
INTRODUCTION
Cardiac myxomas (CMs) are benign cardiac tumors that are characterized by stellate to plump, cytologically bland, mesenchymal cells situated in a myxoid stroma [1, 2]. These tumors often arise as a solitary mass situated in the left atria and have been described to occur more commonly in women than men [3, 4]. Isolated cardiac myxomas (ICM) tend to occur in individuals in the sixth to seventh decade of life and can present with syncope, palpitations, dyspnea or heart failure [5]. Approximately 7% of CMs occur in association with carney complex (CNC), a tumor predisposition syndrome characterized by the development of pigmented cutaneous lesions, myxomas and multiple endocrine neoplasms [6]. Investigation of the pathogenesis of CNC revealed that 70% of affected individuals harbored germline mutations in PRKAR1A, a gene that encodes the regulatory subunit (type I-alpha) of protein kinase A (PKA) [7-10]. Indeed, PRKAR1A mutations have been described in two-thirds of CNC-associated CMs. Loss of function mutations in PRKAR1A lead to increased PKA activation thereby promoting CREB phosphorylation and upregulation of the MAPK and Rb/E25 signaling pathways. This dysregulation is believed to play an important role in tumorigenesis [11]. Genetic aberrations of PRKAR1A have been investigated in ICM [12-14]. Several authors have reported that ICMs do not harbor PRKAR1A mutations and have suggested that the genetic basis of ICM may differ from CNC associated CMs [13, 14]. In contradistinction, a recent study suggested that 31% of ICM may contain inactivating mutations in PRKAR1A [5]. Despite this report, whether PRKAR1A mutations are involved in the pathogenesis of ICM remains controversial and a subject of ongoing debate.
RESULTS AND DISCUSSION
The development of next generation sequencing techniques has facilitated the discovery of previously unidentified cancer driver mutations [15]. We speculated that the discrepancy in the literature regarding the presence of PRKAR1A mutations in ICM could be due to lack of depth and coverage of previously utilized DNA sequencing methods.
In this study, whole exome sequencing (WES) was performed on 8 cardiac myxoma specimens derived from patients without any other manifestation of CNC. The median age of the patients at diagnosis was 51 years (range 44-60 years). Six of the 8 (75%) patients were female. All of the tumors arose within the left atrium, consistent with previous reports [16]. The pathological phenotype of these primary ICM tissues was confirmed by hematoxylin and eosin (H&E) staining (Figure 1).
Figure 1: Representative Hematoxylin and Eosin staining of cardiac myxoma tissue. M1 to M8 indicate the case number from ICM specimens obtained from different patients.
To explore the genetic variation in ICM, we performed whole exome sequencing as previously described [17]. The details of capture statistics and coverage are presented in Supplementary Table 1. After barcode-based sample deconvolution, sequence reads were mapped with BWA tools to the human genome (hg19). Sequence variant calls were performed by GATK after removing potential PCR duplicates. We called, on average 80,425 base substitutions and 8,061 insertions/deletions per tumor. Missense-, nonsense-, or splice-site-altering variants absent from the 1000 Genomes dataset were selected for further consideration. The majority of the identified variants were observed in the intronic and exonic regions, accounting for 44.81% and 33.77% of identified variants, respectively (Supplementary Table 1). These variants included synonymous, non-synonymous, stop-gain, stop-loss, splice-site and frameshift mutations. The majority of detected mutations were non-synonymous, accounting for 750-800 identified mutations. This is in contrast to the 14-22 splice-site mutations that were identified in the ICM from each patient (Figure 2A). Interestingly, we found that the majority of the base pair changes in the transcribed region were C>T (G>A) substitutions (Figure 2B). To identify pathogenic mutations, we excluded single nucleotide polymorphisms (SNP), synonymous mutations, and variants in the untranslated, intronic and intergenic region. We identified 4391 variants, covering 3198 genes across all eight samples. Mutations that were suspected to result in altered gene product activity accounted for 12% of these variants. These included frameshift, stop gain and splice-site mutations (Figure 2C). Additionally, we found 15 inactivating mutations involving PRKAR1A, which have been described in CNC related CM (Figure 2D). Oncogenes such as BRCA1/2, JAK2, NOTCH2, EPHA2 and NCOR2 harbored 2 nonsynonymous mutations, which are extremely rare variants reported in the dbsnp database. Other mutated genes included GXYLT1 (1 frameshift, 1 stop gain and 3 non-synonymous mutations). Furthermore, we found inactivating mutations of ZNF880 and MUC3A in all 8 ICM tissue samples. AIM1L was mutated in 5 of 8 analyzed ICM. However, we found that these variants were germline mutations due to the presence of these mutations in patients matched peripheral blood DNA (data not shown).
