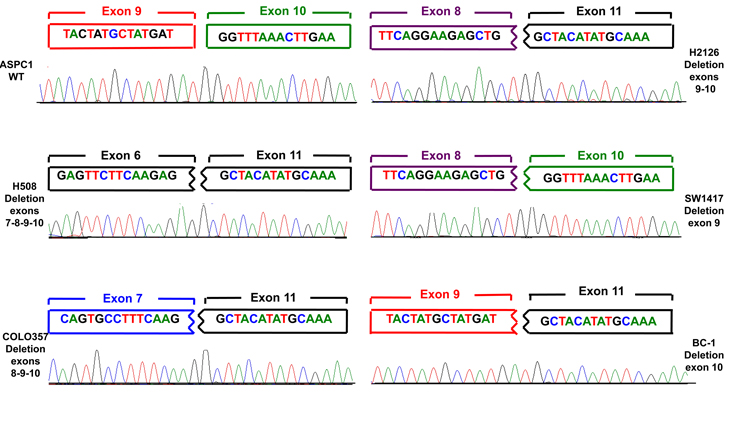
Figure 2: Electropherograms showing a wild-type sequence and four of the 13 different rearrangements observed. For each cell line the 28bp sequence of the anomalous fusion transcript is depicted. The two boxes on the top of each diagram represent the exon at the joint. Colors are as in Figure 1.
At the protein level, the in-frame deletions were predicted to form novel peptide sequences (Table 1). These are presumptive cancer-specific “neo-antigens”.
The same PCR and sequencing analysis from exon 6 to exon 11 was conducted on 48 commercially available cDNAs from different normal human tissues. Reproducible expression of FAM190A transcripts, assessed by multiple independent replicates, was found in 37 samples, 33 of which had a wild-type FAM190A transcript exon structure. Four samples had an exon deletion observed once: in one case exon 9 was lost; in one exon 8; and in two, exon 7. Two samples had a cryptic intronic exon inserted, each observed once. Additional samples of the organs, however, did not confirm the observed deletion or insertion, and these were considered as unconfirmed alternative splice variants.
Table 1: Variations in the FAM190A coding region transcripts.
| Deleted exons |
Consequence |
Predicted non-natural peptide sequence at the fusion joint* |
| 4-5 | Not in frame | SSSSKMNSL~ESFPEINKGRX † |
| 6 | Not in frame | PEFPEPSK~QVQTX |
| 7 | In frame | LKMKRVLQE~DIMKDECSM |
| 7-8 | In frame | LKMKRVLQE~GKVRHLQKA‡ |
| 7-8-9 | In frame | LKMKRVLQE~GLNLKRLET |
| 7-8-9-10 | In frame | LKMKRVLQE~ATYRNRIVS |
| 7 and 9 | In frame | LKMKRVLQE~DIMKDECSMLKLQL KEKDELISQLQEEL~GLNLKRLET§,|| |
| 8 | In frame | LTEEPVPFK~GKVRHLQKA |
| 8-9 | In frame | LTEEPVPFK~GLNLKRLET†† |
| 8-9-10 | In frame | LTEEPVPFK~ATYRNRIVS |
| 9 | In frame | LISQLQEEL~GLNLKRLET§ |
| 9-10 | In frame | LISQLQEEL~ATYRNRIVS** |
| 10 | In frame | TQTELLCYD~ATYRNRIVS |
Before the ~ sign are listed the last nine aminoacidic residues of the exon preceding the deletion. After the ~ sign are listed the first nine aminoacidic residues of the exon following the deletion. An underline indicates the aminoacidic residues that would be created due to a frame-shift. These two not-in-frame deletions would create a truncated predicted polypeptide. †X, stop codon. For HLA-A*0205, the cognate ligand structure – q – g–V– –L matches del7-8‡ and the structure –L– – –L– –L matches the joints produced by del7 and 9§ and del9§. For HLA-A3, the ligand structure –V– – – – I–K– matches del7 and 9||, the structure –L– – –L– –Y– matches del9-10**, and the structure – –F– –L– –K– matches del8-9†† [26].
Structure of FAM190A transcripts in the 5’ non-coding region
5’RACE PCR was performed to determine: 1) the transcript variants expressed in our samples and 2) the structure of the FAM190A transcript in the 5’ non-coding region. In a pancreatic cancer cell line, AsPc1, variant 1 and a novel variant 3 having an alternative first exon were observed. Based on this knowledge, RT-PCR conducted on a second pancreatic cancer cell line, BxPc3, revealed in addition to variant 1, the presence of variant 3 and a novel variant 4 having an alternative first and second exon. Variant 2 was never observed by us in the samples analyzed. In all four variants, the apparent start codon ATG is at the position 91229395 (Table 2).
