
INTRODUCTION
Caveolae are defined as 50-100 nm flask-shaped invaginations of the plasma membrane [1, 2]. They are especially abundant in specific cell types such as endothelial cells, adipocytes, and muscle cells [3, 4]. Caveolae involve in a number of functions including endocytosis, transcytosis of macromolecules like LDL etc [4–6]. Caveolin-1 and cavin-1 are two essential structural components of caveolae [6, 7].
Caveolin-1 is a 21-22 kD integral membrane protein. It contains cytoplasmic N- and C-terminal domain that intercalate the membrane-association domain including the caveolin scaffold domain and the intramembrane domain [6]. The function of caveolin-1 has been intensively studied. Caveolin-1 mediates endocytosis and transcytosis of LDL in endothelial cells. A caveolin-1 deficiency prevents the transcytosis of LDL across endothelial cells [8]. We also previously reported that TNF-α up-regulates expression of caveolin-1 and LDLR via activating NF-κB, and consequently promotes LDL transcytosis across endothelial cells [9]. In addition, caveolin-1 maintains endothelial nitric oxide synthase (eNOS) in an inactive state [10], consequently limiting NO production [11, 12]. NO is an important gas signaling molecule. It not only regulates vascular tone and permeability, but also regulates activity of protein kinases and transcriptional factors. Caveolin-1 deficiency gives rise to a significant NO production, although the expression of eNOS is down-regulated in caveolin-1 siRNA-treated cells or in caveolin-1 knock out mice [13, 14].
Cavin-1 was originally thought as a polymerase I and transcript release factor (PTRF) to promote transcription. More recently, cavin-1 was found localized in caveolae and proved to be an important structure protein of caveolae. Cavin-1 is recruited to caveolae to stabilize caveolin-1 immediately when caveolin-1 oligomers settle down in the cell membrane [15]. The expression of cavin-1 and caveolin-1 are tightly linked; over-expression of cavin-1 lead to a increased expression of caveolin-1, whereas loss of cavin-1 result in a loss of caveolin-1 because of mis-location of caveolin-1 or enhanced lysosomal degradation of caveolin-1 [16]. Furthermore, genetic deletion of cavin-1 in mice was observed impaired caveolae formation [17, 18]. However, it is yet to be fully clarified whether or not cavin-1 plays any role in regulating LDL transcytosis and eNOS activity.
In present study, we tested the effect of cavin-1 on LDL transcytosis in human umbilical vein endothelial cells (HUVECs) transfected with cavin-1 siRNA. To further clarify the molecular mechanisms, the roles of the downstream protein kinases and transcriptional factors of NO, including AMPK, NF-κB and Sp1 were also invested. We found cavin-1 down-regulation inhibited LDL uptake and transcytosis, which were abrogated by eNOS/AMPK inhibitors. In addition, a significant increased NO production was observed; meanwhile, a decreased protein and mRNA expression of eNOS, caveolin-1 and LDLR was observed, which was associated with the increased NO level. NO-induced AMPK activation and NF-κB /Sp1 inhibition may coordinately contribute to these phenomena.
RESULTS
eNOS/AMPK signaling is involved in cavin-1 siRNA-suppressed LDL uptake and LDL transcytosis
Several lines of evidence suggest that caveolae contribute to LDL transcytosis across endothelial cells [5, 7, 15]. However, is not clear whether cavin-1 can affect LDL transcytosis. The uptake of LDL by endothelial cells is an intermediate step of LDL transcytosis. We performed flow cytometry analysis to evaluate FITC-labeled LDL uptake in HUVECs. As shown in Figure 1A, after incubation with FITC-LDL, the mean fluorescent intensity of HUVEC, which reflects the level of LDL uptake, was significantly increased. Treatment with cavin-1 siRNA significantly reduced the mean fluorescent intensity, indicating a decreased LDL uptake. However, both the endothelial nitric oxide synthase (eNOS) inhibitor (L-NAME) and AMPK inhibitor (Compound C) increased the uptake of LDL, compared with that of cavin-1 siRNA-treated cells (Figure 1A and 1B). To evaluate the impact of AMPK on LDL uptake, AMPKα1/2 siRNA was used to specifically suppress the activity of AMPK. Knockdown of cavin-1consistently inhibited the uptake of LDL per cell, which was partially restored by AMPKα1/2 siRNA (Figure 1C and 1D). To further evaluate the impact of AMPK on LDL uptake, cells were incubated with 5-aminoimidazole -4-carboxamide-1-β-d-ribofuranoside (AICAR), a widely used an AMPK activator [19–22]. As shown in Figure 1E and 1F, treatment with AICAR decreased the uptake of LDL in HUVECs in a concentration-dependent manner. Next, we determined the level of LDL transcytosis across HUVECs by using an in vitro model of LDL transcytosis. As shown in Figure 1H and 1I, cavin-1 knockdown caused a significant inhibition of LDL transcytosis, which was completely abrogated by L-NAME and partially abrogated by AMPK inhibition (Compound C or AMPKα1/2 siRNA). In addition, treatment with AICAR decreased LDL transcytosis in a concentration-dependent manner (Figure 1J).
