
INTRODUCTION
Deaths resulting from hepatocellular carcinoma (HCC) rank third in all malignancy-related mortality [1]. In China, liver cancer is the most commonly diagnosed cancer and the leading cause of cancer death in men under the age of 60 years [2]. Surgical resection and liver transplantation are the best options for HCC treatment. However, majority of HCC patients are not eligible for surgical operation due to metastasis, which is also a major obstacle to cure.
Systemic dissection of the molecular mechanisms underlying metastatic progression of HCC is necessary for the development of new diagnostic and therapeutic strategies to prevent and treat metastases. Epithelial-to-mesenchymal transition (EMT) is a key event in metastasis [3, 4]. CR6-interacting factor 1 (CRIF1), also known as Gadd45gip1 or PRG6, regulates cell cycle by inhibiting G1 to S phase progression, and binds to Gadd45 family [5], which plays an important role in genomic stability and regulation of the cell cycle. However, the precise role of CRIF1 in HCC has not been investigated.
In this study, we evaluated the expression of CRIF1 in HCC tissues, and investigated its function in regulating HCC cell proliferation, in vivo tumor growth, EMT, and HCC metastasis.
RESULTS
CRIF1 expression is decreased in HCC patients, and correlates with patients’ survival
First, we analyzed CRIF1 expression in human HCC tissues by immunohistochemistry (IHC). 38 out of 109 HCC tissues (34.9%) had high CRIF1 levels (staining score>6), while 66 of the 109 para-tumor tissues (60.6%) had high CRIF1 levels (chi-square test, P<0.001). CRIF1 was downregulated in HCC tissue (Figure 1A, 1B, 1C, 1D and 1E). Pearson correlation analysis revealed that the CRIF1 protein level was tightly related to tumor differentiation (P = 0.049) and diameter (P = 0.047) (Table 1). Univariate analysis demonstrated that tumor diameter, differentiation, and CRIF1 expression were of statistical significance. As shown in Figure 1F, Decreased CRIF1 expression in HCC tissues was associated with poor post-operative survival (P=0.018). The overall three-year survival rates for the low CRIF1 and high CRIF1 groups were 25.8% and 48.8% respectively (P=0.023).
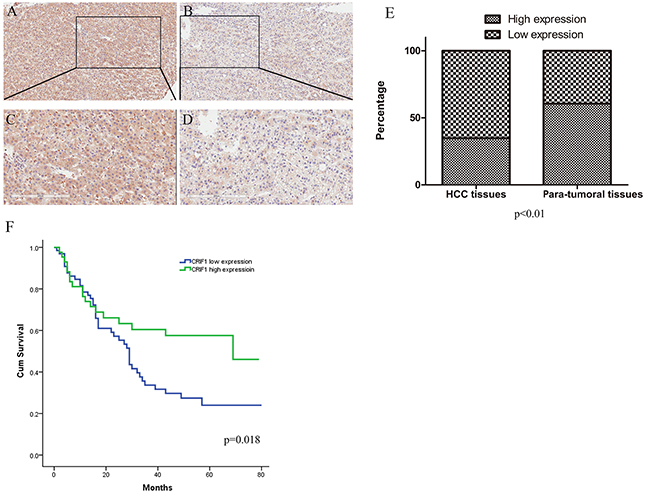
Figure 1: Immunohistochemical staining and prognosis value of CRIF1. Immunohistochemical analysis showed that the CRIF1 protein expression was lower in HCC tissues (B and D) than in paracarcinoma tissues (A and C). (A, B: 100× magnification; C, D: with 200× magnification). (E) CRIF1 was downregulated in the tumor tissues (P<0.01). (F) Its expression level was negatively correlated with the prognosis of HCC patients (P=0.018).
