
INTRODUCTION
Membrane phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) provides the anchor site for PH-domain-containing proteins including protein kinase B (AKT) [1]. Due to close proximity to PH-domain-containing kinases, AKT is activated by phosphorylation on cell membrane [2]. The phosphorylated AKT (pAKT) then phosphorylates a variety of target proteins, leading to an increase in cell survival and proliferation [2]. The homeostasis of PIP3 levels are maintained through phosphoinositide-3-kinase (PI3K) and phosphatase and tensin homolog (PTEN). PI3K phosphorylates phosphatidylinositol-(4,5)-diphosphate (PIP2) to generate PIP3, whereas PTEN dephosphorylates PIP3 to generate PIP2 [3]. Thus, PI3K and PTEN are positive and negative regulators of pAKT, respectively [3]. Genetic alterations of PI3K and PTEN have been found in at least sixteen types of human cancers [4]. Nearly 30-60% prostate cancer cases have either gain-of-function-mutation in PI3K or loss-of-function-mutation or deletion in PTEN [4]. About 45% of prostate cancer cases have increased levels of pAKT, which correlates with the disease severity [5, 6]. The loss of PTEN or increase in pAKT at Ser473 has been used to predict advanced prostate cancer that will fail to respond to treatment [7-9].
Studies have shown that polyunsaturated fatty acid, e.g. arachidonic acid (AA), promotes prostate cancer progression. High dietary AA reduces the time required to convert hormone sensitive to refractory prostate cancer [10]. Mice supplemented with AA in the diet show earlier, more frequent and larger tumor recurrence than controls following the surgical removal of prostate cancer xenograft, which imitates prostatectomy in clinical setting [11]. Dietary AA enhances tumor growth in prostate-specific PTEN-knockout mice [12]. In vitro, AA [13] and its metabolites (i.e., eicosanoids) [14] stimulate the survival and proliferation of human prostate cancer cells. The rate of eicosanoid production is also significantly higher in malignant prostate cancer tissues than benign prostate tissues [15]. In two clinical trials, the rate of increase in prostate specific antigen (PSA) following prostatectomy or radiotherapy was slowed in patients treated with inhibitors of eicosanoid-producing enzymes [16, 17]. Collectively, these studies demonstrate a role of AA and its metabolites in prostate cancer progression.
As a derivative of essential fatty acid linoleic acid, the AA is taken up at the cell membrane and esterified mainly into the sn-2 position of glycerophospholipid [18]. The biological action of AA begins after its hydrolysis by phospholipase A2 (PLA2). Of the PLA2 family, the cytosolic PLA2α (cPLA2α) is the isoform that cleaves AA specifically at the sn-2 position [19]. We [14, 20-23] and others [24-26] have shown that cPLA2α action contributes to survival, proliferation, or motility of prostate cancer cells. Blocking cPLA2α with a gene silencing or pharmacological approach retards proliferation of prostate cancer cells in vitro or in vivo [23].
Since both pAKT and cPLA2α levels are implicated in the prostate cancer, and an understanding of the integration of biochemical pathways involved in cancer progression is a key to the development of improved pharmacological treatment strategies for cancer [27, 28], we aimed to examine the relationship between the oncogenic protein and the lipid modifying enzyme. Specifically, we verified the concordance between pAKT and cPLA2α in prostate tissue of epithelial-specific PTEN-knockout mouse, and tested the hypothesis that pAKT plays a causal role in promoting cPLA2α expression in prostate cancer cells.
RESULTS
Concordant change between pAKT and cPLA2α levels in PTEN-knockout mouse prostate
To determine the relationship between pAKT and cPLA2α in vivo, we firstly confirmed the PTEN status by immunostaining of prostate tissues collected from prostate-epithelial-specific PTEN-KO and PTEN-WT mice (Figure 1a). While PTEN was present in both epithelial and stromal compartments of PTEN-WT mice, this trait was absent in cancer in PTEN-KO mice. However, PTEN remained in stromal cells in the PTEN-KO, confirming a selective deletion of PTEN in prostate epithelium. To determine the relationship between pAKT and cPLA2α levels, we determined by immunoblot the AKT and cPLA2α protein levels in the tissue. While the levels of total AKT were not altered significantly, there was a clear increase in pAKT at Ser473 in PTEN-KO compared to PTEN-WT mice (Figure 1b). Interestingly, the levels of total cPLA2α and pcPLA2α at Ser505 were increased significantly in PTEN-KO mice. Hence, pAKT levels are positively correlated with cPLA2α in rodent prostate cancer induced by PTEN-KO.
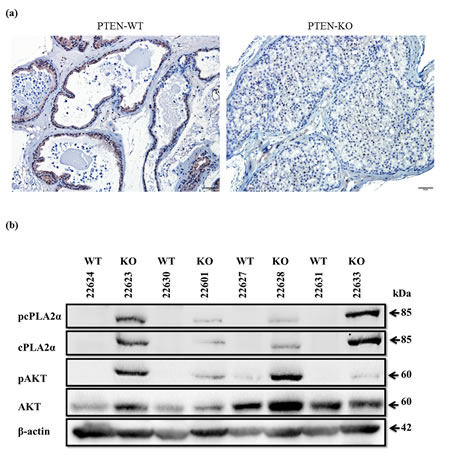
Figure 1: PTEN, AKT and cPLA2α in the prostate of mouse with prostate-epithelial specific knockout (KO) and wide type (WT) of PTEN. (a): Prostate tissue of 6 week-old PTEN KO and WT mice were dissected, fixed, and processed for paraffin blocks. Sections were cut for immunostaining of PTEN. Scale bar: 50 µm. (b): Fresh frozen prostate tissue of 6 week-old PTEN KO and WT were homogenized. The resultant supernatants were used for immunoblot of AKT and cPLA2α.
