
INTRODUCTION
Colorectal cancer (CRC) is one of the most common malignancies of gastrointestinal neoplasms, involving the malignant transformation of normal intestinal epithelial cells resulting from the accumulation of abnormal genetic and epigenetic changes [1]. CRC is diagnosed in an upward trend in China [2].
Epigenetic modifications have been documented contributing to the carcinogenesis of many human cancers [3, 4]. As one predominant modification of epigenetics, DNA methylation, plays a major role during the tumorigenesis of CRC [5]. Aberrant hypermethylation in the gene promoter region puts a damper on its transcription by recruiting methyl-cytosine-binding proteins (MBPs), generating and curtailing the binding ability of transcriptional factors [5, 6]. Thus, the hypermethylation of the gene promoter region is responsible for the crippled function of tumor suppressor genes (TSGs) and for the initiation of various human cancers, including CRC [7].
A growing list of candidate TSGs silenced by hypermethylation in CRC play important roles in CRC tumorigenesis [7, 8]. The importance of gene methylation silencing is highlighted by the growing awareness that DNA methylation can predispose to mutational changes in the early stage of CRC. For instance, O6-methylguanine-DNA methyltransferase (MGMT) functions as a remover of carcinogen-induced mistake adducts from DNA [9]. Previous studies have reported that CRC with epigenetic modified MGMT is prior to heritable alteration in the initial of CRC [10].
In addition, numerous studies have justified clinical values of DNA methylation in early diagnosis of CRC [11, 12]. However, few studies have investigated DNA methylation alterations occurred in the early stage of CRC that may correlate with the later progression of CRC, especially with CRC metastasis. Accordingly, identification of DNA methylation occurred in the early stage of CRC that can inform risk of subsequent metastasis of CRC has the potential to both improve our understanding of cancer progression and impact clinical treatment.
In the present study, we first identified differentially methylated genes in CRC by applying the Illumina humanmethylation450 array and then verified the hypermethylated claudin-11 (CLDN11) in 125 CRC samples. CLDN11 belongs to the claudin family, which constitutes the core components of tight junction and cell adhesion between cells. However, the association of CLDN11 methylation with CRC progression, especially with the metastasis of CRC, was less investigated. Therefore, we assessed the methylation frequency of CLDN11 in 125 CRC samples, and analyzed the associations between aberrantly methylated CLDN11 and the progression of CRC, along with other clinical characteristics, such as gender, age, tumor size and differentiation. Subsequently, the effects of gene methylation exerting on the motility of CRC cells were analyzed. Through these analyses, we attempted to figure out the contribution of methylated CLDN11 to CRC progression, and the associations between CLDN11 methylation and progression free survival (PFS) of CRC.
RESULTS
Methylation array analysis of CRC tissues
To discover epigenetically-modified genes involved in CRC, we profiled genome-wide DNA methylation of three paired CRC tissues and corresponding adjacent tissues using a HM450K array. After quality control, a total of 484,698 CpG sites were analyzed. Differential methylation analysis between CRC tissues and normal tissues was identified a total of 4051 CpG sites (Supplementary Figure 1). Among which, 1017 (25%) and 3034 (75%) were hypermethylated and hypomethylated, respectively (Figure 1). These differentially methylated CpG sites were annotated to 1725 genes, among which the genes of APC, MLH1 and MGMT, frequently reported to be methylated in CRC, were also observed in the current study (data not shown). Then, GO analysis was conducted through DAVID online database (https://david.ncifcrf.gov/) and demonstrated that the methylated genes were mainly functioned as cell adhesion and enriched in the cell adhesion molecule (CAM) pathway (Supplementary Table 1). As a result, we mainly focused on the differential methylated genes in CAM pathway for its critical role played in cell-cell adhesion and cancer metastasis. The observed maximal β value difference between CRC tissues and normal tissues was CLDN11 gene. Figure 2 showed hierarchical cluster analysis of all differentially methylated CpG sites and the two differentially methylated CpG sites of CLDN11 promoter (cg20449692 and cg00894757).
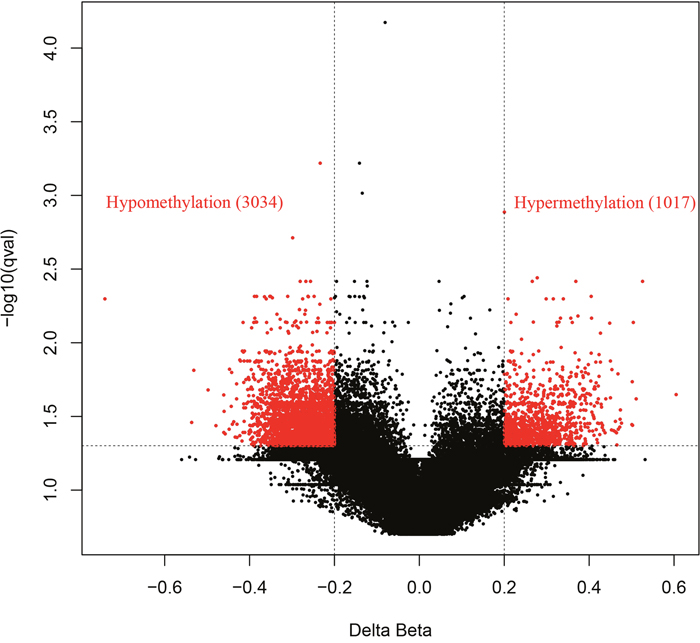
Figure 1: Volcano plot of methylation difference between CRC and normal tissues. A total of 1017 CpG sites hypermethylated in CRC with deltaβ value > 0.2 and FDR P < 0.05 was represented by red point in the right side. A total of 3034 CpG sites hypomethylated in CRC with delta β value < -0.2 and FDR P < 0.05 was represented by red point in the left side.