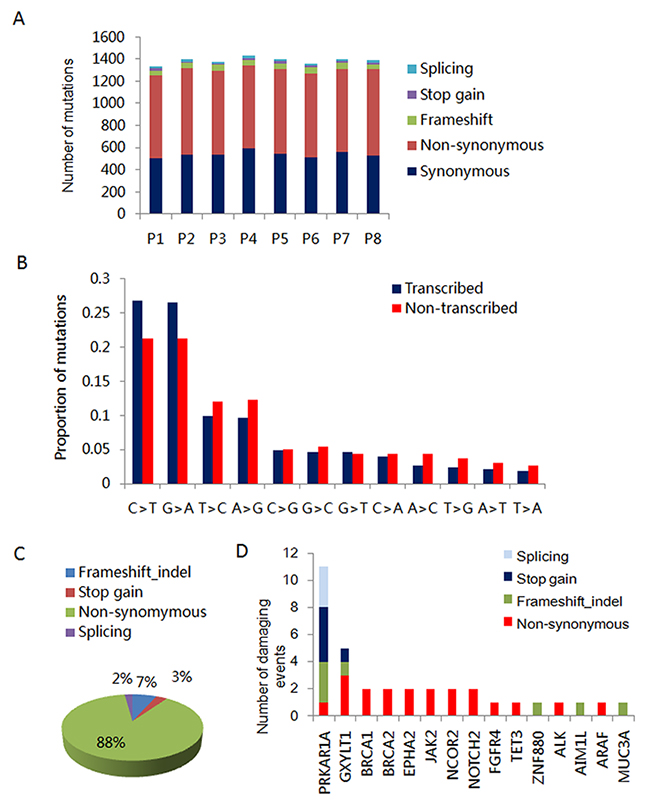
Figure 2: Inactivating mutations of PRKARR1A in ICM. (A) Number of splicing, stop-gain, frameshift, synonymous and non-synonymous mutations in the 8 ICM samples. (B) Relative proportions of base-pair substitutions on the transcribed and non-transcribed strands among the 616 point mutations identified in the exome sequences of 8 ICMs. (C) Percentage of differential types of variants identified by WES in 8 ICMs. (D) Number and type of damaging events for tumor associated genes altered in 8 ICMs.
The loss-of-function mutations of PRKAR1A have been previously identified in CNC and familial CMs. We found 11 inactivating mutations of PRKAR1A in 7 of 8 (86%) analyzed ICM tumors (Table 1). 3 of 8 (37.5%) ICM specimens contained biallelic PRKAR1A mutations, including c.607_610del:p.Leu203fs (pathogenic) + c.C896G:p.Ser299X (pathogenic), c.952delT:p.Leu318fs (pathogenic) + c.769-2 A>G (pathogenic) and c.178-1 G>C (pathogenic) + c. 550+1 G>C (pathogenic). Four of 8 (50%) tumors harbored heterozygous PRKAR1A mutations, including c.523_524insG:p.Tyr175_Val176delinsX (pathogenic), c.C920A:p.Ser307X (pathogenic), c.30delG:p.Glu10fs (pathogenic) and c.C289T:p.Arg97X (pathogenic). In all cases, we could not detect the same genetic alterations in the matched blood samples, suggesting that these variants were somatic mutations (Supplementary Figure 1).