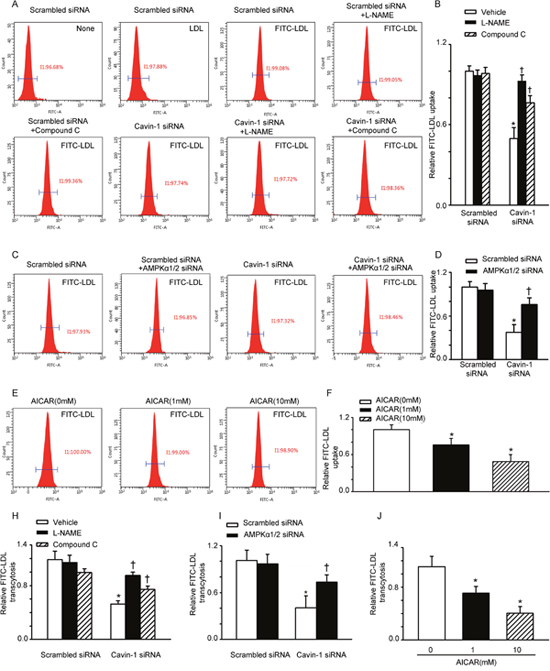
Figure 1: Cavin-1 siRNA suppressed LDL uptake and LDL transcytosis in HUVECs. (A, B) Cells were transfected with scrambled siRNA or cavin-1 siRNA (20 nM) for 48 h, then incubated with L-NAME (50 μM) or Compound C (1 μM ) for 48 h. (C, D) Cells were transfected with scrambled siRNA (20 nM) or cavin-1 siRNA (20 nM) for 3 h, then co-transfected with scrambled siRNA (10 nM) or AMPKα1/2 siRNA (10 nM) for 45 h. (E-F) Cells were treated with indicated concentration of AICAR. (A, C, E) Flow cytometry images of FITC-LDL uptake in HUVECs incubated with FITC-LDL. (B, D, F) Summary bar graph showing the mean FITC-LDL fluorescent intensity in each group. (H, I, J) Quantitative summary of FITC-LDL transcytosis in HUVECs. * P <0.05 vs. scrambled siRNA or AICAR (0 mM); † P <0.05 vs. cavin-1 siRNA, n=4.
Time course for caveolin-1 and eNOS expression after cavin-1 siRNA transfection
LDL transcytosis across endothelial cells is dependent on the function of caveolin-1 [1]. In the present study, we determined that treatment with cavin-1 siRNA inhibited the expression of caveolin-1 after cavin-1 siRNA transefection for 24 h, 36 h, 48 h, and 60 h, which was consistent with the findings presented in previous studies (Figure 2A and 2C) [16]. eNOS is the main regulator of endothelial function by producing NO. Caveolin-1 in caveolae directly interacts with eNOS and regulates nitric oxide (NO) production [12, 23]. NO not only affects vascular tone but also has an impact on gene expression [24]. We further explored the time course for eNOS activity and NO production after cavin-1 siRNA transfection at indicated time points. Compared with scrambled siRNA-treated cells, increased p-eNOS (Ser1177) expression and increased NO production were observed at 24 h, 36 h, 48 h, and 60 h after start of the cavin-1 siRNA transfection (Figure 2B and 2D). The data indicated that cavin-1 siRNA treatment may promote eNOS activation, which was similar to the effects caused by caveolin-1 deficiency [12]. Moreover, we also observed that treatment of cells with cavin-1 siRNA persistently decreased the expression of eNOS from 36 h after start of the transfection.
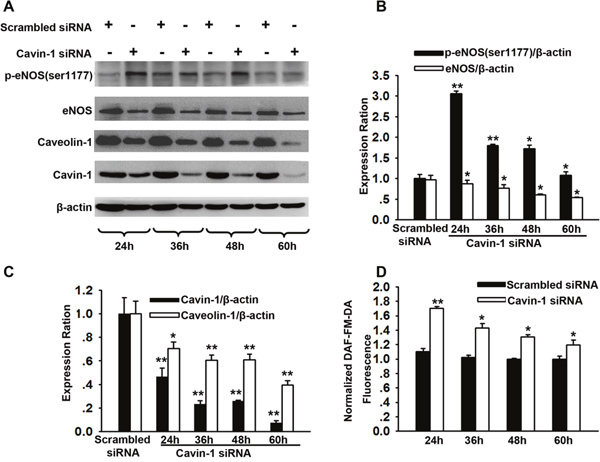
Figure 2: Time course for caveolin-1 and eNOS expression after cavin-1 siRNA transfection. Cells were transfected with cavin-1 siRNA or scrambled siRNA (20 nM) for 24-60 h. (A) The representative western blots image shows the expressions of eNOS, caveolin-1 and cavin-1 at indicated time after siRNA transfection. (B, C) The protein levels of eNOS (B), caveolin-1 (C) and cavin-1 (C) were evaluated by immunoblotting. * P <0.05 or **P <0.01 vs. Scrambled siRNA, n=4. (D) Cavin-1 siRNA enhanced NO production in HUVECs. HUVECs were transfected with cavin-1 siRNA or scrambled siRNA for indicated time and then loaded with DAF-FM-DA dye. After washing with PBS, fluorescence was measured. * P <0.05 or **P <0.01 vs. Scrambled siRNA, n=4.