Table 2: 5’ variations in the FAM190A transcript.
| Variants |
5’-most exon* |
Additional 5’ exon |
||
| Start (bp) | End (bp) | Start | End | |
| #1 | 91048684 | 91048982 | ----------- | ---------- |
| #2‡ | 91156182 | 91156494 | ----------- | ---------- |
| #3 | 91049688 | 91049813 | ----------- | ---------- |
| #4 | 91049688 | 91049813 | 91184749 | 91184873 |
* The genomic positions of the exons are expressed in basepairs according to build GRCh37/hg19
† The exon containing the starting codon ATG
‡ Variant 2 as annotated in the USCS Genome Browser (http://genome.ucsc.edu/), unconfirmed by us.
Additional unconfirmed 5’ variants are reported in the Ensembl database (http://uswest.ensembl.org/index.html).
Structure of the genomic FAM190A
In order to rationalize the transcripts through their genomic structures, we performed PCR analysis at the intron/exon junctions of exons 4 through 10 of 45 genomic DNAs isolated from 37 cancer cell lines and eight pancreatic cancer xenografts. Among these samples, 23 had rearrangements of their transcripts. In nine, the transcript alterations were fully and/or partially explained by homozygous losses of genomic material, which encompassed only the exons deleted in the corresponding transcript. Mechanisms other than genomic deletion might underlie the aberrant transcripts in the 14 remaining cases, such as undiscovered intronic point mutations, small deletions affecting splicing signals, and heterozygous or compound heterozygous genomic deletions.
Mouse FAM190A gene (Fam190a)
Four mouse CFSs have been defined at the molecular level. Of these, Fra6C1 corresponds to the human FRA4F [6]. The murine ortholog of the FAM190A gene, Fam190a, maps to mouse chromosome 6 and has two isoforms which differ in the 5’UTR but encode the same protein (http://www.ncbi.nlm.nih.gov/gene). The human and the mouse transcripts share 82% identity of the nucleotide coding sequence, suggesting that a deletion pattern similar to that observed in the human was possible. To test this possibility, we performed a RT-PCR-based screening analysis on two murine cancer cell lines (CT38, LLC) and one murine embryonic fibroblast line (MEF-P3) along with 36 different normal murine tissues. In 27 samples we produced an amplified fragment, which was of expected size, suggesting that no rearrangements were present. We observed widespread expression of Fam190a in both newborn and adult murine tissues.
Discussion
The contribution of rare and common fragile sites to genome rearrangements and diseases has long been studied. Perhaps owing to an unusual nucleotide composition and high structural flexibility, fragile sites have delayed replication in S phase, a characteristic that may lead to the formation of local replicative gaps and illegitimate chromosomal rearrangements, and result in fixed genomic deletions.
It is still not clear to what extent these play a role in cancer. Recurrent, low-frequency deletions that do not retain the reading frame can affect the coding exons of the FHIT and WWOX genes at the respective fragile sites, and intronic deletions not affecting the structure of the mature mRNA also are seen [14,15]. These patterns have lead to the controversy whether these genes may be either “driver” tumor-suppressor genes or instead reflect the uncovering of “passenger” random changes affecting fragile sites [16,17,18].
In this study we describe the finding of structural defects in the FAM190A transcript in 40% of human cancers and transformed cells. Evidence for widespread rearrangements affecting this region in multiple tumor types suggests that the mutant coding sequences identified might be among the most frequent mutations in human cancer. This high frequency is not readily explained by the mere coincidental location of FAM190A in a fragile region, for the FRA4F site spans about 10 megabasepairs, and the affected region evaluated here is less than 5% of that span. Nor does FAM190A have a deletion pattern in cancers similar to other altered genes evaluated at fragile sites, even if we were to restrict our attention to the exons (or groups of contiguous exons) contained in these genes in which the nucleotide count is a perfect multiple of “3”. The remaining plausible possibility is that the FAM190A changes of cancers is selective, wherein certain particular deletions arising from random processes has become enriched due to providing a growth advantage during neoplastic progression. This selection appears to preferentially act upon gross rearrangements, for whole-exomic and whole-genomic sequencing of human cancers (including ours) [19] has not found sub-exonic subtle mutations of this gene such as missense or nonsense mutations.
The deletions of FAM190A might, in theory, be recessive or dominant during tumorigenesis. Of the rearranged transcripts, 93% remained in-frame at the fusion (intragenic translocation) joint. Thirteen were apparently heterozygous, for a normal transcript co-existed with the mutant form. This suggests that the mutant protein products may retain, provide a new (or gain a) function and they are dominant. Dominant mutant genes selected during oncogenesis are classified as oncogenes.