Table 1: Associations between CRIF1 protein expression level and clinic-pathological features of 109 patients with hepatocellular carcinoma
Variables |
score |
p |
|
|---|---|---|---|
0-6 |
7-12 |
||
Age (≤50, >50) |
17/49 |
13/30 |
0.609 |
Tumor diameter (≤5, >5cm) |
22/44 |
23/20 |
0.047 |
Cirrhosis (with/without) |
40/26 |
24/19 |
0.619 |
Differentiation (I/II/III) |
1/30/35 |
2/25/16 |
0.049 |
AJCC stage (1, 2/3,4) |
8/21/33/4 |
10/17/14/2 |
0.228 |
CRIF1 suppression promotes proliferation of HCC Huh-7 and SK-HEP-1 cells
CRIF1 knockdown promoted cell proliferation and invasive potency, while restoration of CRIF1 had the opposite effect (Figures 2A, 2B and 2C). Flow cytometry revealed that CRIF1 knockdown decreased the number of G1 phase cells and increased the number of G2 and S phase cells (Figure 2D). In accordance, proteins controlling G1/S transition, including cyclin D1, cyclin E1, CDK4, and CDK6 were significantly upregulated after CRIF1 knockdown (Figure 2E).
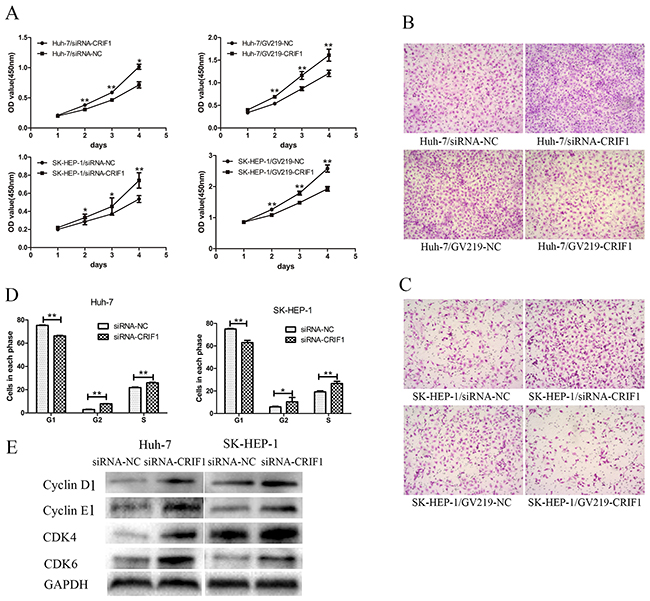
Figure 2: The effects of CRIF1 on phenotype of HCC cell lines. (A) Knockdown of CRIF1 promoted cell proliferation, while enforced expression of CRIF1 inhibited it in Huh-7 and SK-HEP-1. (B, C) The invasive potential of HCC cells was increased in the siRNA-CRIF1 group, and decreased in the GV219-CRIF1 group. (D) CRIF1 knockdown decreased the number of G1 phase cells and increased the number of G2 and S phase cells in hepatocellular carcinoma cell lines. (E) CRIF1 knockdown resulted in elevation of Cyclin D, Cyclin E, CDK4 and CDK6. (**: P<0.01, *:0.01<p<0.05).
CRIF1 inhibits HCC tumorigenicity in mice
To evaluate the role of CRIF1 in promoting HCC tumorigenicity in vivo, mice were subcutaneously injected with HCC cells with CRIF1 knockdown (CRIF1-KD) or normal control (NC) cells. As shown in Figure 3, four weeks after injection, the CRIF1-KD group had significantly larger tumors (1.115 +/- 0.209 cm, n=8) than the CRIF1-NC group (0.496 +/- 0.196 cm, n=8, P<0.001). Xenograft tumors in the CRIF1-KD group had elevated percentage of Ki-67 positive cells compared to the control group (Figures 3C–3F).
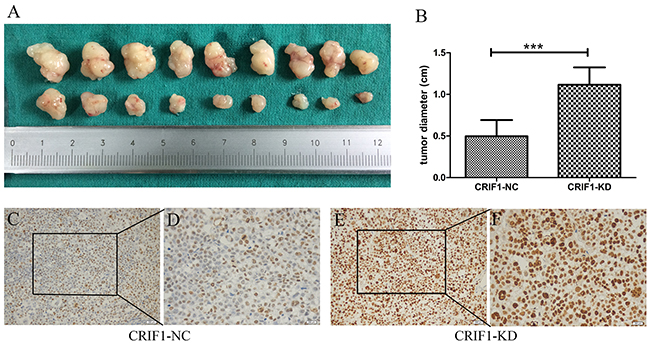
Figure 3: (A, B) Effect of stable CRIF1 on xenograft tumor size 4 weeks after injection of HCC cells. IHC staining of Ki67 for the xenograft tumor of the lenti-CRIF group (C, D) and lenti-NC group (E, F); 200× and 400 ×, magnification).