Decrease in pAKT reduces cPLA2α expression and phosphorylation in prostate cancer cells
To determine if a causal relationship exists between pAKT and cPLA2α, we established a Dox-controlled PTEN expression system in LNCaP cells, which has a frame-shift mutation in PTEN gene resulting in a truncated non-functional PTEN protein [29]. Dox-induced PTEN expression caused a significant decrease in pAKT at Ser473. Concomitantly, phosphorylation of its immediate downstream target GSK3β at Ser9 (Figure 2a) was also diminished. In contrast, total AKT and GSK3β remained unchanged. Control cells transfected with same vector but without PTEN sequence showed no change in pAKT and pGSK3β following Dox treatment. Interestingly, the decrease in pAKT by restoration of PTEN caused reduction of the levels of total cPLA2α and phospho-cPLA2α (pcPLA2α) at Ser505 (Figure 2a, b). Due to the change in configuration following phosphorylation at Ser505, pcPLA2α enhances AA releasing property [30]. In control cells, there was no change in cPLA2α expression or phosphorylation following Dox treatment (Figure 2a, b). As expected, PTEN restoration also reduced the proliferation and increased apoptosis in LNCaP cells compared with control cells which had no functional PTEN (Supplemental Figure 1).
To confirm the effect of PTEN restoration on cPLA2α, we stably transfected another prostate cancer cell line PC-3 with a PTEN-expression construct. PC-3 has a deletion in PTEN gene and thus has no PTEN proteins [31]. Ectopic expression of PTEN caused the reduction of pAKT at Ser473and pGSK3β at Ser9 in PC-3 cells in the absence of alterations in total AKT and GSK3β (Figure 2d). Again, there was a significant decrease in cPLA2α and pcPLA2α at Ser505 in PTEN compared with empty vector transfected PC-3 cells (Figure 2c, d). As expected, PC-3 cell proliferation was reduced after PTEN restoration.
To verify if the regulation of cPLA2α by PTEN is via pAKT, we blocked PI3K enzyme action with LY294002 in PC-3 cells. Indeed, blocking PI3K led to a decrease in levels of pAKT at Ser473 and pGSK3β at Ser9, while there was no change in total AKT and GSK3β (Figure 2e). Similarly, total cPLA2α and pcPLA2α at Ser505 levels were decreased in PC-3 cells treated with PI3K inhibitor compared with vehicle-treated control cells (Figure 2e, f). Taken together, manipulation of pAKT positive regulator (PI3K) or negative regulator (PTEN) changes cPLA2α expression and phosphorylation; suggesting a role of pAKT in the regulation of cPLA2α in prostate cancer cells.
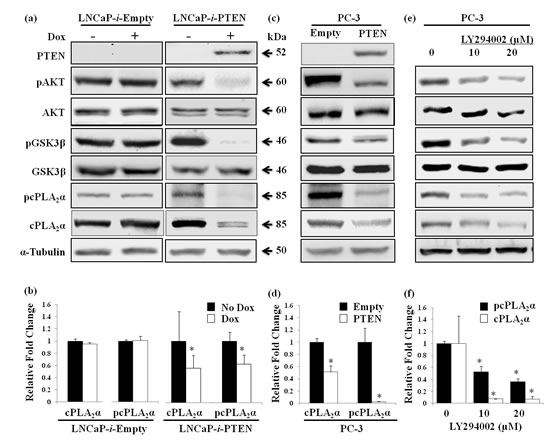
Figure 2: Effect of PTEN expression or PI3K inhibition on cPLA2α protein levels. (a) LNCaP cells carrying tet repressor were stably transfected with either Dox-inducible PTEN (LNCaP-i-PTEN) or the empty vector (LNCaP-i-Empty), and treated with or without Dox (100 ng/mL) for 24 h. The harvested cells were lysed and supernatants were used for immunoblot. (b) Quantified band intensity of 3 independent experiments is represented as the mean ± SD. * P<0.05 versus no Dox treatment. (c) PC-3 cells stably transfected with PTEN or empty vector were lysed for immunoblot. (d) Quantified band intensity of 3 independent experiments is represented as the mean ± SD. * P<0.05 versus empty vector-transfected cells. (e) PC-3 cells treated with PI3K inhibitor (LY294002) for 1 h at indicated doses were harvested at 24 h for immunoblot. (f) Quantified band intensity of 3 independent experiments is represented as the mean ± SD. * P<0.05 versus vehicle-treated control.
Increase in pAKT elevates cPLA2α expression in prostate cancer cells
To determine the effect of an increase in pAKT levels on total cPLA2α and pcPLA2α levels, we transiently transfected LNCaP and PC-3 cells with an expression vector containing Myr-AKT. Due to the addition of the sequence coding for myristoylation signal in AKT construct, the produced AKT protein is able to bind to membrane independent from PIP3 and being phosphorylated. The transfection with Myr-AKT in both LNCaP and PC-3 cells caused an increase in total AKT, pAKT and pGSK3β (Figure 3a). Concomitantly, there was a clear increase in total cPLA2α and pcPLA2α (Figure 3a). Then, we determined the effect of increase in pAKT on cPLA2α in PTEN-positive prostate cancer cells. Myr-AKT was transiently transfected into LNCaP cells in which PTEN had been restored. In the presence of Myr-AKT, the inhibitory effect of PTEN restoration on cPLA2α expression and phosphorylation was abolished (Figure 3b).
To further validate the importance of pAKT on cPLA2α expression and phosphorylation, we introduced AKT inhibitor, AktX, in the presence or absence of Myr-AKT in PC-3 cells. Indeed, blocking AKT phosphorylation with AktX significantly decreased total cPLA2α and pcPLA2α while total AKT was unchanged (Figure 3c). We then confirmed the finding in LNCaP cells with another AKT inhibitor (MK2206). As endogenous cPLA2α levels were relatively low in LNCaP cells, we established a Dox-controlled cPLA2α expression system in LNCaP cells and then introduced MK2206 in the presence of Dox. Blocking AKT phosphorylation with MK2206 significantly decreased total cPLA2α and pcPLA2α while total AKT was unchanged (Figure 3c). These results confirm that phosphorylated form of AKT is required for the effect on cPLA2α protein levels in prostate cancer cells.
