
INTRODUCTION
The single greatest challenge in the treatment of cancer has always been to uncover therapeutic agents that eliminate only tumor cells while sparing normal cells. The era of genomics has given a much deeper understanding of the biology of cancer and the genetic mutations that underlie progression of the disease and has brought with it a new generation of so-called molecular targeted cancer therapeutics that specifically act on the very oncogenic lesions that promote and sustain the disease. These new drugs rely on the fact that maintenance of transformed cells and the pathogenesis of the tumor are inextricably linked to the initial genetic reprogramming that has occurred, a concept referred to as “oncogene addiction” [1]. Nevertheless, most of the more common older-generation chemotherapeutics currently available for the oncologist have broad applicability for diverse neoplastic disease despite suffering from low therapeutic indices in patients. Thus, the notion of using “dirty” compounds for the treatment of cancer has yet to be supplanted in the clinic nor has it lost traction in current drug development, as can be seen in several recently FDA approved chemotherapeutics (e.g. bortezomib or sorafenib). In fact, what seems at first a contradiction to the whole notion of rational designed targeted-therapeutics can be explained in a model elegantly described by Elledge and colleagues as “non-oncogene addiction”[2]. This model puts forth the idea that the vast biological rewiring on which the tumorigenic state of the cell depends on stem from gene products that in and of themselves are not natural oncogenes and whose functions are now “rate-limiting” to the survival and proliferation of the transformed cell. It is this unhinged metabolic burden that renders cancer cells exquisitely dependant on intrinsic stress-relief pathways for their normal existence, offering up unique therapeutic opportunities. One such class of emerging cancer drug targets, protein synthesis inhibitors, will remain the focus of this minireview.
The mRNA translation process may be thought of as occurring in three phases: initiation, elongation, and termination. Initiation, as its name implies, is the process wherein the cell prepares mRNA transcripts for binding to ribosomal subunits and subsequent proper alignment of the ribosome to the correct initiation codon, thus allowing for proper polypeptide synthesis (and gene expression) to ensue. Elongation is polypeptide synthesis, the cycle of amino-acid covalent attachment into polypeptide chains catalyzed by the ribosome, the result of matching the triplet-nucleotide codons embedded in mRNA message to their cognate amino acid-acylated tRNA. Termination is the end stage of polypeptide synthesis, the halting of a transiting ribosome at a stop codon for which there are no corresponding amino acid-acylated tRNAs and thus release of the fully synthesized protein into the cell. Ribosome density measurements along mRNA templates are consistent with the notion that the initiation phase of translation is generally rate-limiting [3]. We refer the reader to other texts for more in-depth analysis of the processes of elongation and termination [4, 5], which are well beyond the purview of this review. What follows is a brief overview of the initiation process, as it elaborates on some of the factors targeted by the inhibitors discussed below (for a recent review and a more thorough analysis see Refs. [6, 7]).
Translation initiation is a multifaceted highly regulated biological process, requiring at least nine eukaryotic initiation factors (eIFs) composed from at least 30 subunits. It can be separated into three distinct steps: i) the binding of initiation factors to mRNA transcripts to prepare for, ii) the binding of the 43S ribosomal complex (which consists of the 40S ribosome subunit in complex with eIF2-GTP-tRNA-Meti ternary complex, eIF3, eIF1, eIF1A and eIF5) to the mRNA message, thus forming the 48S initiation complex and the, iii) joining of the 40S ribosome to the 60S ribosomal subunit, once the 48S ribosome is properly aligned at the start codon of the open-reading frame. For the majority of mRNAs, the binding of 43S ribosomal complexes begins at the 5’-end, where the methyl-7-guanosine (or “cap”) structure is found. Here, a preformed 43S pre-initiation complex is directed to the 5’-end with the help of the eIF4F initiation complex. Once there the ribosome begins “scanning” the 5’-untranslated region (5’-UTR) unidirectionally towards the 3’-end, until it reaches the appropriate initiation codon. Once properly oriented, eIF5 and eIF5B, using the energy from the hydrolysis of two GTP molecules, help replace all the eIFs with the joining large 60S ribosomal subunit, forming an elongation-competent 80S ribosome. Although most mRNAs utilize the cap structure to facilitate recruitment of the 43S ribosomal complex, initiation on a select few cellular and viral mRNAs (typically those that bear no cap-structure or that have long and complex 5’-UTRs) is mediated by an alternative cap-independent mechanism, utilizing a distinct RNA structure termed an internal ribosome entry site (IRES), which directs binding and start codon selection of the 43S complex internally on the mRNA transcript [6, 8].