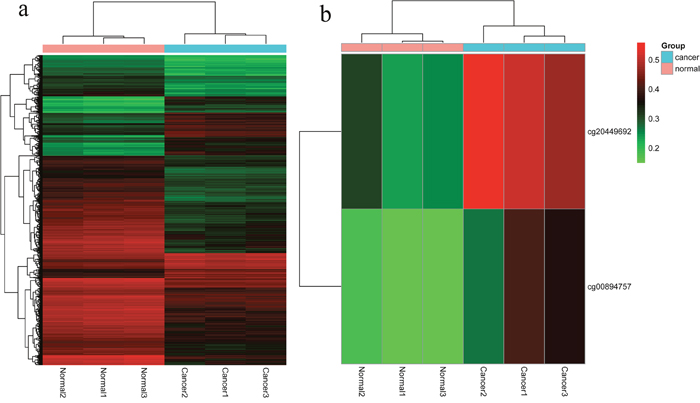
Figure 2: The hierarchical cluster maps of differentially methylated CpG sites in our study cohort. (a) The hierarchical cluster maps of all the significant methylation CpG sites; (b) The hierarchical cluster maps of the two differentially methylated CpG sites of CLDN11 promoter (cg20449692 and cg00894757).
Hypermethylation of CLDN11 in CRC
To verify the methylation level of CLDN11, the fragment of CLDN11 promoter region was amplified for quantitative methylation polymerase chain reaction (qMSP), which neighbor the two differentially methylated CpG sites (including cg20449692 and cg00894757) (Figure 3). A total of 125 bisulphite converted DNA from formalin-fixed and FFPE CRC and normal tissues were analyzed. And the results showed the elevated methylation level of CLDN11 in CRCs than in controls (Figure 4a). Then, methylation difference analyses based on the clinical features were performed; and we found statistically higher methylation level of CLDN11 in CRC with lymph node metastasis when compared with CRC without metastasis (P = 0.01, Figure 4b). Although the methylation difference among CRCs from different clinical stage was not found, the upward trend of mean methylation frequency of CLDN11 in adjoin with the advanced stage of CRC could be identified (Table 1). These results implied the correlation between CLDN11 methylation and the progression of CRC.
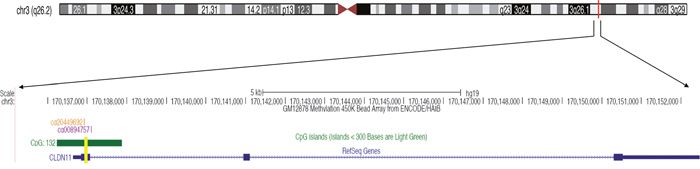
Figure 3: The tested fragment of CLDN11 promoter region. The yellow bar is the tested segment of CLDN11 promoter.
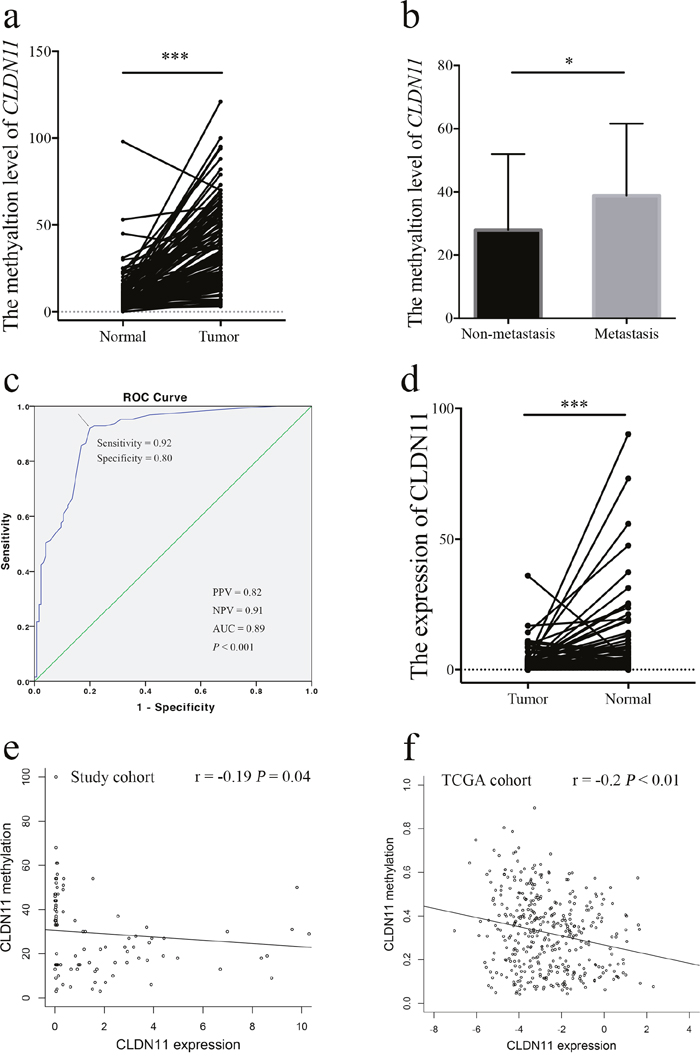
Figure 4: Analysis of CLDN11 gene methylation and expression in CRC patients. (a) Elevated methylation of CLDN11 in CRCs; (b) Elevated methylation of CLDN11 in CRCs with lymph node metastasis. (c) ROC curve for CLDN11 promoter methylation. AUC: area under ROC curve; PPV: positive predict value; NPC: negative predict value; (d) Down-regulated expression of CLDN11 in CRC; (e) Inverse correlation of CLDN11 methylation and expression in our study cohort; (f) Inverse correlation of CLDN11 methylation and expression in TCGA cohort.
Table 1: Associations of CLDN11 methylation with clinical characteristics of CRC
Characteristics |
N |
Methylation% (Mean ± SD) |
P |
|
|---|---|---|---|---|
Gender |
Male |
72 |
33.03 ± 22.41 |
0.68 |
Female |
53 |
34.84 ± 25.96 |
||
Age (y) |
< 60 |
40 |
30.7 ± 22.97 |
0.33 |
≥ 60 |
85 |
35.2 ± 24.41 |
||
Tumor size (cm) |
> 5 |
54 |
35.93 ± 26.63 |
0.45 |
< 5 |
59 |
32.42 ± 22.07 |
||
Location |
Rectum |
60 |
34.44 ± 26.27 |
0.91 |
Colon |
51 |
34.47 ± 22.40 |
||
Differentiation |
Poor |
44 |
35.52 ± 25.37 |
0.052 |
Moderate |
79 |
31.87 ± 21.20 |
||
High |
2 |
72.03 ± 68.65 |
||
Lymph node metastasis |
Positive |
67 |
38.84 ± 22.81 |
0.01 |
Negative |
58 |
27.97 ± 23.98 |
||
Distant metastasis |
Positive |
8 |
40.67 ± 32.33 |
0.41 |
Negative |
117 |
33.32 ± 23.32 |
||
Clinical stage |
I |
12 |
21.32 ± 13.69 |
0.054 |
II |
46 |
29.77 ± 25.80 |
||
III |
59 |
38.54 ± 21.63 |
||
IV |
8 |
40.67 ± 32.33 |
SD stands for standard deviation.