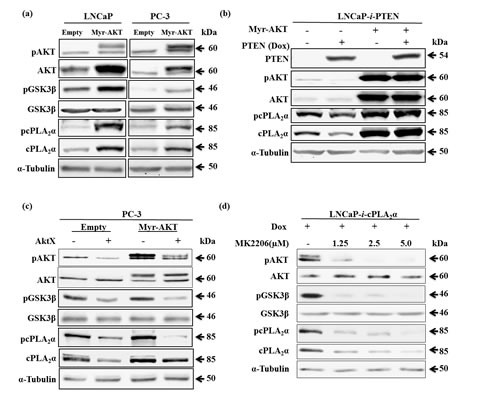
Figure 3: Effect of Myr-AKT expression on cPLA2α protein levels in the presence or absence of PTEN. (a) LNCaP or PC-3 cells transiently transfected with Myr-AKT or empty vector (2 µg, 24 h) were harvested 24 h later. The cell lysates were used for immunoblot. (b) LNCaP-i-PTEN cells transiently transfected with Myr-AKT or empty vector (2 µg, 24 h) were treated with or without Dox (100 ng/mL) for another 24 h. The cell lysates were used for immunoblot. (c) PC-3 cells were transfected with Myr-AKT (2 µg, 24 h) followed by incubation with the AKT phosphorylation inhibitor AktX (5 µM, 1 h). The cells were harvested 24 h later. (d) LNCaP cells stably transfected with Dox-inducible cPLA2α expression system (LNCaP-i-cPLA2α) were treated with Dox (100 ng/mL, 24 h) and then incubated with AKT phosphorylation inhibitor MK2206 at indicated doses for 1 h. The cells were harvested 24 h later. All results are typical of 3 independent experiments.
Regulation of cPLA2α by pAKT is independent of extracellular signal-regulated kinase (ERK1/2)
Previous studies have shown a regulation of cPLA2α phosphorylation at ser505 by mitogen-activated protein (MAP) kinase ERK1/2 [32-34]. To determine if the regulation of cPLA2α by pAKT is related to ERK1/2, we compared the regulation of cPLA2α by these two oncogenic proteins. Blocking MEK, the upstream regulator of ERK1/2, with MEK inhibitor (U0126) in LNCaP cells decreased pERK1/2 and pcPLA2α at Ser505 without changing total cPLA2α protein levels (Figure 4a). However, when LNCaP cells were transfected with Myr-AKT, both total cPLA2α and pcPLA2α protein levels were increased. Moreover, blocking MEK with U0126 in the Myr-AKT transfected LNCaP cells, only the pcPLA2α but not total cPLA2α protein levels were decreased (Figure 4b). Hence, the regulation of cPLA2α by pAKT is via a mechanism different from the MAP kinase ERK1/2.
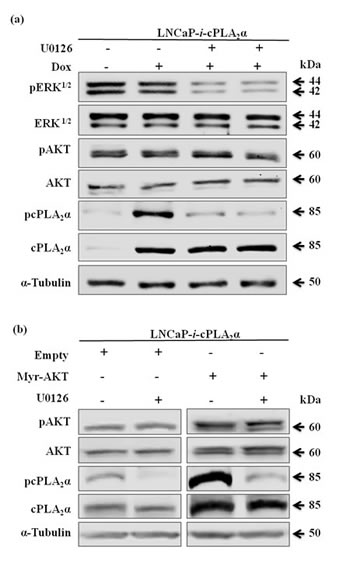
Figure 4: Effect of pAKT on cPLA2α is independent of ERK1/2. (a) LNCaP cells stably transfected with Dox-controlled inducible cPLA2α expression system (LNCaP-i-cPLA2α) were treated with Dox (100 ng/mL, 24 h), followed by incubation with MEK inhibitor U0126 (5 µM, 1 h). The cells were harvested 24 h later. (b) LNCaP-i-cPLA2α cells induced with Dox for 1 h and transfected with Myr-AKT or empty vector (2 µg, 24 h), followed by incubation with U0126 (5 µM, 1 h). The cells were harvested 24 h later. All immunoblotting results are typical of 3 independent experiments.
The decay rate of cPLA2α protein is dependent on pAKT levels
We notice that the magnitude of change in cPLA2α and pcPLA2α in response to pAKT is similar. Hence, pAKT may regulate cPLA2α expression at gene transcription level. To determine if pAKT influences cPLA2α expression at mRNA levels, we measured the steady-state level of cPLA2α mRNA by RT-qPCR. We found no change at mRNA levels of cPLA2α in conditions whether pAKT was increased or decreased (Supplemental Figure 2). Hence, it suggests that regulation of cPLA2α by pAKT occurs at post-transcriptional levels.
We then examined if cPLA2α protein stability was affected by pAKT. After determination of the decay rate of cPLA2α protein in the presence of cycloheximide (Figure 5a,b), PC-3 cells were transfected with Myr-AKT or empty vector followed by incubation with cycloheximide. The decay rate of cPLA2α protein in the presence of Myr-AKT was significantly slowed compared with empty vector-transfected cells (Figure 5c,d). To verify the necessity of pAKT in stabilizing cPLA2α, AKT inhibitor MK2206 was then introduced to Myr-AKT transfected PC-3 cells. Compared with vehicle-treated control cells, AKT inhibitor significantly accelerated the decay rate of cPLA2α protein (Figure 5e,f). It appears that the degradation of cPLA2α is not via proteasome system. Treatment with proteasome inhibitor (MG132), which clearly increased those proteins (p21 and p27) known to be degraded via ubiquitin-proteasome system, had little effect on cPLA2α (Supplement Figure 3).