We found that some of the in-frame rearrangements of the transcript had an obvious basis, for they corresponded to the exons spanned by intragenic homozygous deletions of the genomic DNA. In other instances, a genomic basis was implied, for the prevalence rate of FAM190A transcript alterations was elevated in cancers pre-selected for known heterozygous and homozygous genomic DNA deletions in the neighborhood. In the remaining tumors having no exonic genomic deletions, an undiscovered intronic mutation (similar perhaps to the genomic intronic mutations of the CD22 gene in B-precursor leukemia proposed as causing exon 12 deletions in the transcripts) [20] or an epigenetic mechanism may be the underlying cause.
FAM190A has alternative transcript forms. Transcript variants can physiologically be employed to create tissue regulatory specificity or protein diversity. In particular, the presence of 5’ alternative structures can derive from use of alternative promoters and/or from alternative splicing. The novel 5’ variants we observed in cancer cells may represent a loss of splicing fidelity [21], may subserve a tumorigenic role, or may be shared with certain normal cells.
It will be of interest to explore the functional roles of FAM190A and how these roles may be altered by the intragenic rearrangements. The conservation of the gene among vertebrates, and especially the sharing of exon structure and exonic nucleotide lengths between human and mouse, suggests that mouse models may be fruitful to survey for the normal physiologic roles of FAM190A. Additionally, we noted that some of the fusion joints match the minimum consensus peptide motives presented by the MHC (Table 1). Possibly, the restricted set of fusion joints represent neo-antigens that could be clinically typed by diagnostic antibody panels, targeted by rearrangement-specific therapies, or non-invasively monitored using personalized assays for disease burden [22].
Material and methods
Sample collection
72 human tumor specimens, 39 cell lines and 33 xenografts, were studied. 20 of the cell lines (AsPc1, BT-20, BT-474, CAPAN1, CAPAN2, CFPAC1, COLO357, DLD-1, HeLa, Hs578T, MCF7, MDA-MB-134, MDA-MB-453, MiaPaCa2, Panc-1, P215, PL45, T470, RKO, HEK 293) were randomly chosen from those available to us; the other 19 (AGS, BC-1, BxPc3, COLO205, H508, H727, HT-1376, H1581, H1975, H2126, H2228, KATO III, LNCa-Clone-FGC, LoVo, SW620, SW403, SW780, SW837,and SW1417) were selected for a having a known deletion affecting 4q22 [7]. The cell lines were obtained from European Collection of Cell Cultures (ECACC) (COLO357, P215) and American Type Culture Collection (ATCC).
33 xenografted human cancers of different types were obtained from our described tissue banks [23] under an IRB-approval protocol. Of these samples, 5 were selected for a known deletion affecting 4q22 (PX19, PX19-2R, PX19-3, PX19-4, PX188) [24] and 28 were unselected.
A panel of cDNAs from 48 different human normal tissues, was obtained (TissueScan, OriGene).
Three mouse cell lines (CT-38, LLC, MEF-P3) and a panel of 36 normal samples representing 18 different tissues taken from newborn and adult mice were studied by RT-PCR. The organs included heart, stomach, kidney, liver, lung, brain cerebellum, brain brainstem, brain cortex, pancreas, thymus, spleen, salivary gland, adrenal gland, skin, colon and small intestine.
DNA and RNA isolation
Total RNA was extracted from cell lines and tumors (Trizol, Invitrogen). Purification was performed using columns according to manufacturer’s instructions (Rneasy, Qiagen). RNA quality was assessed by gel electrophoresis of ethidium-bound total RNA. RNA was treated with DNase I (Invitrogen) and retrotranscribed (SuperScript® III, Invitrogen) to form cDNA.
Genomic DNA was extracted according to manufacturer’s instructions (QIAamp, Qiagen). DNA and RNA concentrations were determined using spectrometry (NanoDrop Technologies).
Primer design, PCR, and sequence analysis
The primers were designed using Primer3 (http://frodo.wi.mit.edu/) and synthesized by Integrated DNA Technologies (IDT) (Table S3-5). Designed primers were aligned against the corresponding genome sequence using BLAT (http://genome.ucsc.edu/cgi-bin/hgBlat, assembly Feb 2009, GRCh37/hg19) to confirm specificity. Taq DNA polymerase was used for the PCR reactions.
PCR conditions were as follows: 94 °C for 4 min, 72°C for 10 s, and then 40 cycles of 94 °C for 30 s, 55°C for 30 s, 72°C for 30 s. PCR products were separated on 1% agarose gel in lithium boric acid buffer (LB®, Faster Better Media LLC) [25] to determine presence and size, processed (QIAquick PCR Purification Kit, Qiagen) and analyzed by automated sequencing.
RACE PCR
5’RACE PCR was performed (FirstChoice RLM-RACE, Applied Biosystem) according to manufacturer’s instructions.