Gene expression microarray
A total of 527 differentially expressed genes (fold change>2, p < 0.05) were identified between SK-HEP-1 cells transfected with siRNA-CRIF1 or siRNA-NC. The data were further analyzed with an online database (http://david.abcc.ncifcrf.gov/). The gene ontology analysis (Figure 4A) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Figure 4B) indicated involvement of genes and signaling pathways involved in the regulation of cell death, apoptosis, adhesion, and migration. Among them, MMP3 exhibited the most significant change. Knockdown of CRIF1 in HCC cells increased the mRNA expression of MMP3, Smad6, CDK6, and several other genes, while restoration of CRIF1 decreased the expression of these genes (Figures 4C and 4D).
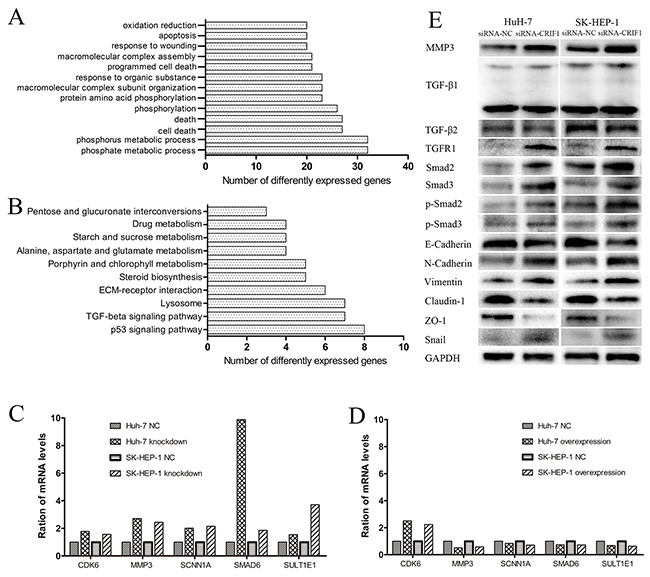
Figure 4: Analysis and validation of the mRNA microarray. (A and B) The gene ontology and KEGG analyses of differentially expressed genes identified by microarray. (C, D) Validation of the most significantly altered genes in the array by RT-PCR. (E) Validation of the TGF-β signaling and EMT-associated proteins by western blot.
TGF-β signaling is activated by CRIF1 knockdown in HCC cells
In KEGG analysis, TGF-β signaling was increased following CRIF1 knockdown. In accordance, we found increased expression of TGF-β receptor I, Smad2, Smad3, phosphorylated Smad2 (p-Smad2) and phosphorylated Smad3 (p-Smad3) (Figure 4E), indicating activation of TGF-β/Smad signaling in CRIF1 suppressed HCC cells.
Since the activation of TGF-β signaling causes EMT and tumor metastasis, we investigated the impact of CRIF1 suppression on EMT in HCC cells. As shown in Figure 4E, we found a decrease of epithelial marker E-cadherin and elevation of mesenchymal markers N-cadherin and vimentin in CRIF1-suppresed cells. Snail, which binds to E-cadherin promoter region to repress transcription, was also upregulated. Meanwhile, tight junction-associated proteins claudin and ZO-1 were downregulated. Double immunofluorescent staining of E-cadherin and N-cadherin confirmed that CRIF1 knockdown promoted EMT (Figure 5). When treated with TGF-β receptor I specific inhibitor SB525334, CRIF1 knockdown-activated TGF-β signaling, invasion, and EMT were abrogated (Figure 6), suggesting that CRIF1 suppression promotes invasion and EMT in Huh-7 and SK-HEP-1 cells through TGF-β/Smad signaling.
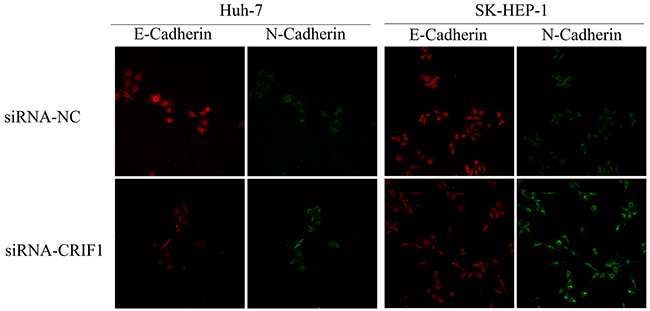
Figure 5: Immunofluorescent double staining of E-Cadherin and N-Cadherin.