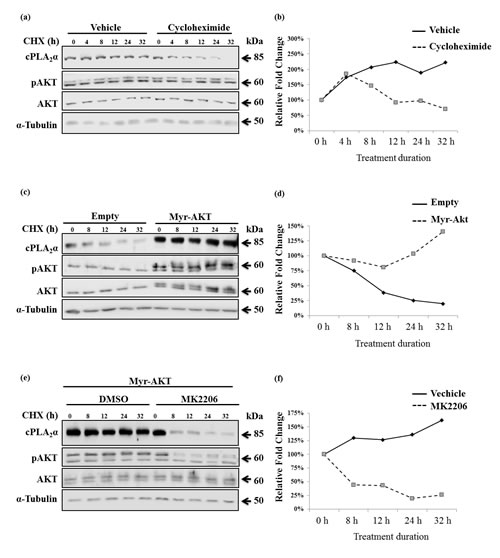
Figure 5: pAKT protects cPLA2α protein from degradation. (a) PC-3 cells were treated with 50 µM cycloheximide (CHX) for the indicated time. The cells were harvested and subjected to immunoblot analysis. (b) PC-3 cells were transfected with empty vector or Myr-AKT (2 µg, 24 h) followed by incubation with CHX (50 µM) for the indicated time; and (c) PC-3 cells were transfected with Myr-AKT (2 µg, 24 h) followed by incubation with MK2206 (5 µM, 1 h). The cells were then treated with 50 µM CHX for the indicated time. All cPLA2α protein levels were normalized by α-tubulin. The ratio of cPLA2α over α-tubulin at time zero of CHX treatment was set as 100%. AKT antibodies detect AKT1 (62 kDa), AKT2 (56 kDa) and AKT3 (62 kDa, expressed mainly in the brain). When the same concentration of PAGE was used, the separation of AKT 1 from 2 is pending on the running time. The Myr-AKT1 migrates closely with the AKT1 on PAGE.
To differentiate whether AKT per se or its target proteins impinge on cPLA2α, we determined the effect of blocking GSK3β (the immediate downstream effector of AKT) on cPLA2α. If this regulation of cPLA2α is downstream of AKT, blocking GSK3β will result in a similar change as PTEN restoration or PI3K inhibition. Interestingly, we found no change in total and pcPLA2α proteins levels despite the successful inhibition of GSK3β (Supplement Figure 4). We then examined the possibility for pAKT and cPLA2α forming a complex. Human embryonic HEK 293 cells were transiently co-transfected with (i) a Myr-AKT construct and a FLAG-cPLA2α construct or a FLAG-empty vector; and (ii) a FLAG-cPLA2α construct and a Myr-AKT construct or a Myr-empty vector, followed by stimulation with EGF for 15 min to increase AKT phosphorylation. We then performed protein co-immunoprecipitation with an anti-FLAG or an anti-AKT antibody. Anti-FLAG antibody co-immunoprecipitated total AKT and more efficiently pAKT. Similarly, anti-AKT antibody co-immunoprecipiated cPLA2α (Figure 6a, b). Taken together, pAKT increases cPLA2α protein expression through a decrease in its protein degradation. The protection of cPLA2α by pAKT is through forming a complex between cPLA2α and pAKT.
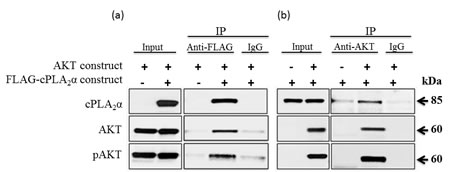
Figure 6: AKT (pAKT) forms a protein complex with cPLA2α. HEK293 cells were co-transfected with (a) a Myr-AKT construct plus FLAG-cPLA2α construct or a FLAG-empty vector; and (b) a FLAG-cPLA2α construct plus a Myr-AKT construct or Myr-empty vector for 36 h. The cells were stimulated with EGF (25 ng/ml, 15 min) to increase AKT phosphorylation. Proteins were extracted and co-immunoprecipitated with a control IgG, an anti-FLAG, or an anti-AKT antibody (Ab, 2 µg each). Eluted proteins were immunoblotted with an anti-cPLA2α, anti-AKT or anti-pAKT Ab.
DISCUSSION
This study has revealed a previously-unrecognized concordant change between pAKT and lipid modifying enzyme cPLA2α protein levels in PTEN-knockout mouse prostate cancer tissues. We have also demonstrated a role of pAKT in protecting cPLA2α from degradation in two human prostate cancer cell lines.
LNCaP is a human lymph node metastasis-derived prostate cancer cell line with a frame-shift mutation in PTEN gene [29]. PC-3 is a bone metastasis-derived prostate cancer cell line with a deletion in PTEN gene [31]. Consequently, pAKT levels are elevated in both cell lines. By determining the response of cPLA2α to the decrease in pAKT, as a result of restoration of PTEN that affects mainly pAKT, we are able to ascertain the effect of a decrease in pAKT on cPLA2α expression and phosphorylation. Further, by blocking enzymatic action of PI3K that also affects mainly pAKT; we can mimic the decrease of pAKT independent of PTEN. It is clear from these genetic (Dox-induced or stable expression of PTEN) and pharmacological (blocking PI3K with LY204002) approaches that a decrease in pAKT leads to a decrease in total and pcPLA2α at Ser505.
To verify the finding aforementioned, we then used Myr-AKT to mimic an increase in pAKT in both LNCaP and PC-3 cells. Due to the inclusion of sequence coding for myristoylation in AKT construct, the produced AKT protein is able to bind to membrane and being phosphorylated there without using PIP3 as the membrane anchor site. We noted a significant increase in cPLA2α expression and phosphorylation in both cell lines following transfection of Myr-AKT. Moreover, Myr-AKT is able to override the suppressive effect of PTEN restoration on cPLA2α expression and phosphorylation.
As Myr-AKT increase both total AKT and pAKT, we introduced inhibitor of AKT phosphorylation in the presence or absence of Myr-AKT in both cell lines. By decreasing only the pAKT, two AKT inhibitors, AktX and MK2206, abolished the stimulatory effect of Myr-AKT on cPLA2α expression and phosphorylation. Together with the cPLA2α response to the decreased pAKT via restoration of PTEN or inhibition of PI3K, it is strongly suggestive that the regulation of cPLA2α is via pAKT not PTEN or PI3K.