Cavin-1 knockdown decreased the expression of caveolin-1, eNOS and LDLR
As mentioned above, knockdown of cavin-1 significantly induced NO production, but reduced eNOS expression. NO regulates gene expression in various cell types and species and induces S-nitration of transcription factors, such as NF-κB and subsequent inhibition of its activity [13]. In addition, NO is an endogenous activator of AMPK and functions via a guanylyl cyclase-mediated and Ca2+-dependent CaMKK pathway [25]. Moreover, it has been reported that AMPK and NF-κB /Sp1 directly or indirectly contribute in regulating the expression and activity of caveolae-residing proteins, such as eNOS, caveolin-1, and the LDL receptor (LDLR) [26–28]. We speculated that activating NO/AMPK signals are critical for cavin-1 to modulate the expression of caveolae-residing proteins, such as eNOS, caveolin-1, and LDLR. To elucidate the role of NO/AMPK signals on cavin-1 siRNA-reduced expression of caveolin-1, LDLR, and eNOS, we evaluated the protein expression of these caveolae-residing proteins in the presence of NO/AMPK signals inhibitors. First, L-NAME was used to inhibit the activities of eNOS. As shown in Figure 3A and 3B, knockdown of cavin-1 suppressed the expression of caveolin-1, LDLR, and eNOS, which was completely reversed by treatment with L-NAME. Furthermore, transfection with cavin-1 siRNA reduced the protein expression of caveolin-1, eNOS, and LDLR, which were partially abrogated by AMPKα1/2 siRNA (Figure 3C and 3D). Under physiological conditions, AICAR treatment reduced the expression of caveolin-1, eNOS, and LDLR (Figure 3G and 3H). Moreover, we also observed that the eNOS inhibitor L-NAME suppressed cavin-1 siRNA– or caveolin-1 siRNA–induced activation of AMPK, as indicated by the expression of p-AMPK (Thr172, Figure 3A-B and 3E-F).
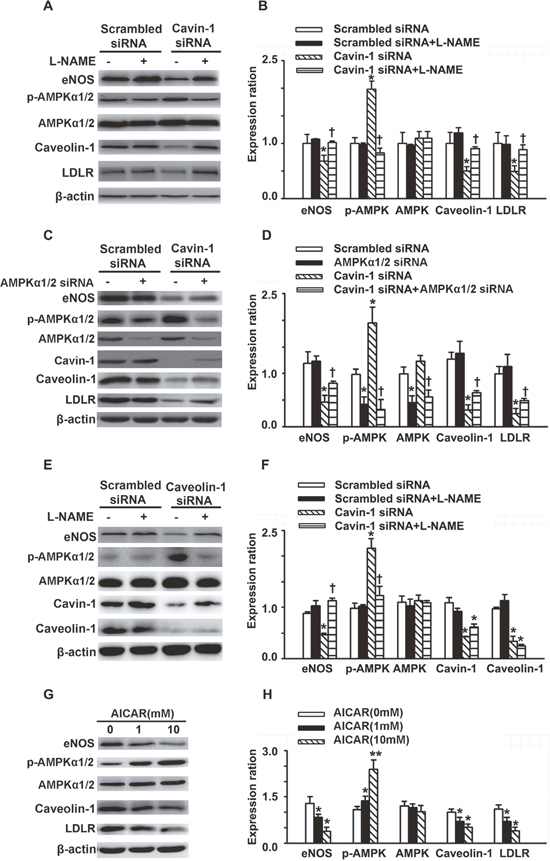
Figure 3: Cavin-1 knockdown decreased the expression of caveolin-1, eNOS and LDLR. (A, B) Cells were transfected with scrambled siRNA or cavin-1 siRNA (20 nM) for 48 h, then incubated with L-NAME (50 μM) for 48 h. (C, D) Cells were transfected with scrambled siRNA (20 nM) or cavin-1 siRNA (20 nM) for 3 h, then cotransfected with scrambled siRNA(10 nM) or AMPKα1/2 siRNA (10 nM) for 45 h. (E, F) Cells were transfected with scrambled siRNA or caveolin-1 siRNA (20 nM) for 48 h, then incubated with L-NAME (50 μM) for 48 h. (G-H) Cells were treated with indicated concentration of AICAR. The protein level of cavolin-1, LDLR, AMPK and eNOS was evaluated by western blotting. (A, C, E, G) The representative images of western blotting. (B, C, D, F) The quantitative analysis of the expression of proteins. *P <0.01 vs. scrambled siRNA or AICAR (0 mM), **P <0.01 vs. AICAR (0 mM), n=4. † P <0.05 vs. Cavin-1 siRNA.