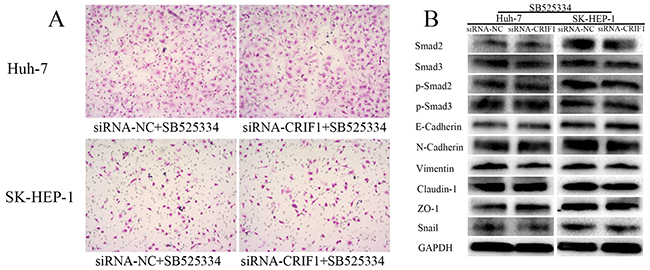
Figure 6: The effects of CRIF1 knockdown was abrogated by TGF-β receptor I specific inhibitor SB525334. (A) Increase in invasive potency of CRIF1 knockdown was abrogated by SB525334. (B) Alteration of TGF-β signaling and EMT-associated proteins were abrogated by SB525334.
DISCUSSION
Metastases account for the great majority of cancer-associated deaths, yet this complex process remains the least understood aspect of cancer biology [6]. Unraveling the mechanism of metastasis will open a new avenue to cancer cure. CRIF1 inhibits cell cycle, and functions as an essential STAT3 coactivator [7]. CRIF1 induces cell cycle arrest in the G1 phase by inhibiting the kinase activity of Cdk2-cyclin complexes, Cdk1-cyclin B1and histone H1 [8, 9]. However, the role of CRIF1 in HCC has not been investigated.
Our data indicate that CRIF1 inhibits HCC cell proliferation, decreases the percentage of G1 phase cells, and inhibits in vivo tumorigenicity of HCC xenografts. In addition, our results are the first to demonstrate that CRIF1 inhibits HCC cell invasiveness, since CRIF1 knockdown increased HCC cells invasive potency, while restoring CRIF1 expression decreased their invasive potency.
Microarray data have indicated that several important genes including MMP3, PTEN, Smad6, and CDK6 are regulated by CRIF1 [10–12]. Among them, MMP3, a member of the matrix metalloproteinase (MMP) family, which is involved in the breakdown of extracellular matrix in processes, such as tissue remodeling and metastasis [13], had the largest fold change. The observed changes in MMP3 protein levels following CRIF1 knockdown or overexpression, may represent one of the mechanisms of how CRIF1 regulates invasiveness of HCC cells.
TGF-β signaling, which plays pivotal roles in tumor metastasis [14], was among the most altered signaling pathways following CRIF1 knockdown. CRIF knockdown increased Smad2, Smad3, p-Smad2 and p-Smad3 expression, suggesting activation of the TGF-β signaling. Expression of TGF-β receptor I was increased after CRIF1 knockdown, implying that TGF-β receptor I plays a driving role in CRIF1-regulated TGF-β signaling.
TGF-β1 has a differential regulatory effect on MMPs. It up-regulates MMP2 and MMP9, down-regulates MMP1 and MMP8 [15–18], and has no effect on MMP3 [19]. In our study, we found that blocking TGF-β signaling did not affect MMP3 expression in HCC cells. TGF-β signaling and MMP3 are involved in EMT [20–24]. MMP3 directly cleaves the extracellular domain of E-cadherin, prompting normal mammary epithelial cells to disaggregate and undergo EMT [25]. Pharmacological inhibition of MMP3 abrogates the TGF-β1-triggered cell invasiveness [26]. In this study, we found elevation of MMP3 and activation of TGF-β signaling following CRIF1 knockdown, so we sought to investigate the role of EMT in CRIF1 regulated cell invasiveness. As expected, CRIF1 knockdown decreased E-cadherin, and increased N-cadherin.