Previous studies have demonstrated that the enzyme responsible for phosphorylation of cPLA2α at Ser505 is the phospho-ERK1/2 (pERK1/2) [34]. We have provided evidence in this study that pAKT influences cPLA2α including its phosphorylation at Ser505. To differentiate the action between pERK1/2 and pAKT on cPLA2α, we determined the effect of blocking MEK, the kinase responsible for phosphorylation of ERK1/2, on cPLA2α and pcPLA2α. Blocking MEK and consequently pERK1/2 only decreases pcPLA2α not total cPLA2α, with or without an increased AKT by Myr-AKT. Hence, the regulation of cPLA2α by pAKT is different from pERK1/2 as pAKT affects both cPLA2α expression and phosphorylation. Considering it is common to have an alteration in PI3K and PTEN in various cancers, it will be interesting to determine whether cPLA2α protein expression and phosphorylation also increase in those cancers.
Regarding the mechanism by which pAKT influences cPLA2α levels, as virtually in all experimental systems we adopted in the study that either increaesed or decreased pAKT, both cPLA2α expression and phosphorylation altered. Thus, the change in phospho-cPLA2α level at Ser505 can be at least partially a consequence of change in cPLA2α expression. Hence, we examined the possibility of pAKT affecting cPLA2α gene experssion. However, we saw no change in the steady-state levels of cPLA2α mRNA in all systems used in this study, including PTEN restoration and ectopic expresson of Myr-AKT. These results suggest that the effect of phospho-AKT on cPLA2α expression and phosphorylation is post-transcriptional. Thus, we determined the decay rate of cPLA2α protein following blockade of protein synthesis. The decay rate of cPLA2α was significantly slowed in Myr-AKT transfected cells compared with that in empty vector-transfected cells. Moreover, this slowed decay rate of cPLA2α protein could be reversed by inhibition of AKT phosphorylation. Hence, it is likely that pAKT promotes cPLA2α protein expression and phosphorylation by increasing cPLA2α protein stability. Moreover, we have obtained evidence based on the co-immunoprecipitation assay that pAKT and cPLA2α form a complex. This is consistent with our finding that blocking GSK3β has no effect on cPLA2α protein levels (Supplement Figure 4), making it unlikely that effectors downstream of AKT regulate cPLA2α. As blocking proteasome with MG132 for 24 hours does not increase cPLA2α protein levels (Supplement Figure 3), further study with extended time is needed to determine the mechanism by which AKT stabilizes cPLA2α from degradation. We searched for the AKT1 phosphorylation site based on cPLA2α protein sequence (PA24A_Human, UniProt # P47712) using the Group-based Prediction System (GPS) software Version 3.0 (http://gps.biocuckoo.org). Four sites were predicted to be potentially phosphorylated by AKT1: Ser2, Thr321, Thr416 and Ser729. Further study is needed to determine if these sites can be phosphorylated by AKT in vitro and in vivo, and the corresponding change in cPLA2α protein stability.
Interestingly, this is not the first evidence to show a regulation of a lipid modifying enzyme by AKT. AKT activation increases fatty acid synthase expression in prostate cancer tissue [35-38]. Fatty acid synthase catalyzes the conversion of acetyl-CoA and malonyl-CoA to fatty acid. Our study indicates that AKT has a similar role in regulating fatty acid cleaving enzyme cPLA2α in prostate cancer. Together with our recent reports on regulation of AKT by cPLA2α and its enzymatic products [20], these findings suggest the presence of feed-forward loop between AKT and lipid modifying enzymes that favours cancer cell proliferation. Not surprisingly, it has been suggested that chemotherapy targeting both AKT signaling and lipid metabolism might be of benefit [39, 40].
In conclusion, we have unravelled a novel regulation of cPLA2α expression and phosphorylation by activation of AKT. In light of the biological significance of cPLA2α on cell survival, proliferation, and metabolism, the role of pAKT in promoting cPLA2α protein stability provides a new node of interaction between oncogenic pathway and lipid metabolism and/or inflammation.
MATERIALS AND METHODS
PTEN-knockout mouse
The prostate epithelial-specific PTEN-knockout mouse was generated by Cre-loxP system. A Probasin-Cre line [41] (kindly provided by Dr. Fen Wang, The Center for Cancer Biology and Nutrition, Houston) was crossed with the PTENflox line [42]. Cre negative littermates on FBV/N genetic background were used as the wide-type control. Male mice were collected at median age of 7 weeks. Individual prostate lobes were dissected free of periprostatic fat and connective tissue and weighed. Lobes were either snap frozen with liquid nitrogen and stored in -80 °C for further protein extraction or fixed in 4% paraformaldehyde over night for histology and immunohistochemistry.
Immunohistochemistry
The mouse prostate tissue was processed for paraffin blocks. Subsequently, paraffin sections were cut and baked at 60 °C for 1 h, after which the sections were deparaffinized, re-hydrated and subjected to antigen retrieval in Tris-EDTA solution [43]. The sections were blocked with 10% horse serum, and then incubated with an antibody to PTEN (Cat. #: 9559, Cell Signaling Technology) overnight at 4 °C. The sections were then washed with TBS and sequentially labeled with a biotinylated secondary antibody and Vectastain ABC kit (Vector Laboratories). The labeled PTEN in the sections was revealed with DAB (DakoCytomation). Thereafter, the sections were counterstained with hematoxyline and coverslipped.