We also conducted a receiver operating characteristic (ROC) analysis for the diagnostic values of methylation CLDN11 in distinguish of CRC, showing that the area under ROC curve (AUC) was 0.89 with 92% sensitivity and 80% specificity (P < 0.001). The positive predict value (PPV) and negative predict value were 82% and 91%, respectively (Figure 4c).
Down-regulation of CLDN11 in CRC
Considering causal relationship between promoter hypermethylation and abated transcriptional activity, we examined the mRNA levels of CLDN11 in 100 CRC tissues and corresponding normal tissues. Our results illustrated lower expression of CLDN11 in CRC tissues compared with normal tissues (Figure 4d). The Pearson’s analysis showed an inverse correlation between methylation and expression (r = -0.19, P = 0.04, Figure 4e). Besides, we abstracted methylation and expression data of CLDN11 from TCGA (https://cancergenome.nih.gov/) (Supplementary Table 2), and the results were in accordance with our data (r = -0.21, P < 0.01, Figure 4f).
High luciferase activity of pGL4-CLDN11 plasmid
To verify the transcriptional activity of the tested fragment of CLDN11, we performed an in vitro luciferase reporter assay. Our results showed that the recombinant plasmid (pGL4-CLDN11), containing the tested sequences of CLDN11, had a higher luciferase activity (Figure 5), implying that the hypermethylation of CLDN11 promoter region might be responsible for its decreased expression.
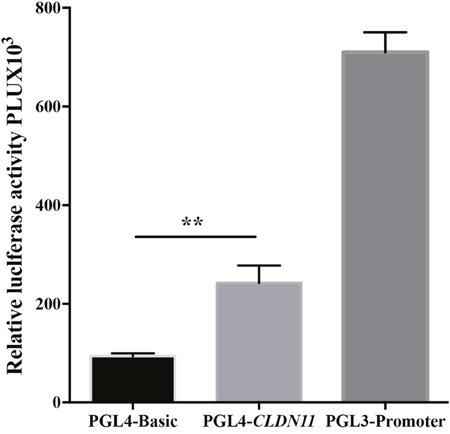
Figure 5: Luciferase activity analysis of fragment of CLDN11 promoter region.
Differential methylation and expression of CLDN11 in CRC cell lines
Next, we checked the methylation and expression distribution of CLDN11 in CRC cell lines (including HCT116, COLO205, SW620 and HT29) and normal colon cell line (NCM460), observing statistically elevated methylation level in HCT116, and remarkably diminutive methylation level in SW620 compared with in NCM460 (Figure 6a). Simultaneously, we found significantly lower CLDN11 expression in HCT116 than in NCM460; and significantly higher CLDN11 expression in SW620 was found (Figure 6b). These results showed that reduced CLDN11 expression is associated with DNA methylation. Therefore, we selected HCT116 cell line stepping into next experiment for its characteristics with statistically high methylation level and low expression of CLDN11.
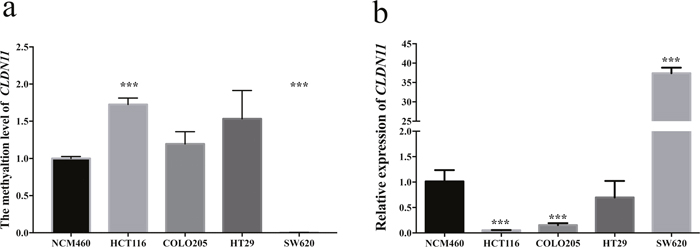
Figure 6: Analysis of CLDN11 methylation and expression in CRC cell lines and normal colon cell line. (a) Statistically elevated methylation of CLDN11 in HCT116 and diminutive methylation of CLDN11 in SW620; (b) Statistically decreased expression of CLDN11 in HCT116 and COLO205, and increased expression of CLDN11 in SW620.
Differential methylation and expression of CLDN11 in HCT116 cells with different concentration of 5-Aza
To furtherly determine whether methylation of CLDN11 promoter region is a potential mechanism of inactivating CLDN11, we measured mRNA levels of CLDN11 in HTCT116 cells treated with 5-Aza. A total of eight different concentrations of 5-Aza were set to treat HCT116 cell line. The methylation status of HCT116 cells was assessed in the meantime. From Figure 7a and Figure 7b, we found that the methylation level of CLDN11 was significantly reduced in cells treated with 9μM of 5-Aza (P = 0.04); and the restored expression of CLDN11 was noticeable. These data implied the negative correlation between methylation and expression of CLDN11.
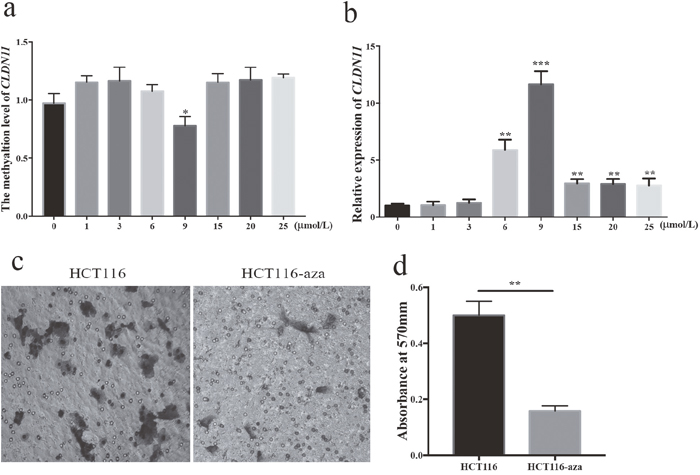
Figure 7: Association of CLDN11 methylation with CRC cell migration. (a) Methylation level of CLDN11 in HCT116 cells treated with seven different concentration of 5-Aza; (b) Expression of CLDN11 in HCT116 cells treated with seven different concentration of 5-Aza; (c) Abated migration ability of HCT116 cells after treated with 5-Aza; (d) High absorbance value of eluent crystal violet solution of HCT116 cell treated with 5-Aza at 570nm.