eIF4F
eIF4F is a heterotrimeric complex composed of: i) eIF4A, a DEAD-box containing ATPase and ATP-dependent RNA helicase required to melt local secondary structure and facilitate access of the ribosome to the mRNA template; ii) eIF4G, a modular scaffolding protein that mediates mRNA binding to the 43S pre-initiation complex, and iii) eIF4E, the cap-binding protein responsible for binding of the eIF4F complex to the mRNA cap structure [5, 9] (Fig. 1). In mammals, there are three isoforms of eIF4A (eIF4AI, II and III) that share ~90% and ~65% identity, respectively, with the most abundant cellular factor eIF4AI [10, 11]. All isoforms are DEAD-box RNA helicase family members but only the paralogs eIF4AI and eIF4AII are found in the eIF4F complex and participate in translation initiation [12, 13]. Similarly, there are two paralogs of eIF4G (eIF4GI and eIF4GII) that share 46% identity, as well as a distantly related protein, p97/DAP5/NAT1, that boasts a similar amino-acid sequence to the C-terminal portion of eIF4G. Despite being able to bind eIF4A and eIF3, p97 cannot bind eIF4E and thus appears to play a role in the cap-independent translation of a specific subset of mRNAs [14]. eIF4E imparts RNA binding specificity to the eIF4F complex by selectively recognizing the cap structure present at the 5’ end of all eukaryotic cellular mRNAs [15-18]. Binding of 43S ribosomal complexes to the cap in order to begin scanning is thought to require eIF4F for most mRNAs. Unstructured mRNAs in in vitro reconstituted systems with purified translation components are able to weakly recruit 43S complexes [19], but the introduction of even a modest amount of RNA secondary structure eliminates all 43S recruitment in the absence of eIF4F. Once cap-bound, the various components of eIF4F act in concert to stabilize it to the mRNA template and prepare the mRNA for binding to 43S complexes. eIF4G, now proximally located on the mRNA template, reinforces the eIF4E-cap interaction [20-22]. eIF4A then unwinds local secondary structure to prepare the template for its interaction with 43S complexes, a process that is greatly improved by the auxiliary eIF4A interacting factors, eIF4B [23] and eIF4H [24]. It is eIF4G that directs 43S ribosome binding through a bridging interaction with eIF3, and finalizes 43S complex binding to the mRNA [25]. Although eIF4F primarily directs 5’-end 43S recruitment, it also brings the 3’-end in close proximity to the 5’-cap, through an interaction between eIF4G and the 3’-end bound poly(A) binding protein (PABP), enabling circularization of the mRNA template [26-28] (Fig. 1). This results in a stimulatory effect on ribosome binding and translation as a whole, either by ensuring that eIF4F remains cap-bound or through the recycling and reinitiation of ribosomes post-termination [27].
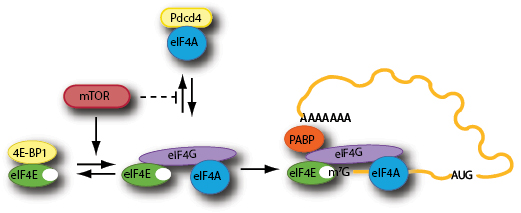
Figure 1: Schematic outline of mTOR regulation of the eIF4F-mRNA binding step.
eIF4F AND oncogenesis - seeing the Forest but not the Trees
Ever since early experiments in model cell culture systems demonstrated that overexpression of eIF4E on its own can drive transformation of fibroblasts [29], there has emerged a large body of literature implicating eIF4F and several other translation initiation factors (e.g. eIF3 subunits, eIF2) in cancer biology [30]. The evidence supporting eIF4E as an oncogene is now quite compelling and has been demonstrated in multiple settings. In both xenograft nude-mouse and more genetically and clinically relevant mouse cancer models, overexpression of eIF4E was shown to both accelerate the onset of tumor formation and aggravate its drug response [29, 31, 32]. Conversely, overexpression of 4E-BP (for 4E-Binding Protein, a competitive inhibitor to the eIF4E-eIF4G interaction) in a p53-/- mouse cancer model slowed progression of cancer in part through a mechanism involving premature senescence [33]. Moreover, in a wide variety of blood and solid tumor samples, elevated expression of eIF4E protein correlates with later stage tumors, more aggressive pathologies and poorer prognosis [34-49], while the opposite appears to be true for 4E-BP [50]. Similarly, eIF4GI overexpression appears to phenocopy some of the oncogenic features of eIF4E: it too can drive transformation of mouse cell lines [51] and increased expression has been found in some overlapping tumor tissues [52-54]. Finally, there have been a few reports which found eIF4A expression to be elevated in some human derived tumor samples, melanoma [55] and hepatocellular carcinoma [56] cells, although the implication of this is as of yet uncertain [57].