Cell culture and chemicals
The bone metastasized prostate cancer cell line, PC-3 (Cat. #: CRL-1435; American Type Culture Collection [ATCC], Manassas, VA), and the lymph node metastasized prostate cancer cell line, LNCaP (Cat. #: CRL-1740; ATCC), were grown in RPMI 1640 supplemented with 10% v/v fetal bovine serum (FBS; AusGeneX, Brisbane, QLD, Australia), penicillin (100 U/mL; Invitrogen, Melbourne, VIC, Australia) and streptomycin (100 µg/mL; Invitrogen). The cells were cultured at 37°C in an incubator providing a humidified environment in the presence of 5% CO2 /95% air. Incubation with various inhibitors began 24 h post seeding. LY294002 (Cat. #: S1105), MK2206 (Cat. #: S1078), U0126 (Cat. #: S1102), and SB216763 (Cat. #: S1075) were purchased from Selleck (Houston, TX). Cycloheximide (Cat. #: C1988), MG132 (Cat. #: C2211), and EGF (Cat. #: E9644) were from Sigma-Aldrich (St. Louis, MO). AktX (Cat. #: 124020) was from Calbiochem (MERCK MILLIPORE, Victoria, Australia).
Stable and transient transfection
LNCaP carrying tet repressor (kindly provided by Dr. P Russell, University of NSW, Australia) were transfected with 20 µg of pcDNA4/TO (Cat. #: V1020-20; Invitrogen) vector with PTEN or cPLA2α sequence using Lipofectamine 2000 (Cat. #: 11668; Invitrogen). The transfected cells were selected by incubation with media containing Blasticidin (5 µg/mL, Invitrogen Cat. #: R210-01) and Geneticin Sulfate (300 µg/mL, Invitrogen Cat. #: 10131) for 10 days. Corresponding control clones were obtained by transfecting cells with the same vector without the PTEN or cPLA2α cDNA sequence. For induction of PTEN or cPLA2α, cells were seeded in 6-well plates and treated with 100 ng/mL of Doxycycline in RPMI/FBS for 24 h. PC-3 stably transfected with PTEN was a gift from Dr Zaklina Kovacevic (The University of Sydney). For transient transfection, LNCaP and PC-3 cells were transfected with 2 µg Myr-AKT (Cat. #: 9008, Addgene, Cambridge, MA) or control vector using Lipofectamine 2000. The cells were incubated for 48 h followed by RNA or protein extraction. Incubation with various inhibitors began 24 h post transfection.
Measurement of cell proliferation and apoptosis
Apolive-Glo Multiplex assay (Cat #: G6410 and G6411) from Promega (Madison, WI) was used to determine the biological effect of PTEN restoration. Briefly the assay determines live cell using a cell membrane permeable fluorogenic peptide which is cleaved by live cell protease activity and generate a fluorescent signal. For the apoptosis, a luminogenic substrate attached to a tetramer peptide is cleaved by Caspase 3/7 activation thereby releasing a glow type luminescent signal.
Protein Co-Immunoprecipitation
Human embryonic HEK 293 cells were transiently transfected with pCMV1-Flag-cPLA2α, pcDNA-Myr-AKT or both with Lipofectamine2000 (Invitrogen) for 36 h. Total protein (0.5 mg) was incubated overnight with 2 µg of anti-Flag, anti-AKT or control IgG antibody. Eluted proteins were immunoblotted with anti-cPLA2α, anti-AKT, and anti-pAKT antibodies. Refer to previous publication for details of methodology [44].
Immunoblotting
PC-3 and LNCaP cells were treated in 6-well plates, and cell lysates were prepared in a lysis buffer as described previously [23]. To detect proteins of interest in mouse prostate tissues, previously frozen tissues were weighed out and added with 20-fold volume of the lysis buffer and homogenized in 1.5-mL Eppendorf tubes on ice. The homogenates were then centrifuged at 12,000 g for 1 min and resultant supernatants were collected and stored at -80 oC until use. Two loading controls, α-tubulin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), were used to avoid overlapping signals from proteins of interest which had a similar molecular weight. Primary antibodies against: cPLA2α (Cat. #: SC-454), phospho-cPLA2α at Ser505 (Cat. #: SC-34391), AKT (Cat. #: SC-8312), and phospho-AKT at Ser473 (Cat. #: SC-7985) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); ERK 1/2 (Cat. #: 9102), phospho-ERK 1/2 (Cat. #: 9106S), GSK3β (Cat. #: 9315) and phospho-GSK3β (Cat. #: 9336S) were bought from Cell Signaling Technology (Danvers, MA); Anti-FLAG-Ab (Cat. #: F3165) were purchased from Sigma; α-tubulin (Cat. #: ab7291) and GAPDH (Cat. #: ab8245) were obtained from Abcam (Boston, MA).
Reverse Transcription-and quantitative real-time PCR
Total RNA was isolated using the UltraClean Tissue & Cells RNA Isolation Kit (Mo Bio Laboratories, CA). The first strand cDNA was synthesized from 500 ng of total RNA with random hexamers and SuperscriptIII (Cat. #: 48190 and P/N 56575, Invitrogen). The primers used for cPLA2α were forward 5’-ATCCTGATGAATTTGAGCGA and reverse 5’CAAGTAGAAGTTCCTTGAACG. The TATA box binding protein (TBP) was used as the housekeeper gene, forward 5’-GAACCACGGCACTGATTTTC, reverse 5’-CCCCACCATGTTCTGAATCT. Quantitative PCR measurements were performed using a SensiMix SYBR Mastermix Kit (Cat. #: QT605, Bioline, Sydney, NSW, Australia) and a RotorGene 6000 PCR machine (Qiagen, Santa Clarita, CA). Conditions for PCR were one cycle of 10 min at 95 oC; 40 cycles of 10 s at 95 oC and 30 s at 60 oC. The Relative Expression Software Tool 2009 (Qiagen) was used to calculate relative changes in cPLA2α normalized to the housekeeping gene. Amplification efficiency was determined using a 5-point dilution curve and was within 100% ± 3% for cPLA2α and TBP.
TCF/LEF promoter assay
The dual luciferase activity of TCF/LEF (TOPO-Flash) promoter assay (Cat. #: E1910) from Promega was used to verify the inhibition of GSK3β by measuring β-catenin activity.