Cavin-1 siRNA inhibited the activity of NF-κB/Sp1, consequently reducing the mRNA expression of caveolin-1, eNOS and LDLR
Previous studies have shown that NF-κB and Sp1 are involved in the transcription of LDLR, caveolin-1, and eNOS [29–32]. In addition, NO has been shown to suppress these transcription factors [31, 33–40]. Therefore, the increased levels of NO induced by cavin-1 knockdown may directly down-regulate NF-κB/Sp1 activity or indirectly down-regulate their activity via activating AMPK, thereby suppressing the transcription of LDLR, caveolin-1, and eNOS. Thus, we sought to evaluate the impact of cavin-1 knockdown-induced eNOS/AMPK signaling pathway activation on the activity of NF-κB/Sp1 and the expression of downstream genes, including LDLR, caveolin-1, and eNOS. As shown in Figure 4A, L-NAME up-regulated the activity of NF-κB/Sp1, when compared with cavin-1 siRNA-treated cells. Similarly, the reduced mRNA levels of LDLR, caveolin-1, and eNOS as caused by cavin-1 siRNA treatment were restored by treatment with L-NAME (Figure 4B). Since AMPK has been reported to inhibit the activity of NF-κB/Sp1 , Compound C and AMPKα1/2 siRNA were used to identify the role of AMPK activation in this process. As shown in Figure 4A and 4C, AMPK inhibition (as mediated by Compound C and AMPKα1/2 siRNA treatment), partially restored the decreased activity of NF-κB/Sp1 caused by cavin-1 siRNA. Moreover, cavin-1 siRNA reduced the mRNA levels of LDLR, caveolin-1, and eNOS, which were partially restored by AMPK inhibitors (Compound C and AMPKα1/2 siRNA) (Figure 4D). In addition, treatment with AICAR decreased the activity of NF-κB/Sp1, consequently inhibiting mRNA expression levels of caveolin-1, eNOS, and LDLR (Figure 4E and 4F).
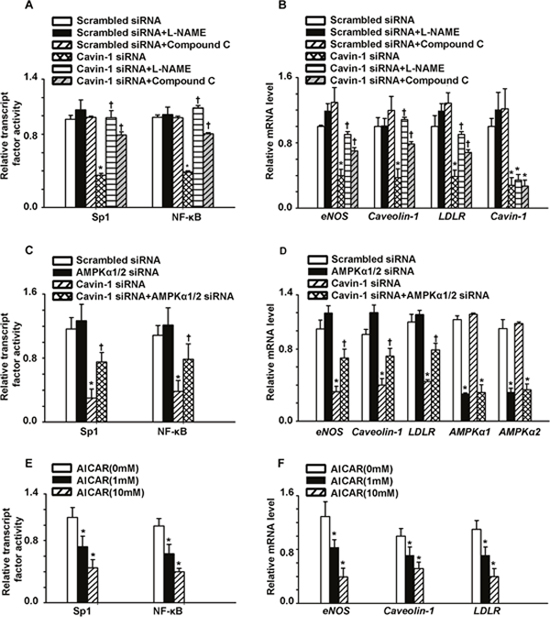
Figure 4: Cavin-1 knockdown inhibited the activity of NF-κB/Sp1, consequently reducing the mRNA expression of caveolin-1, eNOS and LDLR. (A, B) Cells were transfected with scrambled siRNA or cavin-1 siRNA (20 nM) for 48 h, then incubated with L-NAME (50 μM) or Compound C (1 μM ) for 48 h. (C, D) Cells were transfected with scrambled siRNA (20 nM) or cavin-1 siRNA (20 nM) for 3 h, then cotransfected with scrambled siRNA (10 nM) or AMPKα1/2 siRNA (10 nM) for 45 h. (E, F) Cells were treated with indicated concentration of AICAR. (B, D, F) The mRNA level was evaluated by qPCR. (A, C, E) The activity of NF-κB or Sp1 was detected. * P < 0.05 vs. scrambled siRNA; † P < 0.05 vs. cavin-1 siRNA; n=4.
DISCUSSION
Expression of cavin-1 and caveolin-1 are closely linked [14, 16, 18, 23, 41]. The loss of fine balance is expected to result in impaired caveolae and dysfunction of caveolae-related signaling, e.g. LDL transcytosis and eNOS. In present study, we first demonstrated that cavin-1 is essential for caveolae-mediated LDL transcytosis in endothelial cells, and cavin-1 knockdown increased eNOS activity and NO production. Reacting to the increased NO, the activity of NF-κB/Sp1 is inhibited by NO or AMPK, consequently suppressed expression of caveolae-residing proteins such as LDLR, caveolin-1 and eNOS.
Cavin-1 is one of the essential components of caveolae, and is found most abundantly in endothelial cells [42–44]. Functionally, cavin-1 stabilizes caveolae by anchoring caveolin-1 to cytoskeleton via a C-terminal region. Changes of cavin-1 expression result in a comparable change of caveolin-1 expression [18, 44]. Indeed, cavin-1 deficiency leads to loss of morphologically identifiable caveolae [23]. Loss of cavin-1 release caveolae components including caveolin-1 into plasma and increase their lysosomal degradation [16]. Here, consistent with previous studies [45], we also found down-regulation of caveolin-1 expression in cavin-1 knockdown. The interaction of caveolin-1 with eNOS is intensively proved to maintain eNOS in an inactive state [46, 47]. We showed that knockdown of cavin-1 disrupted the association of caveolin-1 with eNOS, thereby enhanced eNOS activity and NO production (Figure 2B and 2D). However, we accidently found the expression of eNOS was significantly decreased from 36 h to 60 h (Figure 2B). Given that NO is an important gas signal molecule, it is necessary to ensure that it is produced the right amount in the right position. Shen et al reported that NO suppressed VEGF-induced increase in eNOS expression, indicating a physiological role for NO in feedback inhibiting eNOS expression [48]. Moreover, Isabella M. Grumbach confirmed that a negative feedback mechanism involving nitric oxide and NF-κB modulates eNOS transcription.Thus, there may be a negative feedback mechanism that down-regulates eNOS expression to limit the NO production when responding to an increased NO level caused by cavin-1 knockdown.