And yet, even with all of the aforementioned data demonstrating eIF4E (or more generally eIF4F) as a bona fide oncogene, the precise molecular mechanism governing its tumorigenicity has remained somewhat elusive. Although it might seem intuitive at first that cancer cells would benefit greatly from increased rates of protein of synthesis (and in fact they do), in the case of eIF4E this is not really the case, as the levels that elicit oncogenesis are somewhat modest (~2-3 fold) [31] and probably not high enough to alter overall protein abundance. More likely, the mechanism by which increased eIF4F activity contributes to transformation is via the increase in translation of a limited set of pro-oncogenic mRNA transcripts [58, 59]. One of the reasons for this pronounced selectivity is thought to be due to the differential requirements that some mRNAs have towards eIF4F activity [60]. mRNAs that have long, G-C rich and structured 5’-UTR nucleotide sequences, are generally poorly translated (presumably due to lowered 48S complex formation rates due to unfavorable eIF4F-cap interaction [61-63]), under most transient and normal growth conditions of the cell, where initiation factors in general, and eIF4F in particular, are limiting. In essence, they cannot outcompete the other more efficiently translated messages (those that have short and relatively unstructured 5’-UTRs) for ribosomes [60]. This all changes once the levels and activity of eIF4F rises: those messages that were once outcompeted will have their translation rates disproportionately stimulated, stemming from an increase in the rate-constant of ribosome binding and initiation due to the relative decrease in thermal stability at their 5’-ends mediated by increased eIF4F levels. Perhaps not surprisingly, a great many pro-growth, stress-associated or cell-cycle regulated transcripts whose protein levels are tightly controlled and are kept at a relatively low homeostatic level generally bear the hallmarks of a poorly translated message. In fact, simple overexpression of eIF4E can increase the levels of a wide variety of messages encoding growth and survival regulators (e.g. PDGF, FGF-2, VEGF), signal transducers (e.g. Pim-1, Ras), and components of the cell cycle and apoptotic machinery (e.g. cyclin D1, c-myc, RNR2, ODC, survivin and Mcl-1) [60, 64-68]. And this list is expanding given recent efforts at gene profiling of polysomal bound mRNAs that are under eIF4F regulation [69, 70]. Future technologies, such as those involving novel RNA-sequencing methodologies [3] will no doubt uncover an even more complex translational regulatory system. Thus targeting translation initiation, and in particular the eIF4F complex, would seem a viable and novel chemotherapeutic avenue.
INHIBITING eIF4F – NOT ALL SUBUNITS ARE EQUAL
In recent years, there has been renewed interest in the development of pharmacological agents that disrupt these key steps of translation, in particular agents that block eIF4F activity. This has been based on a few early proof-of-principle experiments that showed that downregulation of eIF4E via antisense oligonucleotides [71, 72] or direct inhibition from peptides that compete for eIF4G binding [73] can stop transformation, cause cell-cycle arrest, and elicit apoptosis. But much of the attention paid of late to eIF4E biology as it pertains to cancer, especially from oncologists and pharmaceutical companies, relates to the anti-neoplastic potential of the macrolide rapamycin (and its analogs) and its cellular target, the master kinase mTOR. Note that a full treatment of the literature and published research on the subject of mTOR is well beyond the purview of this text and we encourage the reader to look at a recent review for a more complete understanding of the extensive and important role that mTOR plays in the cell [74]. Nevertheless, it is important to emphasize one key downstream target and effector of mTOR function, namely eIF4F (Fig 1).
mTOR is a master regulator of cellular homeostasis, sensing inputs from growth factor withdrawal, amino acid imbalance, energy depletion and even oxygen tension and altering gene expression and metabolism in order for the cell to adjust, and one of those key metabolic and gene expression pathways that it regulates is protein synthesis, primarily through its target 4E-BP, one of the earliest known targets shown to be directly phosphorylated by mTOR [9, 75] . In most cells, there exists three 4E-BP isoforms (4E-BP1, 2 and 3) and all are phosphorylated on multiple serine and threonine residues by mTOR. 4E-BPs act as small molecular mimics to eIF4G, bearing the canonical Tyr-X-X-X-X-Leu-ϕ eIF4E binding consensus motif (where ϕ represents a hydrophobic residue) [76]. Under nutrient replete and growth-factor abundant conditions, where mTOR activity is turned on, 4E-BP is hyperphosphorylated and can no longer bind to eIF4E allowing for robust translation initiation. However, upon growth factor withdrawal or nutrient starvation, mTOR activity is reduced, leading to the dephosphorylation of 4E-BP, allowing for tight binding to eIF4E (since they are no longer sterically hindered by the phosphate charges), blocking access to eIF4G and thereby inhibiting translation. Although much of the regulation of protein synthesis derived from signaling through mTOR acts predominantly through the 4E-BPs [77], mTOR can also modulate translation initiation and eIF4F through phosphorylation of p70 S6 kinase (or S6K) and its downstream substrates Pdcd4 [78] and eIF4B [79]. Pdcd4 was first identified as tumor suppressor in the clonally derived JB6 cell line that had lost the ability for neoplastic transformation [80]. It was subsequently found to bind to both eIF4A and eIF4G and thus inhibits eIF4A helicase activity while preventing its association with eIF4G, restricting translation [81]. Following activation of mTOR, Pdcd4 is phosphorylated on Ser67 by S6K1, which in turn promotes phosphorylation of Ser71 and Ser76 allowing for binding to βTRCP (F-box proteins) and ubiquitin-mediated degradation [78]. This releases eIF4A and concomitantly stimulates cap-dependent translation [78]. eIF4B is also a substrate of S6K, whose phosphorylation can also affect translation, but its effects are far from obvious [79, 82].