Acknowledgements
Funded by The Emerald Foundation, The Everett and Marjorie Kovler Professorship in Pancreatic Cancer Research, and NIH grants CA62924, CA134292, and CA128920. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Glover TW. Common fragile sites. Cancer Lett. 2006; 232: 4-12.
2. Kannan K, Munirajan AK, Bhuvarahamurthy V, Mohanprasad BK, Shankar P, Tsuchida N, Shanmugam G. FHIT gene mutations and single nucleotide polymorphism in Indian oral and cervical squamous cell carcinomas. Oral Oncol. 2000; 36: 189-193.
3. Kannan K, Krishnamurthy J, Feng J, Nakajima T, Tsuchida N, Shanmugam G. Mutation profile of the p53, fhit, p16INK4a/p19ARF and H-ras genes in Indian breast carcinomas. Int J Oncol. 2000; 17: 1031-1035.
4. Iliopoulos D, Guler G, Han SY, Druck T, Ottey M, McCorkell KA, Huebner K. Roles of FHIT and WWOX fragile genes in cancer. Cancer Lett. 2006; 232: 27-36.
5. Richards RI. Fragile and unstable chromosomes in cancer: causes and consequences. Trends Genet. 2001; 17: 339-345.
6. Rozier L, El-Achkar E, Apiou F, Debatisse M. Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: possible association with an inherited disease and cancer. Oncogene. 2004; 23: 6872-6880.
7. Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, Fu B, Swamy S, Dalgliesh GL, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010; 463: 893-898.
8. Nancarrow DJ, Handoko HY, Smithers BM, Gotley DC, Drew PA, Watson DI, Clouston AD, Hayward NK, Whiteman DC. Genome-wide copy number analysis in esophageal adenocarcinoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2008; 68: 4163-4172.
9. Calhoun ES, Hucl T, Gallmeier E, West KM, Arking DE, Maitra A, Iacobuzio-Donahue CA, Chakravarti A, Hruban RH, Kern SE. Identifying allelic loss and homozygous deletions in pancreatic cancer without matched normals using high-density single-nucleotide polymorphism arrays. Cancer Res. 2006; 66: 7920-7928.
10. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback R, Ligon AH, Cho YJ, Haery L, Greulich H, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010; 463: 899-905.
11. Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, Vogelstein B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992; 89: 2965-2969.
12. Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, Richards WG, Sugarbaker D, Chen F, Rubin MA, Janne PA, Girard L, Minna J, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005; 65: 5561-5570.
13. Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal SA, Leroy C, Jia M, Menzies A, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010; 467:1109-13.
14. Driouch K, Prydz H, Monese R, Johansen H, Lidereau R, Frengen E.Alternative transcripts of the candidate tumor suppressor gene, WWOX, are expressed at high levels in human breast tumors. Oncogene. 2002; 21: 1832-1840.
15. Sozzi G, Huebner K, Croce CM. FHIT in human cancer. Adv Cancer Res. 1998; 74: 141-166.
16. Wang L, Darling J, Zhang JS, Qian CP, Hartmann L, Conover C, Jenkins R, Smith DI. Frequent homozygous deletions in the FRA3B region in tumor cell lines still leave the FHIT exons intact. Oncogene. 1998; 16: 635-642.
17. Hilgers W, Kern SE. Molecular genetic basis of pancreatic adenocarcinoma. Genes Chromosomes Cancer. 1999; 26: 1-12.
18. Le Beau MM, Drabkin H, Glover TW, Gemmill R, Rassool FV, McKeithan TW, Smith DI. An FHIT tumor suppressor gene? Genes Chromosomes Cancer. 1998; 21: 281-289.
19. Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009; 324: 217.
20. Uckun FM, Goodman P, Ma H, Dibirdik I, Qazi S CD22 EXON 12 deletion as a pathogenic mechanism of human B-precursor leukemia. Proc Natl Acad Sci U S A. 2010; 107: 16852-16857.
21. Lee MP, Feinberg AP. Aberrant splicing but not mutations of TSG101 in human breast cancer. Cancer Res. 1997; 57: 3131-3134.
22. Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, Antipova A, Lee C, McKernan K, De La Vega FM, Kinzler KW, Vogelstein B, Diaz LA Jr, Velculescu VE. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010; 2: 20ra14.
23. Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994; 8: 27-32.
24. Iacobuzio-Donahue CA, van der Heijden MS, Baumgartner MR, Troup WJ, Romm JM, Doheny K, Pugh E, Yeo CJ, Goggins MG, Hruban RH, Kern SE. Large-scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome-wide allelic loss. Cancer Res. 2004; 64: 871-875.
25. Brody JR, Calhoun ES, Gallmeier E, Creavalle TD, Kern SE. Ultra-fast high-resolution agarose electrophoresis of DNA and RNA using low-molarity conductive media. Biotechniques. 2004; 37: 598, 600, 602.
26. Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995; 41: 178-228.