Statistical Analysis
The statistical software NCSS (v12.0; Kaysville, UT) was used for analysis. One-Way ANOVA was implemented to determine the difference between individual groups of data. Fisher’s LSD Multiple-Comparison Test was used to determine whether the difference between individual groups (P<0.05) was considered significant.
ACKNOWLEDGEMENTS
We thank Roland Gamsjaeger for critical reading of the manuscript. This work was supported by IPRS Scholarship (S Vignarajan, the University of Sydney; and C. Xie, The University of Western Sydney); The University of Western Sydney Internal Research Grant Scheme (Q. Dong) and a Seed Grant from the School of Science and Health, The University of Western Sydney (Q. Dong). T. Liu is an Australian Research Council Future Fellow.
REFERENCES
1. Engelman JA, Luo J, Cantley LC, Engelman JA, Luo J and Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature Reviews Genetics. 2006; 7(8):606-619.
2. Manning BD, Cantley LC, Manning BD and Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007; 129(7):1261-1274.
3. Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ and Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annual Review of Biochemistry. 2001; 70:535-602.
4. Chalhoub N and Baker SJ. PTEN and the PI3K pathway in cancer. Annu Rev Pathol Mech Dis 2009; 4:127-150.
5. Malik SN, Brattain M, Ghosh PM, Troyer DA, Prihoda T, Bedolla R, Kreisberg JI, Malik SN, Brattain M, Ghosh PM, Troyer DA, Prihoda T, Bedolla R and Kreisberg JI. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clinical Cancer Research. 2002; 8(4):1168-1171.
6. Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, Shelley SA, Jove R, Tsichlis PN, Nicosia SV and Cheng JQ. AKT1/PKB alpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. American Journal of Pathology. 2001; 159(2):431-437.
7. Kreisberg JI, Malik SN, Prihoda TJ, Bedolla RG, Troyer DA, Kreisberg S, Ghosh PM, Kreisberg JI, Malik SN, Prihoda TJ, Bedolla RG, Troyer DA, Kreisberg S and Ghosh PM. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Research. 2004; 64(15):5232-5236.
8. McCall P, Witton CJ, Grimsley S, Nielsen KV and Edwards J. Is PTEN loss associated with clinical outcome measures in human prostate cancer? British Journal of Cancer. 2008; 99(8):1296-1301.
9. Choucair K, Ejdelman J, Brimo F, Aprikian A, Chevalier S and Lapointe J. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer. 2012; 12:543.
10. McEntee MF, Ziegler C, Reel D, Tomer K, Shoieb A, Ray M, Li X, Neilsen N, Lih FB, O’Rourke D, Whelan J, McEntee MF, Ziegler C, Reel D, Tomer K, Shoieb A, et al. Dietary n-3 polyunsaturated fatty acids enhance hormone ablation therapy in androgen-dependent prostate cancer. American Journal of Pathology. 2008; 173(1):229-241.
11. Kelavkar UP, Hutzley J, Dhir R, Kim P, Allen KG, McHugh K, Kelavkar UP, Hutzley J, Dhir R, Kim P, Allen KGD and McHugh K. Prostate tumor growth and recurrence can be modulated by the omega-6:omega-3 ratio in diet: athymic mouse xenograft model simulating radical prostatectomy. Neoplasia (New York). 2006; 8(2):112-124.
12. Berquin IM, Min Y, Wu R, Wu J, Perry D, Cline JM, Thomas MJ, Thornburg T, Kulik G, Smith A, Edwards IJ, D’Agostino R, Zhang H, Wu H, Kang JX, Chen YQ, et al. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. Journal of Clinical Investigation. 2007; 117(7):1866-1875.
13. Ghosh J and Myers CE. Arachidonic acid stimulates prostate cancer cell growth: critical role of 5-lipoxygenase. Biochemical & Biophysical Research Communications. 1997; 235(2):418-423.
14. Patel MI, Kurek C, Dong Q, Patel MI, Kurek C and Dong Q. The arachidonic acid pathway and its role in prostate cancer development and progression. Journal of Urology. 2008; 179(5):1668-1675.
15. Chaudry AA, Wahle KW, McClinton S and Moffat LE. Arachidonic acid metabolism in benign and malignant prostatic tissue in vitro: effects of fatty acids and cyclooxygenase inhibitors. International Journal of Cancer. 1994; 57(2):176-180.
16. Pruthi RS, Derksen JE, Moore D, Carson CC, Grigson G, Watkins C and Wallen E. Phase II trial of celecoxib in prostate-specific antigen recurrent prostate cancer after definitive radiation therapy or radical prostatectomy. Clinical Cancer Research. 2006; 12((7 Pt 1)):2172-2177.
17. Smith MR, Manola J, Kaufman DS, Oh WK, Bubley GJ and Kantoff PW. Celecoxib versus placebo for men with prostate cancer and a rising serum prostate-specific antigen after radical prostatectomy and/or radiation therapy.[see comment]. Journal of Clinical Oncology. 2006; 24(18):2723-2728.
18. Leslie CC. Regulation of arachidonic acid availability for eicosanoid production. Biochemistry & Cell Biology. 2004; 82(1):1-17.
19. Ghosh M, Tucker DE, Burchett SA and Leslie CC. Properties of the Group IV phospholipase A2 family. Progress in Lipid Research. 2006; 45(6):487-510.
20. Hua S, Yao M, Vignarajan S, Witting P, Hejazi L, Gong Z, Teng Y, Niknami M, Assinder S, Richardson D and Dong Q. Cytosolic phospholipase A2 sustains pAKT, pERK and AR levels in PTEN-null/mutated prostate cancer cells. Biochimica et Biophysica Acta. 2013; 1831(6):1146-1157.
21. Dong. (2009). Phospholipase A2. In: Schwab M, ed. Cancer Encyclopedia: Spinger UK).
22. Dong Q, Patel M, Scott KF, Graham GG, Russell PJ and Sved P. Oncogenic action of phospholipase A2 in prostate cancer. Cancer Letters. 2006; 240(1):9-16.