Restored expression of CLDN11 inhibited migration of HCT116 cells
Subsequently, we carried out a migration assay to analyze the effect of CLDN11 methylation in CRC cells to cell migration ability, showing that the migration of HCT116 cells was dramatically decreased when CLDN11 methylation was converted with demethylation agent (Figure 7c and Figure 7d). The Transwell results testified that hypermethylation of CLDN11 promoter region in CRC cells committed the metastasis of cells.
Survival analysis on TCGA cohort
Regrettably, we are absent of the survival data of all included patient in the current study. Therefore, to analyze the clinical implication of CLDN11 methylation to CRC, we obtained the progression free survival (PFS) data, along with the methylation data of CRC from TCGA (Supplementary Table 3). Based on the mean methylation of two CpG sites of CLDN11, we classified all CRC patients as hypermethylation group (methylation data more than mean methylation) and hypomethylation group (methylation data less than mean methylation). Subsequently, we performed Kaplan-Meier survival and log-rank test, observing that patients with hypermethylation level of CLDN11 harbored low progression free survival (PFS) (P = 0.041) (Figure 8), which indicated CLDN11 methylation could serve as a useful prognosis biomarker for CRC.
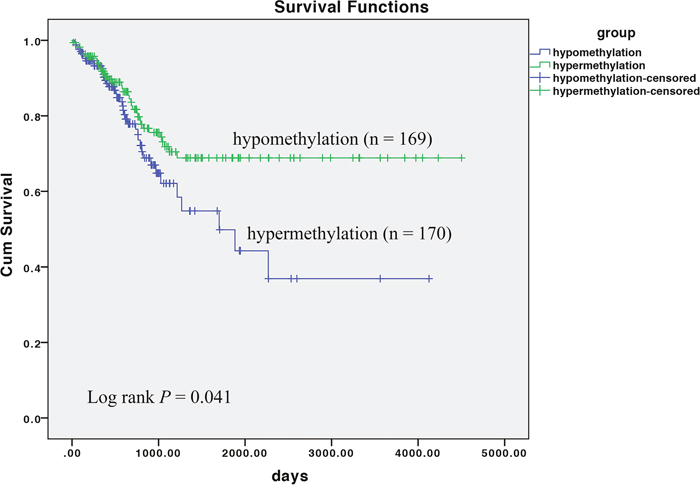
Figure 8: Association of CLDN11 methylation with progression free survival (PFS) of CRC.
DISCUSSION
In the recent decades, outgoing studies have demonstrated that tumor cells are activated by genetic and epigenetic changes, but also routinely use epigenetic processes to render their escape from host surveillance [13]. These epigenetically changes occurred at the early stages of malignancies, and are involved the throughout tumor process including invasion [14, 15]. Both genetic and epigenetic alterations in cancer result in de novo acquisition of developmental programs of cell with unlimited self-renewal potential [16].
Next-generation sequencing provides a new perspective of viewing epigenetic modification. A large-scale of cancer investigations have been identified one of key player of epigenetic changes, DNA methylation across a variety of human cancers, including colorectal cancer (CRC) [17, 18], and clarified how DNA methylation influent the process of cancer. DNA methylation is a relatively stable epigenetic change, mediating the silencing of certain genes [19]. Active gene promoters harbor the absence of DNA methylation in the CpG island located in more than 60% of tumor suppressor genes. Abnormal gains of DNA methylation in the unmethylated gene promoter CpG island associated with the loss of tumor suppressor function [19], as genetic mutation. Whereas, DNA methylation occurred at early stages of malignancies, even before mutations [20]. Therefore, emerging studies have identified multiple aberrant methylation genes and implicated candidates for the development of specific markers for cancer detection and diagnosis [21, 22].
In the current study, genome-wide DNA methylation of CRC and normal control was measured by Illumina Infinium HumanMethylation450 array, targeting 485,512 CpG sites and covering 99% of RefSeq genes of the genome [23]. After analysis of the methylation array data, we observed a unanimous phenomenon of the GO analysis results of differential methylated genes between CRC and normal tissues (Figure 1 and Supplementary Table 1). Both the cellular location, biological process and KEGG pathway enrichment demonstrated methylation abnormality of genes in the cell adhesion molecules (CAMs) pathway (Supplementary Table 1). Among these genes in CAMs, the maximal differential methylated gene is CLDN11 (Figure 2). Subsequent qMSP and RT-PCR analysis testified the high methylation of CLDN11 in CRC, along with loss of expression (Figure 4).
CLDN11 belongs to claudin family, of which there are 27 members. The claudins are responsible for the formation of tight junction (TJ) and cell adhesion, whose N and C-termini reside in the cytosol possesses two extracellular loop regions [24]. The cytoplasmic adaptor proteins of CLDN11 are the ZO proteins termed as ZO-1, -2 and -3, linking to the actin cytoskeleton and involving the regulation of cell motility [24]. Traditionally, TJ is intercellular structures that functions as a fence and barrier of epithelial cells, regulating and controlling the lateral diffusion of proteins within the lipid bilayer [25]. However, in the recent years, the role of TJs played in cell proliferation, transformation, and metastasis suppression, become much more intriguing. Numerous carcinomas are related with various environment stimulus and carcinogens. Therefore, our body needs a selective process to avoid these factors. Owing to the location of TJs, they are responsible for this selection and deregulation of TJs could destroy the maintenance of normal cell resulting in abnormal cellular biology, which is associated with various human disease, as well as cancers [26].