To confirm our hypothesis that CRIF1 regulates HCC EMT through TGF-β signaling, we treated CRIF1-KD and CRIF1-NC cells with SB525334, a TGF-β receptor I (ALK5) specific inhibitor [27–29]. Since blocking the TGF-β receptor inhibited the HCC cell invasiveness and EMT, these data indicate that CRIF1 regulates cell invasion via TGF-β-activated EMT and MMP3 expression. EMT and MMP3 have been considered as candidate targets for cancer therapy [30–35]. For example, galunisertib, TGF-β receptor I inhibitor, has been associated with strong improvement of HCC in the phase II trial [36].
Together, our results reveal a novel function of CRIF1 in regulating EMT and MMP3 expression in HCC cells, and suggest that CRIF1 may be an ideal target for inhibiting HCC metastasis.
MATERIALS AND METHODS
Clinical sample preparation
This study was approved by the Ethical Committee of the First Affiliated Hospital of Zhejiang University School of Medicine, and conducted in accordance with the ethical principles of the Declaration of Helsinki. All clinical samples used in this study were obtained from patients after informed consents. A total of 109 paired specimens of HCC were randomly enrolled in this study from HCC patients undergoing hepatectomy from 2010 to 2012 at the First Affiliated Hospital of Zhejiang University School of Medicine.
Reagents and cell culture
The human HCC cell lines Huh-7 and SK-HEP-1 were purchased from Chinese Academy of Sciences Shanghai cell bank and cultured in complete growth medium, as recommended by the manufacturer. Cells were maintained at 37°C in a humidified incubator with 5% CO2. The TGF-β RI specific inhibitor SB525334 was purchased from Sellleck (Selleck Chemicals, Houston, TX, USA), and used at a concentration of 1 μM.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from tissues or cells with Trizol reagent (Invitrogen, USA). qRT-PCR was carried out using the master SYBR Green kit (Takara Bio, Japan) according to the manufacturer’s instruction by ABI Prism 7500 fast sequence detection system (applied Biosystem, USA). All reactions were performed in triplicates, and analyzed by using the 2-ΔΔCt method. The results were normalized to mRNA expression of GAPDH, which was used as internal control.
Western blotting
Total proteins were extracted from tissues and cells. Anti-GAPDH antibody (1:5000, A5441, Sigma), anti-CRIF1 (1:2000, 16260-1-AP, Proteintech), anti-cyclin D1 (1:2000, ab137875, Abcam), anti-cyclin E1 (1:1000, ab52189, Abcam), anti-CDK4 (1:1000, ab137675, Abcam), anti-CDK6 (1:1000, ab151247, Abcam), anti-MMP3 (1:1000, ab52915, Abcam), anti-TGF-β1 (1:500, ab9758, Abcam), anti-TGF-β2 (1:100, ab167655, Abcam), TGF-β receptor 1 (1:1000, ab155258, Abcam), anti-Smad2 (1:1000, 3122, CST), anti-Smad3 (1:1000, 9513, CST), anti-p-Smad2 (1:1000, 3108, CST), anti-p-Smad3 (1:1000, 9520, CST), anti-E-Cadherin (1:2000, ab133597, Abcam), and anti-N-Cadherin (1:1000, ab19348, Abcam) antibodies were used. Immunodetection was performed using an EZ-ECL chemiluminescence detection kit (BeitHaemek, Israel).
Immunohistochemistry
Immunohistochemistry (IHC) was performed on HCC tissues using anti-CRIF1 (1:100, ab111254, Abcam) and anti-Ki67 (1:50, ab833, Abcam) antibodies. Immunostaining of CRIF1 was semi-quantitatively scored by multiplying the values of the mean staining intensity and the percentage of CRIF1-positive cells. The intensity of staining was scored as absent (0), weakly positive (1), moderately positive (2), or strongly positive (3). The percentage of CRIF1-positive cells was scored as no cells (0), less than 10% of cells (1), 11–50% of cells (2), 51–80% of cells (3), and more than 80% of cells (4). Finally, cases were grouped as low expression (scores 0–6) or high expression (scores 7–12).