It is reported that NO can regulate the transcription via direct s-nitration of transcript factors [13]. We assume that NO may mediate the decrease in eNOS expression induced by cavin-1 knockout. Hence, we tested the mRNA and protein expression of eNOS, caveolin-1 and LDLR with or without L-NAME (an eNOS inhibitor) in HUVECs transfected with cavin-1 siRNA. As we expected, L-NAME treatment up-regulated the decreased mRNA and protein expression of eNOS, caveolin-1 and LDLR induced by cavin-1 or caveolin-1 knockdown. Previous study has reported that AMPK is also a down-stream signaling molecule of NO [37]. Thus we also measured the activity of AMPK and found that L-NAME treatment inhibited AMPK activation induced by cavin-1 or caveolin-1 knockdown. Furthermore, cavin-1 siRNA suppressed expression of eNOS, caveolin-1 and LDLR, which were partially restored by AMPK inhibitors (Compound C and AMPKα1/2 siRNA). In addition, AICAR, a tool drug employed to activate AMPK, suppressed the expression of eNOS, caveolin-1 and LDLR in physiological condition. In summary, the loss of cavin-1 inhibited the expression of eNOS, caveolin-1 and LDLR, while activating AMPK by increasing eNOS-derived NO.
NF-κB /Sp1, two transcript factors, mediate the transcription of eNOS, caveolin-1 and LDLR [9, 13, 40, 49]. NO cannot only suppress the activity of NF-κB /Sp1, but also inhibit both activities via activating AMPK [28, 50]. Here, we showed that cavin-1 siRNA suppressed the activity of NF-κB/Sp1, which was abrogated by L-NAME. However, AMPK inhibition (as mediated by Compound C and AMPKα1/2 siRNA treatment), partially restored the decreased activity of NF-κB/Sp1 caused by cavin-1 siRNA. As summarized in Figure 5, all of these dated elucidated that cavin-1 knockout increased NO production, which then induced an AMPK/eNOS/NF-κB/Sp1 circuit loop to regulate caveolae residing proteins’ expression, e.g. LDLR, caveolin-1, eNOS, thereby to modulate caveolae-mediated LDL transcytosis in endothelial cells.
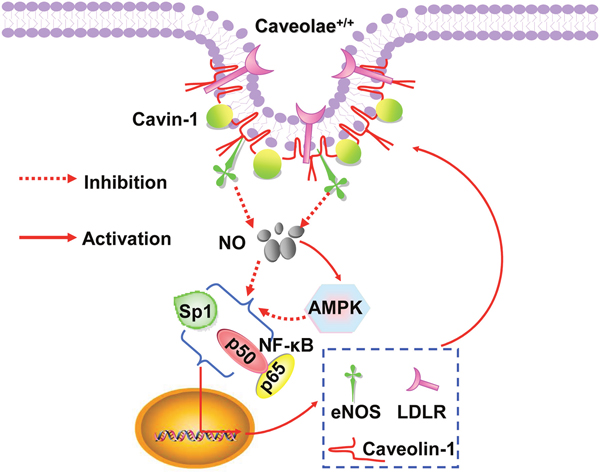
Figure 5: Schematic diagram summarized the molecular mechanism underlying the cavin-1-mediated regulation of downstream proteins.
MATERIALS AND METHODS
Primary culture of human umbilical vein endothelial cells
The collection of human umbilical cords was approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China) and conducted in accordance with the Declaration of Helsinki (2008) and all applicable national and local regulations. All subjects provided written informed consent prior to the initiation of the study. HUVECs were isolated as described [51]. The cells were cultured in endothelial cell medium (ECM, Sciencell, Carlsbad, CA) at 37°C in an incubator with a humidified atmosphere of 5% CO2. Cells were used between passages 2 and 4.
RNA interference
HUVECs grown to confluence in 40%~50% were transfected with 20 nM cavin-1 siRNA (GGAGGTTGAGGAGGTTATT), caveolin-1 siRNA(AACGAGAAGCAAGTGTACGAC) or AMPK α1/2 siRNA (Santa Cruz Biotech-nology, CA, USA) using Hyperfect transfection reagent (Qiagen, Hilden, Germany). siRNAs were synthesized by RIBOBIO (Guangzhou, China). To evaluate the effect of NO, L-NAME at the concentration of 50 μM was added at the beginning of siRNA transfection, Complete medium containing 50 μM L-NAME was replenished every 24 h. AICAR (Selleckchem, Houston, TX, USA).