Additionally, claudin family also contributes to the adhesion between cells. Currently, the resultant of destroyed cell adhesion by the loss of claduin is critical to the metastasis of cancer cells [27, 28]. The dissociation of cancer cells from surrounding cancer cells and normal cells is the initial process of cancer cells metastasis and invasion [28]. It has been reported that recovery of various claudin proteins expression, such as CLDN3, CLDN4, and CLDN7, can reduce the malignant behavior of cancer cells and reduce the invasiveness in breast cancer [28]. Interestingly, claudin expression is in a tissue specific manner [29]. Victoria and his colleagues have demonstrated that one of the principal reasons of increased relatively increased expression of CLDN1 in CRC is the hypomethylation of its promoter region [30], which is also identified in our methylation array data (Supplementary Figure 2). Whereas, the different phenomenon was observed in breast cancer, hypermethylation and reduced expression of CLDN1, suggesting that the expression of CLDN1 varies in different tissues [31]. The down-regulation of CLDN7 is found in metastatic breast cancer [32] and invasive ductal carcinomas [33]. Whereas, the transcriptional activity of CLDN3 and CLDN4 is increased in ovarian cancer [34]. The expression of CLDN11 also varies differently in different tissues. It has been reported increased expression of CLDN11 in gastric cancer [35]. Nevertheless, in the current study, we observed the decreased expression of CLDN11 in CRC. Besides, we reversed methylation level of CLDN11 in CRC cell lines treated with 5-Aza. The deoxycytidine analog 5-aza-2’-deoxycytidine (5-Aza, also named decitabine) has been widely used as a DNA methylation inhibitor to experimentally induced gene expression, which now is postulated to have clinical activity in myelodysplastic syndrome (MDS), chronic myelogeneous leukemia (CML), acute myelogenous leukemia(AML) [36–38]. Interestingly, the effect of demethylation and restored expression of CLDN11 is significant when CRC cells were treated at low-dose of 5-Aza, rather than at high-dose. It was believed that low-dose schedules of 5-Aza could be served as antitumor drug to inhibitor DNA methylation and to reactivate gene expression [39, 40]. However, the covalent enzyme-DNA adduct, rather than DNA demethylation, is main reason for the toxicity of 5-Aza at high dose in cultured cell lines [41], which means the effect of demethylation and inversion of gene is slight when the tumor cells is cultured at high dose of 5-Aza. This might be one of the principle reason why slight demethylation and increased expression of CLDN11 in CRC cells treated with high dose of 5-Aza in our study. As shown in Figure 7, the results implied that DNA methylation contributes to the deduced expression of CLDN11 in CRC. However, researches on a correlation between CLDN11 methylation and CRC have been limited so far. In our study, our results and TCGA cohort showed a correlation between CLDN11 methylation and the metastasis and poor survival of CRC (Figure 4 and Figure 8). An in vitro experiment also portrayed the aggrandized migration ability of human colonic cancer cells with hypermethylation of CLDN11 (Figure 7).
Previous studies have elaborated the expression of CLDN3 and CLDN4 effect on the metastatic ability of breast cancer cells [42]. In cervical cancer cells, CLDN1 overexpression could promote invasion and metastasis [43]. On the other hand, claudin-low breast cancers have potential to metastasis and chemoresistant [44].
In a conclusion, our investigation shows the epigenetic modification mechanism of loss of CLDN11 expression in CRC. Besides, our results and TCGA cohort datasets represent the association between CLDN11 methylation and metastasis and unsatisfactory survival of CRC, which implies the potential of methylated CLDN11 in prognosis of CRC.
MATERIALS AND METHODS
Patients
At an initial discovery set, DNA from three matched patients CRC with corresponding normal tissues was applied the genome-wide DNA methylation analysis on HumanMethylation450 platform. All patients were enrolled in Gastrointestinal Surgery in Affiliated Hospital of Ningbo University and diagnosed by pathology. Before operation and tissue collection, all patients were informed and signed the consent forms. The adjacent normal tissue was at least 5 cm away from the tumor. All the samples were stored in liquid nitrogen at -80°C after surgery. On an independent validation set, a total of 125 paired formalin-fixed and paraffin-embedded (FFPE) CRC and cutting edge tissues were collected from Diagnostic Pathology Central of Ningbo. All patients were pathological diagnosed.
DNA and total RNA extraction
Genomic DNA was extracted by QIAamp DNA Mini Kit or DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) as recommended by the manufacturer. Total RNA from FFPE tissues were isolated using RNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The concentration and quality of DNA, as well as of RNA, was measured by ultramicro nucleic acid ultraviolet tester (NANODROP 1000, Wilmington, USA).
Bisulfite treatment
Extracted DNA was bisulphite-converted with ZYMO EZ DNA Methylation-Gold Kit according to the manufacturer’s protocol (Zymo Research, Orange, CA, USA). The bisulphite-modified DNA was resuspended in 10 μl of TE buffer for the following methylation analysis.
Illumina humanmethylation450 beadchip
The bisulphite converted DNA of three paired CRC and adjacent normal tissues were used for hybridization on the HumanMethylation450 BeadChip (HM450K, Illumina, San Diego, CA, USA). The bead array data were read by Illumina’s Genome Studio program (Methylation Module). And the raw methylation data was exported from Genome Studio and analyzed using Bioconductor minfi packages.
Quantitative methylation polymerase chain reaction (qMSP)
Bisulphite converted DNA as described above was amplified by fluorescence-based real-time quantitative methylation-specific PCR (qMSP) using SYBR Green Master Mix (Roche Diagnostics) with Roche 480 II RT-PCR instrument (Roche Diagnostics). The qMSP primers for CLDN11 were designed by MethPrimer (www.urogene.org/methprimer/) [45]. Each reaction involved 1x PCR buffer and 0.5 μM forward primer and reverse primer in a total volume of 10 μl. The PCR reaction were conducted as follows: 95°C for 10 min, amplification for 45 cycles of 95°C for 20 sec, 60°C for 30 sec and 72°C for 30 sec. The primer of ACTB were simultaneously amplified of each sample as internal control [46] and the full methylated DNA from healthy person (Qiagen, Hilden, Germany) served as a positive methylation control. The methylation levels were calculated as the difference in Ct value between CLDN11 and ACTB using the following formula: 2[ΔΔCt (Positive control)- ΔΔCt (Samples) ] ×100 [46], and ΔΔCt = Ct (CLDN11)- Ct (ACTB).