Table 1: Somatic mutations of PRKAR1A in 8 ICM patients
Sample |
Age |
Sex |
Diagnosis |
PRKAR1A mutations |
Variant classification |
|---|---|---|---|---|---|
M1 |
48 |
F |
Myxoma |
c.523_524insG:p.Tyr175_Val176delinsX |
Pathogenic |
M2 |
46 |
F |
Myxoma |
c.C920A:p.Ser307X |
Pathogenic |
M3 |
53 |
F |
Myxoma |
c.30delG:p.Glu10fs |
Pathogenic |
M4 |
46 |
M |
Myxoma |
c.952delT:p.Leu318fs |
Pathogenic |
c.769-2 A>G |
Pathogenic |
||||
M5 |
44 |
M |
Myxoma |
c.178-1 G>C |
Pathogenic |
c. 550+1 G>A |
Pathogenic |
||||
M6 |
53 |
F |
Myxoma |
c.607_610del:p.Leu203fs |
Pathogenic |
c.C896G:p.Ser299X |
Pathogenic |
||||
M7 |
56 |
F |
Myxoma |
c.C289T:p.Arg97X |
Pathogenic |
M8 |
60 |
F |
Myxoma |
Not detected |
- |
Next, we investigated the effect of these inactivating variants on PRKAR1A expression in primary tumor tissues by immunohistochemistry (IHC). In agreement with the mutation analysis, PRKAR1A expression was not identified in the 7 tumors that contained somatic PRKAR1A mutations, suggesting the abrogation of PRKAR1A function in ICM (Supplementary Figure 2). To further assess the PRKAR1A mutation in ICM, we investigated the mutation status of PRKAR1A in an additional 61 ICM tumors. We amplified the 10 exons of PRKAR1A by PCR, and performed targeted Sanger DNA sequencing. In this cohort, 39 of 61 (64%) ICM specimens harbored inactivating PRKAR1A mutations (Table 2). Similar to our WES data, no hotspot mutation was found in these samples. In addition, most of the PRKAR1A mutations were distributed across Exons 1-5. While expression of PRKAR1A varied across these samples, we speculate that some of them may have resulted in the truncation of the PRKAR1A protein due to the fact that most of the identified mutations were frameshift and nonsense mutations. For instance, case No.9 showed PRKAR1A positive due to the antibody, which may still be able to recognize 200-260 aa of truncated PRKAR1A protein.
Table 2: Analysis of PRKAR1A mutations in 61 FFPE ICMs by Sanger sequencing. “+” indicates the positive expression of PRKAR1A in these FFPE tissue samples, while “-” indicates negative expression of this protein. All the variants were classified according to ACMG criteria
Sample |
PRKAR1A mutations |
Variant classification |
IHC |
Sample |
PRKAR1A mutations |
Variant classification |
IHC |
|---|---|---|---|---|---|---|---|
1 |
Not detected |
+/- |
32 |
c.421-440del:p.Leu141fs |
Pathogenic |
- |
|
2 |
c.8delC:p.Ser3fs |
Pathogenic |
+ |
33 |
Not detected |
- |
|
3 |
c.C73G:p.His25Asp |
Likely pathogenic |
- |
34 |
Not detected |
- |
|
4 |
c.C205T:p.Gln69X |
Pathogenic |
- |
35 |
Not detected |
- |
|
5 |
Not detected |
+ |
36 |
c.8delC:p.Ser3fs |
Pathogenic |
- |
|
6 |
c.162delG:p.Glu85fs;162_163insG, p.Glu85fs, c.912_913insT:p.Leu271fs |
Pathogenic |
- |
37 |
c.290delG:p.Arg97fs |
Pathogenic |
- |
7 |
Not detected |
+ |
38 |
Not detected |
+ |
||
8 |
c.A1115C:p.Gln372Pro |
Likely pathogenic |
- |
39 |
c.162delT:p.Phe54fs |
Pathogenic |
+ |
9 |
c.267delA:p.Pro89fs, c.A367T:p.Lys123X |
Pathogenic |
+ |
40 |
Not detected |
+ |
|
10 |
c.482delG:p.Gly161fs |
- |
41 |
c.478delG:p.Ala160fs |
Pathogenic |
++ |
|
11 |
c.A1093T:p.Ile365Phe |
Likely pathogenic |