Western blot assay
Western blot assay was carried out as described previously [52]. The following primary antibodies were used at 1:1000 dilutions: rabbit anti-p-eNOS ser1177, anti-eNOS, anti–p-AMPKα thr172, anti-AMPKα, anti-caveolin-1, and anti-β-actin purchased from Cell Signaling Technology (Beverly, MA); Anti-cavin-1 and anti-LDLR purchased from Proteintech (China). The goat anti-rabbit and goat anti-mouse secondary antibodies from Abbkine (Redlands, CA, USA) were used at 1:10 000 dilution.
NF-κB and Sp1 activity assay
NF-κB and Sp1 activity was assayed by an ELISA-based method as described previously [53]. Total cell extracts were collected and quantified with BCA reagent (Pierce, Rockford, IL). The cell extracts were incubated in a streptavidin-coated 96-well plate (Roche Diagnostics Corporation, Indianapolis, IN 46256), which had been coated with 3’end biotinylated oligonucleotide containing the conservative binding site NF-κB or Sp1. The sequences were shown in Supplementary Table 1. Activated transcription factors from extracts specifically bound to the respective immobilized oligonucleotide. NF-κB or Sp1 activity was detected with NF-κB or Sp1 antibody (1:500, Proteintech, China) and secondary antibody conjugated to horseradish peroxidase (1:10 000, Abbkine, CA). NF-κB and Sp1 activity was finally determined as absorbance values measured with a microplate reader at a wavelength of 450 nm.
RNA extraction and quantitative PCR
Total RNA was extracted from HUVECs using the TRIzol reagent (Invitrogen), and 2 μg of RNA was used to synthesize the first strand cDNA using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). cDNA (2 μl) was amplified using specific primers (Supplementary Table 1) and SYBR Green qPCR kit (TOYOBO, Osaka, Japan).The specific primers are listed in Supplementary Table 1. Expression levels were normalized to β-actin.
Measurement of intracellular NO production
The cell-permeable dye 4,5-diaminofluorescein diacetate (DAF-FM-DA, Beyotime Institute of Biotechnology, China) was used to measure the intracellular NO levels. The experiment was conducted as described previously [54].
LDL uptake in HUVECs
FITC-LDL was prepared as described previously [55]. HUVECs were seeded on 12-well plate (Roche Diagnostics Corporation, Indianapolis, IN 46256) and were transfected with specific siRNA for 45h. And then, the transfected cells were incubated with serum-free Opti-MEM containing FITC-LDL (50 μg/ml) for 3 h. Cells were harvested using 0.125% typsin (with out EDTA) and the uptake of LDL were measured by flow cytometry (Mindry, Bricyte E6). Meanwhile, background fluorescence of HUVEC treated with naïve LDL was subtracted from the mean FITC-LDL fluorescent intensity of each sample, representing the uptake of FITC-LDL.
Measurement of LDL transcytosis
The amount of LDL transcytosis was measured as described previously [55]. Briefly, HUVECs were seeded on polyester membrane of costar transwell (6.5 mm diameter and 0.4 μm pore size) and the integrity of the cell monolayer was tested by a method described previously [56]. Two inserts of cell monolayers with equal integrity were assigned into the same group: the non-competitive insert and the competitive insert. HUVECs grown to confluence in 30-40% were transfected with siRNA for 45h. The non-competitive insert was incubated with FITC-LDL (50 μg/ml) for 3h to determine the total amount of trans-endothelial LDL. Paracellular transport was determined by incubation with 50 μg/ml FITC-LDL and 6-fold excess unlabeled LDL (300 μg/ml) in competitive insert. Samples were then collected from the outer chambers and further dialyzed against PBS to remove the free FITC due to degradation or metabolism in the cell. The FITC fluorescent intensity was measured by a fluorescence spectrophotometer (Tecan, Ininite F200PRO) with excitation and emission wavelengths of 490nm and 520nm, Meanwhile, background fluorescence of serum-free Opti-MEM was subtracted from the value of each sample. The amount of LDL transcytosis is the difference in the fluorescent intensity between the non-competitive insert and the competitive insert.
Statistical analysis
All data were expressed as the mean± SEM from at least three separate experiments. Unpaired Student t-tests with Bonferroni correction were used to analyze individual group statistical comparisons, and one-way ANOVA with post hoc testing was used to evaluate multiple group comparisons. Statistical significance is defined as P <0.05.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORTS
This study was supported by Grants from the National Natural Science Foundation of China (81373413, 8157343, and 81470458), Grants from the Ministry of Education of China (NCET-10-0409) and Grants from the Fundamental Research Funds for the Central Universities (2015ZHYX006, 2016YXZD023 and 0118550041). This work was supported by Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST.
REFERENCES
1. Sun SW, Zu XY, Tuo QH, Chen LX, Lei XY, Li K, Tang CK, Liao DF. Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells. Acta Pharmacol Sin. 2010; 31: 1336-42.
2. Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002; 54: 431-67.
3. Pilch PF, Souto RP, Liu L, Jedrychowski MP, Berg EA, Costello CE, Gygi SP. Cellular spelunking: exploring adipocyte caveolae. J Lipid Res. 2007; 48: 2103-11.
4. Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007; 8: 185-94.
5. Frank PG, Pavlides S, Lisanti MP. Caveolae and transcytosis in endothelial cells: role in atherosclerosis. Cell Tissue Res. 2009; 335: 41-7.
6. Parton RG, Hanzal-Bayer M, Hancock JF. Biogenesis of caveolae: a structural model for caveolin-induced domain formation. J Cell Sci. 2006; 119: 787-96.
7. Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013; 14: 98-112.
8. Frank PG, Pavlides S, Cheung MW, Daumer K, Lisanti MP. Role of caveolin-1 in the regulation of lipoprotein metabolism. Am J Physiol Cell Physiol. 2008; 295: C242-48.
9. Zhang Y, Yang X, Bian F, Wu P, Xing S, Xu G, Li W, Chi J, Ouyang C, Zheng T, Wu D, Zhang Y, Li Y, Jin S. TNF-α promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: crosstalk between NF-κB and PPAR-γ. J Mol Cell Cardiol. 2014; 72:85–94.
10. Bernatchez PN, Bauer PM, Yu J, Prendergast JS, He P, Sessa WC. Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc Natl Acad Sci USA. 2005; 102:761–66.
11. García-Cardeña G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC, and GarciaCardena G. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997; 272:25437–40.
12. Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, Sessa WC. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000; 6: 1362-67.
13. Grumbach IM, Chen W, Mertens SA, Harrison DG. A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription. J Mol Cell Cardiol. 2005; 39: 595-603.
14. Aravamudan B, VanOosten SK, Meuchel LW, Vohra P, Thompson M, Sieck GC, Prakash YS, Pabelick CM. Caveolin-1 knockout mice exhibit airway hyperreactivity. Am J Physiol Lung Cell Mol Physiol. 2012; 303:L669–81.
15. Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol. 2003; 23: 1161-68.
16. Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J Cell Biol. 2010; 191:615–29.
17. Liu L, Pilch PF. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem. 2008; 283: 4314-22.
18. Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, Walser P, Abankwa D, Oorschot VM, Martin S, Hancock JF, Parton RG. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008; 132: 113-24.
19. Zheng T, Yang X, Wu D, Xing S, Bian F, Li W, Chi J, Bai X, Wu G, Chen X, Zhang Y, Jin S. Salidroside ameliorates insulin resistance through activation of a mitochondria-associated AMPK/PI3K/Akt/GSK3β pathway. Br J Pharmacol. 2015; 172:3284–301.
20. Wu D, Yang X, Zheng T, Xing S, Wang J, Chi J, Bian F, Li W, Xu G, Bai X, Wu G, Jin S. A Novel Mechanism of Action for Salidroside to Alleviate Diabetic Albuminuria: Effects on Albumin Transcytosis across Glomerular Endothelial Cells. Am J Physiol Endocrinol Metab. 2016; 310:E225-37.
21. Iglesias MA, Ye JM, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, Cooney GJ, Kraegen EW. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes. 2002; 51: 2886-94.
22. Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF, Goodyear LJ. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006; 55: 2067-76.
23. Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008; 8: 310-7.
24. Pfeilschifter J, Eberhardt W, Beck KF. Regulation of gene expression by nitric oxide. Pflugers Arch. 2001; 442: 479-86.
25. Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003; 278: 31629-39.
26. Mauro L, Pellegrino M, Giordano F, Ricchio E, Rizza P, De Amicis F, Catalano S, Bonofiglio D, Panno ML, Andò S. Estrogen receptor-α drives adiponectin effects on cyclin D1 expression in breast cancer cells. FASEB J. 2015; 29:2150–60.
27. Kim HG, Hien TT, Han EH, Hwang YP, Choi JH, Kang KW, Kwon KI, Kim BH, Kim SK, Song GY, Jeong TC, Jeong HG. Metformin inhibits P-glycoprotein expression via the NF-κB pathway and CRE transcriptional activity through AMPK activation. Br J Pharmacol. 2011; 162:1096–108.
28. Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced NF-kB activation via AMPK activation in vascular endothelial cells. Hypertension. 2006; 47: 1183-88.
29. Zhang R, Min W, Sessa WC. Functional analysis of the human endothelial nitric oxide synthase promoter Sp1 and GATA factors are necessary for basal transcription in endothelial cells. J Biol Chem. 1995; 270: 15320-6.
30. Gan Y, Shen YH, Wang J, Wang X, Utama B, Wang J, Wang XL. Role of histone deacetylation in cell-specific expression of endothelial nitric-oxide synthase. J Biol Chem. 2005; 280: 16467-75.
31. Spiecker M, Peng HB, Liao JK. Inhibition of endothelial vascular cell adhesion molecule-1 expression by nitric oxide involves the induction and nuclear translocation of IkappaBalpha. J Biol Chem. 1997; 272:30969–74.
32. Dasari A, Bartholomew JN, Volonte D, Galbiati F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1–mediated activation of two GC-rich promoter elements. Cancer Res. 2006; 66: 10805-14.