CRIF1 siRNA interference, and recombinant plasmid and lentivirus infection
siRNA-CRIF1 and siRNA-NC (negative control) were synthesized by Invitrogen. The siRNA sequence for CRIF1 was ACACCUAGUAGGCUGCGUGCCUGUC. Hu-7 and SK-Hep-1cells were cultured in 6-well plates in MEM. When cells were about 30% confluent, they were transfected with siRNA- CRIF1 or siRNA-NC using transfection reagent (Lipofectamine 2000, Invitrogen, Carlsbad, CA). The CRIF1-recombinant and NC vectors were constructed by GeneChem. Stable CRIF1 knockdown (CRIF1-KD) and its negative control (CRIF1-NC) cell lines were obtained by infection of lentivirus targeting CRIF1 and the negative control. The efficiency of transfections was validated by qRT-PCR and western blotting. Fluorescence of CRIF1-KD and CRIF1-NC cells was observed after transfection and puromycin screening (Supplementary Figure 1).
Cell proliferation assay
Cell proliferation was measured using the Cell Counting Kit-8 (Dojindo, Japan) assay. Briefly, pretreated HCC cells (transfected with siRNA or GV219) were placed in 96-well plates (normally 4000 cells per well) and cultured for indicated times. The absorbance was measured at 450 nm using Absorbance Reader (BIO-TEK ELX800); the experiments were repeated 3 times.
Tumor invasion assay
Polycarbonate membranes Transwell (Corning, USA) and Matrigel (BD, USA) were used for the migration assay. After different treatments, a total of 5×104 Hu-7 and SK-Hep-1cells were suspended in 200 μL of serum-free medium and seeded into the upper chamber, while 600 μL of MEM containing 10% FBS was added to the lower chamber. After incubation at 37°C, the cells on the upper membrane were removed with cotton swabs. The membranes were fixed in paraformaldehyde solution and stained with 0.5% crystal violet. Cells on the lower surface of the membrane were counted in randomly selected fields with microscope (100×). The experiment was independently repeated three times.
Xenograft tumorigenicity assay
All animal experiments were performed according to the Zhejiang University Animal Care Facility and National Institutes of Health guidelines.
Six-week-old BALB/c male nude mice (average weight 21 g) were randomly divided into two groups (8 mice each). Pretreated SK-Hep-1cells (4×106 cells in 0.1ml PBS) were injected subcutaneously into the nude mice. Tumor volumes were measured after mice were sacrificed.
Cell cycle and apoptosis assays
For cell cycle analysis, cells were harvested and fixed in ice-cold 75% ethyl alcohol at 4° C overnight. After incubation with a DNA PREP kit solution (Beckman Coulter, Fullerton, CA) in the dark for 30 min, cell apoptosis was detected by FACS. Data were analyzed using ModFit LT3.1 software. Apoptosis was detected in HCC cells not expressing GFP (Green Fluorescent Protein) using an FITC Annexin V Apoptosis Detection Kit II (BD, San Jose, CA), and in cells expressing GFP using an Annexin V Apoptosis Detection Kit APC (eBioscience, SanDiego, CA). Cells were analyzed using a flow cytometer (LSR II; BD Bioscience). All experiments were conducted in triplicates.
Statistical analysis
All values are expressed as mean ± standard deviation. Student’s t-test was applied to evaluate statistical significance. The variable was converted to a logarithm if it did not conform to a normal distribution. X2 test was used to detect the difference between categorical variables. The endpoint for follow-up was patient death. Overall survival rates were calculated using the Kaplan–Meier method. Log-rank tests were performed for univariate analysis. P-values less than 0.05 were considered statistically significant. All statistical analyses were performed using SPSS 11.0software (SPSS, Chicago, IL).
Abbreviations
CRIF1: CR6-interacting factor 1
HCC : hepatocellular carcinoma
EMT : Epithelial-to-mesenchymal transition
MMP: matrix metalloproteinase
ACKNOWLEDGMENTS
We thank Jili Cao, Yuhui Liu, and Chao Wang for their technical help. We also thank Haijun Guo for his assistance in preparation of patient clinical information.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
FUNDING
This work was supported by the National Natural Science Funds for Distinguished Young Scholar of China [grant numbers 81625003]; Cheung Kong
Scholars Program Foundation of Chinese Ministry of Education.
This work was supported by the Projects of Medical and Health Technology Program in Zhejiang Province (2017RC002).
REFERENCES
1. Schutte K, Bornschein J, Malfertheiner P. Hepatocellular carcinoma--epidemiological trends and risk factors. Dig Dis. 2009; 27: 80-92. https://doi.org/10.1159/000218339.
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016; 66: 115-32. https://doi.org/10.3322/caac.21338.