+/- |
42 |
Not detected |
- |
|
12 |
Not detected |
- |
43 |
Not detected |
+ |
||
13 |
c.A872G:p.Glu291Gly |
Likely pathogenic |
+ |
44 |
c.C124T:p.Arg42X |
Pathogenic |
- |
14 |
c.1059delT:p.Pro353fs |
Pathogenic |
- |
45 |
c.289C>Tp.Arg97X |
Pathogenic |
- |
15 |
Not detected |
+ |
46 |
c.1059delT:p.Pro353fs |
Pathogenic |
- |
|
16 |
c.421-440del:p.Leu141fs |
Pathogenic |
+ |
47 |
c.T89A:p.Leu30Gln, c.502+2T>G |
Pathogenic |
+ |
17 |
c.767delT:p.Leu256X, c.892-2A>T |
Pathogenic |
- |
48 |
c.200-213del:p.Asn67fs |
Pathogenic |
+ |
18 |
Not detected |
+ |
49 |
Not detected |
+ |
||
19 |
c.201-213del:p.Asn67fs |
Pathogenic |
+ |
50 |
c.290delG:p.Arg97fs |
Pathogenic |
- |
20 |
c.219_220insGTAAGGCACT:p.Arg74fs |
Pathogenic |
+ |
51 |
Not detected |
+ |
|
21 |
c.421-440del:p.Leu141fs |
Pathogenic |
- |
52 |
c.10_11insTG:p.Gly4fs |
Pathogenic |
+ |
22 |
Not detected |
- |
53 |
Not detected |
- |
||
23 |
c.8delC:p.Ser3fs |
Pathogenic |
- |
54 |
c.783delG:p.Lys261fs |
Pathogenic |
+ |
24 |
c.550-2A>G |
Pathogenic |
+ |
55 |
c.10_11insGT:p.Gly4fs |
Pathogenic |
- |
25 |
Not detected |
+ |
56 |
c.550-2A>G |
Pathogenic |
+ |
|
26 |
c.G569A:p.Trp190X, c.737-738del:p.Tyr246fs |
Pathogenic |
- |
57 |
Not detected |
- |
|
27 |
c.453delT:p.DAsp151fs |
Pathogenic |
- |
58 |
c.478delG:p.Ala160fs, c.T619A:p.Tyr207Asn |
Pathogenic |
- |
28 |
Not detected |
+ |
59 |
c.251-300del:p.Pro84fs |
Pathogenic |
- |
|
29 |
c.C124T:p.Arg42X |
Pathogenic |
+ |
60 |
c.C196T:p.Gln66X |
Pathogenic |
+ |
30 |
Not detected |
+ |
61 |
Not detected |
+ |
||
31 |
c.349-1G>A |
Pathogenic |
- |
Overall our results suggest that ICM may contain PRKARA1A mutations in approximately two-thirds of cases. The remaining samples may contain mutations at levels below our detection limit due to sample procurement. On the other hand, we speculate that there may be other driver mutations in the non-PRKAR1A mutated ICM specimens that have yet to be identified. Therefore, we examined the whole exome sequencing results from the patient whose ICM did not contain a PRKAR1A mutation. In this specimen, we found that the patient presented with a nonsense mutation in MXI1, a tumor suppressor gene that has been reported to be frequently inactivated in prostate cancer [18, 19]. This mutation, however, was determined to be a germline mutation, supported by the presence of this mutation in matched blood sample. Interestingly, this patient also carried NOTCH2, EPHA2, NCOR2, FGFR4, and ARAF germline missense mutations. These non-SNP variants were not found in the other patients with PRKAR1A mutations. Unfortunately, we could not identify any clinical or pathologic differences between PRKAR1A mutated and non-mutated ICM patients. Although this is only one sample, this raises the possibility that ICM that do not harbor PRKAR1A mutations may be genetically heterogeneous with multiple genetic variants contributing to tumorigenesis. Thus, further work is necessary to define the clinical and pathologic differences between PRKAR1A-mutated and PRKAR1A non-mutated isolated cardiac myxomas. Our work, reported herein, clearly defines that PRKAR1A mutations are present in a substantial proportion of isolated cardiac myxomas.