Reverse transcription-polymerase chain reaction (RT-PCR)
The cDNA was generated using the Reverse Transcription Kit (Promega, Madison city, WI, USA). Real-time RT-PCR method was performed to assess the expression level of CLDN11 with SYBR Green Master Mix (Roche Diagnostics) on a Roche 480 II RT-PCR instrument (Roche Diagnostics). The relative expression of CLDN11 in each sample was calculated with the 2-ΔΔCt method, that is -ΔΔCt = Ct [(CLDN11)-Ct (GAPDH) (normal samples)]-[Ct (CLDN11)-Ct (GAPDH) (tumor samples)], in which the GAPDH was used as internal control [47]. And the same method was used to determine the relative expression levels of CLDN11 of all the cell lines in the current study. Each sample was measured three times. The sequences of primers in the current study were listed in Table 2.
Table 2: Sequences of qMSP and RT-PCR primers
Characteristics |
Sequence |
|
|---|---|---|
qMSP |
CLDN11-Forward |
5'-ATAAGTTGATAGGAGAATCGAATCG-3' |
CLDN11-Reverse |
5'-AACGAAAATAACCCTAAACACGTT-3' |
|
ACTB-Forward |
5'-TGGTGATGGAGGAGGTTTAGTAAGT-3' |
|
ACTB-Reverse |
5'-AACCAATAAAACCTACTCCTCCCTTAA-3' |
|
RT-PCR |
CLDN11-Forward |
5'-TATGTGGAGTGAGTGGGCCA-3' |
CLDN11-Reverse |
5'-AACCAGATGGTGGCAACAAC-3' |
|
GAPDH-Forward |
5'-ACCCACTCCTCCACCTTTGAC-3' |
|
GAPDH-Reverse |
5'-TGTTGCTGTAGCCAAATTCGTT-3' |
Cell culture
Human colorectal cancer cell lines HCT116, COLO205, HT29 and SW620, as well as human normal colon epithelial cell line NCM460 were obtained from Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Cells were cultured at 37°C in a humidified atmosphere of 5% CO2 with RPMI-1640 Medium (HyClone, Logan, Utah) containing 10% fetal bovine serum (FBS) (ExCell Biology, Shanghai, China) with 50 U/ml penicillin and 50g/ml streptomycin (HyClone, Logan, Utah). Exponentially growing cells were used for experiments.
Construction of recombinant plasmids
A fragment of CLDN11 promoter region was synthesized which included the tested segment of CLDN11 by qMSP. The recombinant plasmid termed as pGL4-CLDN11 concatenated the fragment of CLDN11 and pGL4 Basic vector (Promega, Madison, WI, USA). The pRL-SV40 vector (Promega) with Ranilla luciferase gene was applied as an internal control and the pGL3-Promoter vector (Promega) was used as a positive control in the current study.
Transfection and reporter gene activity assay
The human HEK293T cells in exponential growth phase were cultured in 24-well plates (5×104/well) in 500μl RPMI-1640 with 10% FBS. After 12h culture, cells of 70% attachment were transfected with pGL4-CLDN11, pGL4-Basic, pRL-SV40 and pGL3-Promoter vector using the Lipofectamin 2000 reagent according to the transfection protocol recommended by Invitrogen (Invitrogen Corp., Carlsbad, CA, USA). After 24 h of 293T cells transfection, Renilla and firefly luciferase activity was detected by SmartSpec Plus spectrophotometer (Bio-Red, Hercules, CA, USA). Reporter gene activity was measured by following the manufacturer’s protocol (Dual-Luciferase Reporter Assay Systems, Promega).
Treatment of HCT116 cells with 5-aza-2’-deoxycytidine
The human colorectal cancer cell lines HCT116 were cultured into a 24-well plate and exposed to eight different concentrations of 5-aza-2’-deoxycytidine (5-Aza) for 3 days (including 0μM, 1 μM, 3 μM, 6 μM, 9 μM, 15 μM, 20 μM and 25 μM) (Sigma-Aldrich, St. Louis, MO, USA). The cells treated with 5-Aza were harvested and used for detection of CLDN11 methylation and expression.
Transwell migration assays
Transwell migration assay was performed with 24-well matrigel-coated chambers (8-μm pore size) (Corning Costar, NY, USA). Briefly, cells were serum-starved for 12h and subsequently dissociated with trypsin. After washed by PBS for twice, cells were resuspeded in serum-free medium and a total of 5×104 cells were added to the upper chamber adding complete medium on the bottom well. After 48h seeding, medium was aspirated and the cells that were unable to migrate were removed from the surface of the upper chamber. Therefore, the migrating cells were fixed by formaldehyde and stained by crystal violet solution. Images of three different fields of view were captured from each membrane under a microscope. The membranes were dissolved with 10% acetic acid, and absorbance was detected by SpectraMax 190 (Molecular DevicesA). Each assay was independently performed three times.
Extraction data from TCGA portal
From The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/) portal, we downloaded the Illumina HumanMethylation 450K methylation frequencies and RNA-seq expression of CLDN11, as well as the progression free survival (PFS) data of 395 CRC patients. For methylation data of CLDN11, we abstracted the level 3 methylation data of the two CpG sites (including cg20449692 and cg00894757), which were identified differentially methylated in our CRC samples. Based on the mean methylation frequencies of these two CpG sites of CLDN11, 339 CRC patients with PFS data were classified as hypermethylation group and hypomethylation group, and then the effect of CLDN11 methylation level to CRC outcome was analyzed. A total of 394 CRCs with both methylation and expression data of CLDN11 were used to verify the association between expression and methylation.
Statistical analysis
The methylation data from Illumina humanmethylation450K were analyzed using R 3.1.2 software (https://www.r-project.org/). The raw data were imported into R software using Bioconductor minfi [48]. Probes with a detection P value > 0.01 and those measuring SNPs or mapping in sex chromosomes were removed. Raw data was background corrected by subtracting control probes, normalized using subset-quantile within array normalization (SWAN) [49] to correct the discrepancies between type I and type II probes, and then computed methylated and unmethylated intensities. The methylation levels of CpG sites were calculated as β-value (β = intensity [methylated]/intensity [methylated+unmethylated+100]). Subsequently, M-values were calculated by transforming β-value for the following formula: M = log2(β/(1-β)) [50]. Then, the differential methylated CpG-sites (using M-values) between CRC and normal controls were identified. Benjamini-Hochberg false-discovery rate (FDR) method adjusted P value of each probe was calculated. FDR adjusted P value less than 0.05 and the delta β value either more than 0.2 (termed as hypermethylation) nor less than -0.2 (termed as hypomethylation) were used as the cut-off values, highlighting by red points in the volcano plot. Subsequently, genes of all the hypermethylation probes were annotated based on the reference genes of Illumina arrays, and performed the Gene Ontology (GO) using web-based DAVID (https://david.ncifcrf.gov/).