3. Xu M, Jin H, Xu CX, Sun B, Song ZG, Bi WZ, Wang Y. miR-382 inhibits osteosarcoma metastasis and relapse by targeting Y box-binding protein 1. Mol Ther. 2015; 23: 89-98. https://doi.org/10.1038/mt.2014.197.
4. Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L, Larsson O, Muller WJ, Miller WH, et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2015; 34: 2032-42. https://doi.org/10.1038/onc.2014.146.
5. Chung HK, Yi YW, Jung NC, Kim D, Suh JM, Kim H, Park KC, Song JH, Kim DW, Hwang ES, Yoon SH, Bae YS, Kim JM, et al. CR6-interacting factor 1 interacts with Gadd45 family proteins and modulates the cell cycle. J Biol Chem. 2003; 278: 28079-88. https://doi.org/10.1074/jbc.M212835200.
6. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017; 168: 670-91. https://doi.org/10.1016/j.cell.2016.11.037.
7. Kwon MC, Koo BK, Moon JS, Kim YY, Park KC, Kim NS, Kwon MY, Kong MP, Yoon KJ, Im SK, Ghim J, Han YM, Jang SK, et al. Crif1 is a novel transcriptional coactivator of STAT3. EMBO J. 2008; 27: 642-53. https://doi.org/10.1038/sj.emboj.7601986.
8. Ran Q, Hao P, Xiao Y, Xiang L, Ye X, Deng X, Zhao J, Li Z. CRIF1 interacting with CDK2 regulates bone marrow microenvironment-induced G0/G1 arrest of leukemia cells. PLoS One. 2014; 9: e85328. https://doi.org/10.1371/journal.pone.0085328.
9. Zhang X, Ran Q, Li Z, Liu Y, Liang X, Chen X. Cell cycle arrest of Jurkat cells by leukemic bone marrow stromal cells: possible mechanisms and involvement of CRIF1. Transplant Proc. 2011; 43: 2770-3. https://doi.org/10.1016/j.transproceed.2011.05.048.
10. Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: Multiple Functions in Human Malignant Tumors. Front Oncol. 2015; 5: 24. https://doi.org/10.3389/fonc.2015.00024.
11. Yoshida K, Murata M, Yamaguchi T, Matsuzaki K. TGF-beta/Smad signaling during hepatic fibro-carcinogenesis (review). Int J Oncol. 2014; 45: 1363-71. https://doi.org/10.3892/ijo.2014.2552.
12. Li A, Blow JJ. The origin of CDK regulation. Nat Cell Biol. 2001; 3: E182-4. https://doi.org/10.1038/35087119.
13. Munshi HG, Stack MS. Reciprocal interactions between adhesion receptor signaling and MMP regulation. Cancer Metastasis Rev. 2006; 25: 45-56. https://doi.org/10.1007/s10555-006-7888-7.
14. Chiovaro F, Martina E, Bottos A, Scherberich A, Hynes NE, Chiquet-Ehrismann R. Transcriptional regulation of tenascin-W by TGF-beta signaling in the bone metastatic niche of breast cancer cells. Int J Cancer. 2015; 137: 1842-54. https://doi.org/10.1002/ijc.29565.
15. Kim ES, Kim MS, Moon A. TGF-beta-induced upregulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int J Oncol. 2004; 25: 1375-82.
16. Stuelten CH, DaCosta Byfield S, Arany PR, Karpova TS, Stetler-Stevenson WG, Roberts AB. Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-alpha and TGF-beta. J Cell Sci. 2005; 118: 2143-53. https://doi.org/10.1242/jcs.02334.
17. Chen H, Li D, Saldeen T, Mehta JL. TGF-beta 1 attenuates myocardial ischemia-reperfusion injury via inhibition of upregulation of MMP-1. Am J Physiol Heart Circ Physiol. 2003; 284: H1612-7. https://doi.org/10.1152/ajpheart.00992.2002.
18. Kim ES, Sohn YW, Moon A. TGF-beta-induced transcriptional activation of MMP-2 is mediated by activating transcription factor (ATF)2 in human breast epithelial cells. Cancer Lett. 2007; 252: 147-56. https://doi.org/10.1016/j.canlet.2006.12.016.