33. Zhang J, Wang S, Wesley RA, Danner RL. Adjacent sequence controls the response polarity of nitric oxide-sensitive Sp factor binding sites. J Biol Chem. 2003; 278: 29192-200.
34. Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-κB DNA binding by nitric oxide. Nucleic Acids Res. 1996; 24: 2236-42.
35. Peng HB, Libby P, Liao JK. Induction and stabilization of I κ B α by nitric oxide mediates inhibition of NF-κ B. J Biol Chem. 1995; 270:14214–19.
36. Liu J, Bi X, Chen T, Zhang Q, Wang SX, Chiu JJ, Liu GS, Zhang Y, Bu P, Jiang F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015; 6: e1827.
37. Zhang J, Xie Z, Dong Y, Wang S, Liu C, Zou MH. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem. 2008; 283: 27452-61.
38. Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001; 108:1341–48.
39. Berendji-Grün D, Kolb-Bachofen V, Kröncke KD. Nitric oxide inhibits endothelial IL-1[beta]-induced ICAM-1 gene expression at the transcriptional level decreasing Sp1 and AP-1 activity. Mol Med. 2001; 7:748–54.
40. Sellak H, Yang X, Cao X, Cornwell T, Soff GA, Lincoln T. Sp1 transcription factor as a molecular target for nitric oxide–and cyclic nucleotide–mediated suppression of cGMP-dependent protein kinase-Iα expression in vascular smooth muscle cells. Circ Res. 2002; 90: 405-12.
41. Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001; 293: 2449-52.
42. Briand N, Dugail I, Le Lay S. Cavin proteins: new players in the caveolae field. Biochimie. 2011; 93: 71-77.
43. Bai L, Deng X, Li J, Wang M, Li Q, An W, A D, Cong YS. Regulation of cellular senescence by the essential caveolar component PTRF/Cavin-1. Cell Res. 2011; 21: 1088-101.
44. Hayer A, Stoeber M, Bissig C, Helenius A. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic. 2010; 11: 361-82.
45. Verma P, Ostermeyer-Fay AG, Brown DA. Caveolin-1 induces formation of membrane tubules that sense actomyosin tension and are inhibited by polymerase I and transcript release factor/cavin-1. Mol Biol Cell. 2010; 21: 2226-40.
46. Collins BM, Davis MJ, Hancock JF, Parton RG. Structure-based reassessment of the caveolin signaling model: do caveolae regulate signaling through caveolin-protein interactions? Dev Cell. 2012; 23: 11-20.
47. Hoop CL, Sivanandam VN, Kodali R, Srnec MN, van der Wel PC. Structural characterization of the caveolin scaffolding domain in association with cholesterol-rich membranes. Biochemistry. 2012; 51:90–99.
48. Shen BQ, Lee DY, Zioncheck TF. Vascular endothelial growth factor governs endothelial nitric-oxide synthase expression via a KDR/Flk-1 receptor and a protein kinase C signaling pathway. J Biol Chem. 1999; 274: 33057-63.
49. Li C, Briggs MR, Ahlborn TE, Kraemer FB, Liu J. Requirement of Sp1 and estrogen receptor α interaction in 17β-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology. 2001; 142:1546–53.
50. Zhao S, Wu J, Zheng F, Tang Q, Yang L, Li L, Wu W, Hann SS. β-elemene inhibited expression of DNA methyltransferase 1 through activation of ERK1/2 and AMPKα signalling pathways in human lung cancer cells: the role of Sp1. J Cell Mol Med. 2015; 19: 630-41.
51. Cheung AL. Isolation and culture of human umbilical vein endothelial cells (HUVEC). Curr Protoc Microbiol. 2007; Appendix 4:Appendix 4B.
52. Xing SS, Yang XY, Zheng T, Li WJ, Wu D, Chi JY, Bian F, Bai XL, Wu GJ, Zhang YZ, Zhang CT, Zhang YH, Li YS, Jin S. Salidroside improves endothelial function and alleviates atherosclerosis by activating a mitochondria-related AMPK/PI3K/Akt/eNOS pathway. Vascul Pharmacol. 2015; 72: 141-52.
53. Jin S, Lu D, Ye S, Ye H, Zhu L, Feng Z, Liu S, Wang D, Hu Q. A simplified probe preparation for ELISA-based NF-kappaB activity assay. J Biochem Biophys Methods. 2005; 65: 20-9.
54. Xing S, Yang X, Li W, Bian F, Wu D, Chi J, Xu G, Zhang Y, Jin S. Salidroside stimulates mitochondrial biogenesis and protects against H2O2-induced endothelial dysfunction. Oxid Med Cell Longev. 2014; 2014: 904834.
55. Bian F, Yang X, Zhou F, Wu PH, Xing S, Xu G, Li W, Chi J, Ouyang C, Zhang Y, Xiong B, Li Y, Zheng T, et al. C-reactive protein promotes atherosclerosis by increasing LDL transcytosis across endothelial cells. Br J Pharmacol. 2014; 171: 2671-84.
56. Cankova Z, Huang JD, Kruth HS, Johnson M. Passage of low-density lipoproteins through Bruch’s membrane and choroid. Exp Eye Res. 2011; 93:947–55.