23. Patel MI, Singh J, Niknami M, Kurek C, Yao M, Lu S, Maclean F, King NJ, Gelb MH, Scott KF, Russell PJ, Boulas J and Dong Q. Cytosolic phospholipase A2-alpha: a potential therapeutic target for prostate cancer. Clinical Cancer Research. 2008; 14(24):8070-8079.
24. Hughes-Fulford M, Tjandrawinata RR, Li C-F and Sayyah S. Arachidonic acid, an omega-6 fatty acid, induces cytoplasmic phospholipase A2 in prostate carcinoma cells. Carcinogenesis. 2005; 26(9):1520-1526.
25. Pineiro R, Maffucci T and Falasca M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene. 2011; 30(2):142-152.
26. Ross S, Essary B, de la Houssaye BA, Pan Z, Mikule K, Mubarak O and Pfenninger KH. Thrombin causes pseudopod detachment via a pathway involving cytosolic phospholipase A2 and 12/15-lipoxygenase products. Cell Growth & Differentiation. 2000; 11(1):19-30.
27. Assinder SJ, Dong Q, Kovacevic Z and Richardson DR. The TGF-beta, PI3K/Akt and PTEN pathways: established and proposed biochemical integration in prostate cancer. Biochem J. 2009; 417(2):411-421.
28. Assinder SJ, Dong Q, Mangs H and Richardson DR. Pharmacological targeting of the integrated protein kinase B, phosphatase and tensin homolog deleted on chromosome 10, and transforming growth factor-beta pathways in prostate cancer. Mol Pharmacol. 2009; 75(3):429-436.
29. Li J, Yen C, Liaw D, Padsypanina K, Bose S, Wang S, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner S, Giovanella B, Ittmann M, tycko B, Hibshoosh H, Wigler M, et al. PTEN a putative tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997; 275:1943-1947.
30. Tucker DE, Ghosh M, Ghomashchi F, Loper R, Suram S, John BS, Girotti M, Bollinger JG, Gelb MH, Leslie CC, Tucker DE, Ghosh M, Ghomashchi F, Loper R, Suram S, John BS, et al. Role of phosphorylation and basic residues in the catalytic domain of cytosolic phospholipase A2alpha in regulating interfacial kinetics and binding and cellular function. Journal of Biological Chemistry. 2009; 284(14):9596-9611.
31. Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ and Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998; 58(13):2720-2723.
32. Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A and Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993; 72(2):269-278.
33. Leslie CC. Properties and regulation of cytosolic phospholipase A2. [Review] [77 refs]. Journal of Biological Chemistry. 1997; 272(27):16709-16712.
34. Gordon RD, Leighton IA, Campbell DG, Cohen P, Creaney A, Wilton DC, Masters DJ, Ritchie GA, Mott R, Taylor IW, Bundell KR, Douglas L, Morten J and Needham M. Cloning and expression of cystolic phospholipase A2 (cPLA2) and a naturally occurring variant. Phosphorylation of Ser505 of recombinant cPLA2 by p42 mitogen-activated protein kinase results in an increase in specific activity. Eur J Biochem. 1996; 238(3):690-697.
35. Bandyopadhyay S, Pai SK, Watabe M, Gross SC, Hirota S, Hosobe S, Tsukada T, Miura K, Saito K, Markwell SJ, Wang Y, Huggenvik J, Pauza ME, Iiizumi M and Watabe K. FAS expression inversely correlates with PTEN level in prostate cancer and a PI 3-kinase inhibitor synergizes with FAS siRNA to induce apoptosis. Oncogene. 2005; 24(34):5389-5395.
36. Swinnen JV, Heemers H, Deboel L, Foufelle F, Heyns W and Verhoeven G. Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway. Oncogene. 2000; 19(45):5173-5181.
37. van den Bergh RC, Essink-Bot ML, Roobol MJ, Wolters T, Schroder FH, Bangma CH, Steyerberg EW, van den Bergh RCN, Essink-Bot M-L, Roobol MJ, Wolters T, Schroder FH, Bangma CH and Steyerberg EW. Anxiety and distress during active surveillance for early prostate cancer. Cancer. 2009; 115(17):3868-3878.
38. van den Bergh RC, Roemeling S, Roobol MJ, Aus G, Hugosson J, Rannikko AS, Tammela TL, Bangma CH and Schroder FH. Outcomes of men with screen-detected prostate cancer eligible for active surveillance who were managed expectantly. European Urology. 2009; 55(1):1-8.
39. Shiragami R, Murata S, Kosugi C, Tezuka T, Yamazaki M, Hirano A, Yoshimura Y, Suzuki M, Shuto K and Koda K. Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. Int J Oncol. 2013; 43(2):431-438.
40. Wang HQ, Altomare DA, Skele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A and Testa JR. Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene. 2005; 24(22):3574-3582.
41. Jin CL, McKeehan K and Wang F. Transgenic mouse with high Cre recombinase activity in all prostate lobes, seminal vesicle, and ductus deferens. Prostate. 2003; 57(2):160-164.
42. Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X and Wu H. Cre/LoxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002; 32(2):148-149.
43. Pileri SA, Roncador G, Ceccarelli C, Piccioli M, Briskomatis A, Sabattini E, Ascani S, Santini D, Piccaluga PP, Leone O, Damiani S, Ercolessi C, Sandri F, Pieri F, Leoncini L and Falini B. Antigen retrieval techniques in immunohistochemistry: comparison of different methods. Journal of Pathology. 1997; 183(1):116-123.
44. Sun Y, Liu PY, Scarlett CJ, Malyukova A, Liu B, Marshall GM, MacKenzie KL, Biankin AV and Liu T. Histone deacetylase 5 blocks neuroblastoma cell differentiation by interacting with N-Myc. Oncogene. 2014; 33(23):2987-2994.