MATERIALS AND METHODS
Ethics statement
Blood and tumor samples from ICM patients with were obtained under the approval of the Ha’er Bin Medical University Internal Review Board. Written informed consent was obtained from all patients.
Clinical samples
A total of 8 paired ICM tissues and blood samples were recruited from the Affiliated Hospital of Harbin Medical University (Harbin, China). 61 FFPE ICM tissue samples were obtained from Zhongshan Hospital of ShangHai.
Whole exome sequencing
Genomic DNA was isolated and sequenced using standard protocols for next generation sequencing (Compass Biotechnology, Co. Ltd. Beijing, China). Briefly, the Agilent SureSelect Human All Exon 60 Mb kit (Agilent Technologies, Santa Clara, CA, USA) was utilized to capture all exons. Shotgun libraries were established by shearing DNA and ligating sequencing adaptors bearing a 6 base-pair index, followed by polymerase chain reaction (PCR). Libraries were hybridized to SureSelect Human All Exon V6 solution-based probes, amplified, pooled, and sequenced on Illumina Hiseq X10 (PE 150 bp). The final targeted region was 60,456,963 base pairs, including approximately 20,000 genes (230,418 exons and splice junctions) in the genome. The average mean fold coverage was 203X. 97.9% of target bases were covered at least at once and 97.5% were covered at least 20X.
Validation of variants by Sanger sequencing
The germline and somatic variants identified by WES were further analyzed by Sanger Sequencing. Briefly, primers were designed by generunner software to amplify the genomic region using the extracted DNA from ICM tissues and blood samples. The PCR product was sent for automatic DNA sequencing.
Hematoxylin and eosin (H&E) staining
ICM primary tissues were fixed in 4% paraformaldehyde for 24h, washed in PBS and were embedded in paraffin. Two-micrometer sections were stained with hematoxylin and eosin following standard procedures.
Immunohistochemistry
For immunohistochemistry (IHC), the antigen retrieval step was performed with EDTA Buffer (1mM EDTA, 0.05% Tween 20, pH 8.0) in a pressure cooker. Endogenous peroxide activity was blocked by 3% peroxidase, and the slides were further blocked by 5% BSA to prevent nonspecific binding. Primary antibodies directed against PRKAR1A (Abcam, ab139695) incubated at an optimal dilution (1:100). The secondary antibody was donkey anti-rabbit IgG H&L (Abcam, ab6802, diluation 1:1000). For detection, the Polink-2 Plus horseradish peroxidase (HRP) Polymer Detection system (PV-9001; GBI Labs, Mukilteo, WA, USA) was used. Hematoxylin dye was used as counter stain. The slides were examined with an Olympus BX61 microscope with cell Sens Standard Software Version 1.6 (Olympus Corporation, Tokyo, Japan).
Author contributions
JH performed the DNA extraction as well as sequencing analysis, MJS extracted the genomic DNA, performed PCR and targeted sequencing analysis. YYH, EYL, CSW and LG performed the pathological assay for ICMs. DC performed the bioinformatic analysis. MS and KP wrote the manuscript. ZPZ and YL designed all the experiments and wrote the manuscript.
ACKNOWLEDGMENTS
The study has been supported by the Hundred Talents Program of CAS and Innovative Research Grant to Scientific Research Center for Translational Medicine at Dalian Institute of Chemical Physics.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Kacerovska D, Sima R, Michal M, Hes O, Roucka P, Zarybnicka M, Hora M, Chudacek Z, Kazakov DV. Carney complex: a clinicopathologic and molecular biological study of a sporadic case, including extracutaneous and cutaneous lesions and a novel mutation of the PRKAR1A gene. J Am Acad Dermatol. 2009; 61: 80-7. https://doi.org/10.1016/j.jaad.2008.11.015.
2. Pucci A, Gagliardotto P, Zanini C, Pansini S, di Summa M, Mollo F. Histopathologic and clinical characterization of cardiac myxoma: review of 53 cases from a single institution. Am Heart J. 2000; 140: 134-8. https://doi.org/10.1067/mhj.2000.107176.