19. Casey TM, Eneman J, Crocker A, White J, Tessitore J, Stanley M, Harlow S, Bunn JY, Weaver D, Muss H, Plaut K. Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-beta 1) increase invasion rate of tumor cells: a population study. Breast Cancer Res Treat. 2008; 110: 39-49. https://doi.org/10.1007/s10549-007-9684-7.
20. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009; 9: 265-73. https://doi.org/10.1038/nrc2620.
21. Ye Z, Li J, Han X, Hou H, Chen H, Zheng X, Lu J, Wang L, Chen W, Li X, Zhao L. TET3 inhibits TGF-beta1-induced epithelial-mesenchymal transition by demethylating miR-30d precursor gene in ovarian cancer cells. J Exp Clin Cancer Res. 2016; 35: 72. https://doi.org/10.1186/s13046-016-0350-y.
22. Wu RS, Hong JJ, Wu JF, Yan S, Wu D, Liu N, Liu QF, Wu QW, Xie YY, Liu YJ, Zheng ZZ, Chan EC, Zhang ZM, et al. OVOL2 antagonizes TGF-beta signaling to regulate epithelial to mesenchymal transition during mammary tumor metastasis. Oncotarget. 2017; 8: 39401-39416. https://doi.org/10.18632/oncotarget.17031.
23. Ratz L, Laible M, Kacprzyk LA, Wittig-Blaich SM, Tolstov Y, Duensing S, Altevogt P, Klauck SM, Sultmann H. TMPRSS2:ERG gene fusion variants induce TGF-beta signaling and epithelial to mesenchymal transition in human prostate cancer cells. Oncotarget. 2017; 8: 25115-30. https://doi.org/10.18632/oncotarget.15931.
24. Kaowinn S, Kim J, Lee J, Shin DH, Kang CD, Kim DK, Lee S, Kang MK, Koh SS, Kim SJ, Chung YH. Cancer upregulated gene 2 induces epithelial-mesenchymal transition of human lung cancer cells via TGF-beta signaling. Oncotarget. 2017; 8: 5092-110. https://doi.org/10.18632/oncotarget.13867.
25. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006; 6: 392-401. https://doi.org/10.1038/nrc1877.
26. Wu YH, Chang TH, Huang YF, Huang HD, Chou CY. COL11A1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. Oncogene. 2014; 33: 3432-40. https://doi.org/10.1038/onc.2013.307.
27. Ying WZ, Aaron KJ, Sanders PW. Transforming growth factor-beta regulates endothelial function during high salt intake in rats. Hypertension. 2013; 62: 951-6. https://doi.org/10.1161/HYPERTENSIONAHA.113.01835.
28. Li Y, Zhao Z, Xu C, Zhou Z, Zhu Z, You T. HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett. 2014; 355: 130-40. https://doi.org/10.1016/j.canlet.2014.09.007.
29. Alzayadneh EM, Chappell MC. Angiotensin-(1-7) abolishes AGE-induced cellular hypertrophy and myofibroblast transformation via inhibition of ERK1/2. Cell Signal. 2014; 26: 3027-35. https://doi.org/10.1016/j.cellsig.2014.09.010.
30. Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004; 3: 1011-22. https://doi.org/10.1038/nrd1580.
31. Wilson C, Nicholes K, Bustos D, Lin E, Song Q, Stephan JP, Kirkpatrick DS, Settleman J. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget. 2014; 5: 7328-41. https://doi.org/10.18632/oncotarget.2397.
32. Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S, Baba H, Mori M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010; 101: 293-9. https://doi.org/10.1111/j.1349-7006.2009.01419.x.
33. Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006; 6: 227-39. https://doi.org/10.1038/nrc1821.
34. Overall CM, Kleifeld O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer. 2006; 94: 941-6. https://doi.org/10.1038/sj.bjc.6603043.
35. Jayachandran A, Dhungel B, Steel JC. Epithelial-to-mesenchymal plasticity of cancer stem cells: therapeutic targets in hepatocellular carcinoma. J Hematol Oncol. 2016; 9: 74. https://doi.org/10.1186/s13045-016-0307-9.
36. de Gramont A, Faivre S, Raymond E. Novel TGF-beta inhibitors ready for prime time in onco-immunology. Oncoimmunology. 2017; 6: e1257453. https://doi.org/10.1080/2162402X.2016.1257453.