The following analyses were carried out using Statistical Program for Social Sciences (SPSS) software 13.0 (SPSS, Inc., Chicago, IL, USA), and represented by GraphPad software (InStat). The paired samples and two independent samples t-test were applied under the appropriate condition to compare the difference of methylation and expression, luciferase activity, and absorbance rate in our study cohort. ROC analysis was used to study the diagnostic power of gene methylation in distinguishing of CRC. The correlation of methylation and expression was computed by the Pearson rank test, representing using R software. In TCGA cohort, we collected the PFS data of CRC, analyzed the impact of methylation level on the PFS by Kaplan-Meier method, plotted Kaplan-Meier survival curves and compared using the log-rank test. Statistical significance was defined as P < 0.05.
Abbreviations
CRC: colorectal cancer; CLDN11: claudin-11; 5-Aza: 5-aza-2’-deoxycytidine; PFS: progression free survival; MBP: methyl-cytosine-binding protein; TSG: tumor suppressor gene; MGMT: O6-methylguanine-DNA methyltransferase; FFPE: formalin-fixed and paraffin-embedded; HM450K: HumanMethylation450 BeadChip; qMSP: fluorescence-based real-time quantitative methylation-specific polymerase chain reaction; RT-PCR: reverse transcription-polymerase chain reaction; FBS: fetal bovine serum; TCGA: The Cancer Genome Atlas; FDR: false-discovery rate; GO: Gene Ontology; SPSS: Statistical Program for Social Sciences; CAM pathway: cell adhesion molecule pathway; ROC: receiver operating characteristic; AUC: area under ROC curve; PPV: positive predict value; NPC: negative predict value; TJ: tight junction.
ACKNOWLEDGMENTS
This work acknowledges the support and help from Professor Junming Guo of Medical School of Ningbo University.
CONFLICTS OF INTEREST
The authors declare no conflicts of interests regarding the publication of this article.
FUNDING
This work was supported by the Zhejiang Provincial Natural Science Foundation of China [grant number LY16H160005]; the Project of Scientific Innovation Team of Ningbo [grant number 2015B11050]; the Ningbo Natural Science Foundation [grant number 2014A610235 and 2017A610236].
REFERENCES
1. Jiang Y, Lin DH, Xu JP, Chen WX, Zheng SJ, Song L. Genotype GG of rs895819 functional polymorphism within miR-27a might increase genetic susceptibility to colorectal cancer in Han Chinese population. J Clin Lab Anal. 2016; 30:351-355.
2. Chen W, Zheng R, Zhang S, Zeng H, Xia C, Zuo T, Yang Z, Zou X, He J. Cancer incidence and mortality in China, 2013. Cancer Lett. 2017.
3. Croes L, Op de Beeck K, Pauwels P, Vanden Berghe W, Peeters M, Fransen E, Van Camp G. DFNA5 promoter methylation a marker for breast tumorigenesis. Oncotarget. 2017; 8:31948-31958. https://doi.org/10.18632/oncotarget.16654.
4. Wang BH, Li YY, Han JZ, Zhou LY, Lv YQ, Zhang HL, Zhao L. Gene methylation as a powerful biomarker for detection and screening of non-small cell lung cancer in blood. Oncotarget. 2017; 8:31692-31704. https://doi.org/10.18632/oncotarget.15919.
5. Chen CH, Yan SL, Yang TH, Chen SF, Yeh YH, Ou JJ, Lin CH, Lee YT, Chen CH. The relationship between the methylated septin-9 DNA blood test and stool occult blood test for diagnosing colorectal cancer in Taiwanese people. J Clin Lab Anal. 2017.
6. Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999; 99:451-454.
7. Wierzbicki PM, Adrych K, Kartanowicz D, Stanislawowski M, Kowalczyk A, Godlewski J, Skwierz-Bogdanska I, Celinski K, Gach T, Kulig J, Korybalski B, Kmiec Z. Underexpression of LATS1 TSG in colorectal cancer is associated with promoter hypermethylation. World J Gastroenterol. 2013; 19:4363-4373.
8. Durso DF, Bacalini MG, do Valle IF, Pirazzini C, Bonafe M, Castellani G, Caetano Faria AM, Franceschi C, Garagnani P, Nardini C. Aberrant methylation patterns in colorectal cancer: a meta-analysis. Oncotarget. 2017; 8:12820-12830. https://doi.org/10.18632/oncotarget.14590.
9. Alonso S, Dai Y, Yamashita K, Horiuchi S, Dai T, Matsunaga A, Sanchez-Munoz R, Bilbao-Sieyro C, Diaz-Chico JC, Chernov AV, Strongin AY, Perucho M. Methylation of MGMT and ADAMTS14 in normal colon mucosa: biomarkers of a field defect for cancerization preferentially targeting elder African-Americans. Oncotarget. 2015; 6:3420-3431. https://doi.org/10.18632/oncotarget.2852.
10. Esteller M, Risques RA, Toyota M, Capella G, Moreno V, Peinado MA, Baylin SB, Herman JG. Promoter hypermethylation of the DNA repair gene O(6)-methylguanine-DNA methyltransferase is associated with the presence of G:C to A:T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 2001; 61:4689-4692.
11. Song L, Yu H, Jia J, Li Y. A systematic review of the performance of the SEPT9 gene methylation assay in colorectal cancer screening, monitoring, diagnosis and prognosis. Cancer Biomark. 2017; 18:425-432.
12. Warren JD, Xiong W, Bunker AM, Vaughn CP, Furtado LV, Roberts WL, Fang JC, Samowitz WS, Heichman KA. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011; 9:133.
13. Tomasi TB, Magner WJ, Khan AN. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol Immunother. 2006; 55:1159-1184.