3. Law DA, Dulaney JJ, Graeber G. Left atrial myxoma: a case presentation and review of the literature. W V Med J. 1995; 91: 95-7.
4. Carney JA. Differences between nonfamilial and familial cardiac myxoma. Am J Surg Pathol. 1985; 9: 53-5.
5. Maleszewski JJ, Larsen BT, Kip NS, Castonguay MC, Edwards WD, Carney JA, Kipp BR. PRKAR1A in the development of cardiac myxoma: a study of 110 cases including isolated and syndromic tumors. Am J Surg Pathol. 2014; 38: 1079-87. https://doi.org/10.1097/PAS.035707R1035707R10202.
6. Milunsky J, Huang XL, Baldwin CT, Farah MG, Milunsky A. Evidence for genetic heterogeneity of the Carney complex (familial atrial myxoma syndromes). Cancer Genet Cytogenet. 1998; 106: 173-6.
7. Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. 2006; 1: 21. https://doi.org/10.1186/1750-1172-1-21.
8. Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, Rene-Corail F, Stergiopoulos S, Bourdeau I, Bei T, Clauser E, Calender A, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. 2009; 94: 2085-91. https://doi.org/10.1210/jc.2008-2333.
9. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000; 26: 89-92. https://doi.org/10.1038/79238.
10. Casey M, Vaughan CJ, He J, Hatcher CJ, Winter JM, Weremowicz S, Montgomery K, Kucherlapati R, Morton CC, Basson CT. Mutations in the protein kinase A R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J Clin Invest. 2000; 106: R31-8. https://doi.org/10.1172/JCI10841.
11. Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kunstman JW, Korah R, Suttorp AC, Dietrich D, Haase M, Willenberg HS, Stalberg P, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet. 2014; 46: 613-7. https://doi.org/10.1038/ng.2956.
12. Singh S, Tripathy MP, Mohanty BB, Biswas S. Sporadic Multicentric Right Atrial and Right Ventricular Myxoma Presenting as Acute Pulmonary Thromboembolism. Heart Views. 2016; 17: 19-22. https://doi.org/10.4103/1995-705X.182642.
13. Fogt F, Zimmerman RL, Hartmann CJ, Brown CA, Narula N. Genetic alterations of Carney complex are not present in sporadic cardiac myxomas. Int J Mol Med. 2002; 9: 59-60.
14. Mabuchi T, Shimizu M, Ino H, Yamguchi M, Terai H, Fujino N, Nagata M, Sakata K, Inoue M, Yoneda T, Mabuchi H. PRKAR1A gene mutation in patients with cardiac myxoma. Int J Cardiol. 2005; 102: 273-7. https://doi.org/10.1016/j.ijcard.2004.05.053.
15. Crisan A, Goya R, Ha G, Ding J, Prentice LM, Oloumi A, Senz J, Zeng T, Tse K, Delaney A, Marra MA, Huntsman DG, Hirst M, et al. Mutation discovery in regions of segmental cancer genome amplifications with CoNAn-SNV: a mixture model for next generation sequencing of tumors. PLoS One. 2012; 7: e41551. https://doi.org/10.1371/journal.pone.0041551.
16. Shapiro LM. Cardiac tumours: diagnosis and management. Heart. 2001; 85: 218-22.
17. Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T, Grarup N, Guo Y, Hellman I, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010; 42: 969-72. https://doi.org/10.1038/ng.680.
18. Prochownik EV, Eagle Grove L, Deubler D, Zhu XL, Stephenson RA, Rohr LR, Yin X, Brothman AR. Commonly occurring loss and mutation of the MXI1 gene in prostate cancer. Genes Chromosomes Cancer. 1998; 22: 295-304.
19. Eagle LR, Yin X, Brothman AR, Williams BJ, Atkin NB, Prochownik EV. Mutation of the MXI1 gene in prostate cancer. Nat Genet. 1995; 9: 249-55. https://doi.org/10.1038/ng0395-249.