14. Ma SS, Srivastava S, Llamosas E, Hawkins NJ, Hesson LB, Ward RL, Ford CE. ROR2 is epigenetically inactivated in the early stages of colorectal neoplasia and is associated with proliferation and migration. BMC Cancer. 2016; 16:508.
15. Wang W, Ji G, Xiao X, Chen X, Qin WW, Yang F, Li YF, Fan LN, Xi WJ, Huo Y, Wen WH, Yang AG, Wang T. Epigenetically regulated miR-145 suppresses colon cancer invasion and metastasis by targeting LASP1. Oncotarget. 2016; 7:68674-68687. https://doi.org/10.18632/oncotarget.11919.
16. Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013; 339:1567-1570.
17. Chen JJ, Wang AQ, Chen QQ. DNA methylation assay for colorectal carcinoma. Cancer Biol Med. 2017; 14:42-49.
18. Radpour R, Barekati Z, Kohler C, Lv Q, Burki N, Diesch C, Bitzer J, Zheng H, Schmid S, Zhong XY. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One. 2011; 6:e16080.
19. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011; 11:726-734.
20. Okugawa Y, Grady WM, Goel A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. 2015; 149:1204-1225 e1212.
21. Kandimalla R, van Tilborg AA, Kompier LC, Stumpel DJ, Stam RW, Bangma CH, Zwarthoff EC. Genome-wide analysis of CpG island methylation in bladder cancer identified TBX2, TBX3, GATA2, and ZIC4 as pTa-specific prognostic markers. Eur Urol. 2012; 61:1245-1256.
22. Renard I, Joniau S, van Cleynenbreugel B, Collette C, Naome C, Vlassenbroeck I, Nicolas H, de Leval J, Straub J, Van Criekinge W, Hamida W, Hellel M, Thomas A, et al. Identification and validation of the methylated TWIST1 and NID2 genes through real-time methylation-specific polymerase chain reaction assays for the noninvasive detection of primary bladder cancer in urine samples. Eur Urol. 2010; 58:96-104.
23. Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, Fan JB, Shen R. High density DNA methylation array with single CpG site resolution. Genomics. 2011; 98:288-295.
24. Singh AB, Uppada SB, Dhawan P. Claudin proteins, outside-in signaling, and carcinogenesis. Pflugers Arch. 2017; 469:69-75.
25. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. 2016; 17:564-580.
26. Sawada N. Tight junction-related human diseases. Pathol Int. 2013; 63:1-12.
27. Osanai M, Takasawa A, Murata M, Sawada N. Claudins in cancer: bench to bedside. Pflugers Arch. 2017; 469:55-67.
28. Tabaries S, Siegel PM. The role of claudins in cancer metastasis. Oncogene. 2017; 36:1176-1190.
29. Singh AB, Sharma A, Dhawan P. Claudin family of proteins and cancer: an overview. J Oncol. 2010; 2010:541957.
30. Hahn-Stromberg V, Askari S, Ahmad A, Befekadu R, Nilsson TK. Expression of claudin 1, claudin 4, and claudin 7 in colorectal cancer and its relation with CLDN DNA methylation patterns. Tumour Biol. 2017; 39:1010428317697569.
31. Di Cello F, Cope L, Li H, Jeschke J, Wang W, Baylin SB, Zahnow CA. Methylation of the claudin 1 promoter is associated with loss of expression in estrogen receptor positive breast cancer. PLoS One. 2013; 8:e68630.
32. Sauer T, Pedersen MK, Ebeltoft K, Naess O. Reduced expression of Claudin-7 in fine needle aspirates from breast carcinomas correlate with grading and metastatic disease. Cytopathology. 2005; 16:193-198.
33. Kominsky SL, Argani P, Korz D, Evron E, Raman V, Garrett E, Rein A, Sauter G, Kallioniemi OP, Sukumar S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene. 2003; 22:2021-2033.
34. Rangel LB, Agarwal R, D'Souza T, Pizer ES, Alo PL, Lancaster WD, Gregoire L, Schwartz DR, Cho KR, Morin PJ. Tight junction proteins claudin-3 and claudin-4 are frequently overexpressed in ovarian cancer but not in ovarian cystadenomas. Clin Cancer Res. 2003; 9:2567-2575.
35. Lin Z, Zhang X, Liu Z, Liu Q, Wang L, Lu Y, Liu Y, Wang M, Yang M, Jin X, Quan C. The distinct expression patterns of claudin-2, -6, and -11 between human gastric neoplasms and adjacent non-neoplastic tissues. Diagn Pathol. 2013; 8:133.
36. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004; 429:457-463.
37. Leone G, Voso MT, Teofili L, Lubbert M. Inhibitors of DNA methylation in the treatment of hematological malignancies and MDS. Clin Immunol. 2003; 109:89-102.
38. Santini V, Kantarjian HM, Issa JP. Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann Intern Med. 2001; 134:573-586.
39. Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS, Cortes J, Kantarjian HM. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004; 103:1635-1640.
40. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980; 20:85-93.
41. Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2'-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A. 1994; 91:11797-11801.
42. Agarwal R, D'Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res. 2005; 65:7378-7385.
43. Zhang WN, Li W, Wang XL, Hu Z, Zhu D, Ding WC, Liu D, Li KZ, Ma D, Wang H. CLDN1 expression in cervical cancer cells is related to tumor invasion and metastasis. Oncotarget. 2016; 7:87449-87461. https://doi.org/10.18632/oncotarget.13871.
44. Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009; 69:4116-4124.
45. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002; 18:1427-1431.
46. Eads CA, Lord RV, Kurumboor SK, Wickramasinghe K, Skinner ML, Long TI, Peters JH, DeMeester TR, Danenberg KD, Danenberg PV, Laird PW, Skinner KA. Fields of aberrant CpG island hypermethylation in Barrett's esophagus and associated adenocarcinoma. Cancer Res. 2000; 60:5021-5026.
47. Sikand K, Singh J, Ebron JS, Shukla GC. Housekeeping gene selection advisory: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and beta-actin are targets of miR-644a. PLoS One. 2012; 7:e47510.
48. Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014; 30:1363-1369.
49. Maksimovic J, Gordon L, Oshlack A. SWAN: subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012; 13:R44.
50. Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010; 11:587.