The question arises as to which of these two interactions (4E:4E-BP and eIF4A:Pdcd4) is rate-limiting (and therefore more important) in vivo for eIF4F assembly. The answer may be that it depends on context or that both are important but affect translation initiation in different ways. Comparing relative abundance of factors (eIF4A is the most abundant translation initiation factor present at three copies per ribosome [83] and eIF4E is the least abundant present at 0.26 copies per ribosome [83]) does not address this issue since subcellular localization could render abundant factors rate-limiting. The relative stoichiometry of eIF4E and 4E-BPs versus that of Pdcd4 and eIF4A has not been reported and may differ depending on cell type. Another point to consider is that the cap structure is required to stimulate translation initiation, but its presence is not an absolute requirement for translation initiation in vitro or in vivo [84-92]. De Gregorio et al. [93] have shown that eIF4G mutants lacking the eIF4E binding site can activate translation on uncapped mRNA reporters in vitro and that the central core domain of eIF4G (containing one eIF4A and eIF3 binding site) is sufficient to mediate ribosome recruitment [94]. The 5’ end specificity that is observed upon translation of uncapped transcripts may be imparted because the mRNA is masked by RNA binding proteins [89]. These results suggest a model whereby blocking eIF4E from entering into the eIF4F complex could still allow for translation because in principle, eIF4G:eIF4A mediated, 5’-end directed translation initiation could still occur, albeit at a reduced rate. Removal of the eIF4A subunit from the eIF4G:eIF4A dimers or from the eIF4F complex would result in stronger repression or translation inhibition of a different set of mRNA transcripts. Indeed, we propose that differences in translation inhibition observed between cell lines exposed to rapamycin [95-98] in part may be a consequence of which step (4E:4E-BP or eIF4A:Pdcd4) is predominantly affected upon mTOR inhibition.
Clearly, a cancer cell would benefit greatly from hyperactive mTOR activity and this is what is observed in many forms of cancer. In fact, mutations in upstream regulators of mTOR (e.g. PTEN, Akt, PI3K etc.) are some of the most frequently observed (see for example Ref [99]). Thus, cancers driven by hyperactive mTOR signaling would be hypersensitive to treatment with rapamycin, and this is often the case in cell culture [100] and mouse models [98, 101]. In the clinic, however, rapamycin treatment has had much more modest success. Although the rapamycin analog everolimus (RAD001) was recently approved by the FDA for the treatment of advanced stage renal cell carcinoma [102], overall clinical outcome with rapamycin analogs are unpredictable and in general are only marginally effective as monotherapy [103]. The reasons behind rapamycin’s struggles in the clinic are not yet clear, but might relate to its ability to reactivate PI3K/Akt signaling upstream of mTOR, negating a well known mTOR negative-feedback loop [104-106]. In addition, recent data has suggested that rapamycin’s ability to inhibit mTOR kinase activity (and downstream 4E-BP phosphorylation) is only partial, suggesting that catalytic-site inhibitors might prove more effective [107, 108]. Additionally, eIF4E has been shown to be a genetic modifier of the rapamycin response, with increased levels of eIF4E imparting rapamycin resistance [109]. As well, the status of 4E-BP1 also appears to contribute to mTOR’s oncogenic repertoire. Recent publications have suggested that 4E-BP1 is vital for the regulation of cell proliferation by mTOR [77] and 4E-BP1 inactivation contributes to growth in mouse tumor models [33, 77, 110]. Moreover, the expression of a nonphosphorylatable, constitutively active 4E-BP1 suppressed growth of tumors driven by PI3K and K-Ras mutations [111]. Taken together, these studies demonstrate that development of novel compounds and strategies targeting eIF4F-dependent translation directly would be a feasible therapeutic strategy in the treatment of cancer.
eIF4E Cap-Analog Inhibitors.
Oldest among inhibitors of eIF4F are synthetic nucleotide cap-analogs, those that compete for binding with nascent capped transcripts to eIF4E. These have been used extensively now for over 30 years in the field and have been instrumental in the identification of specific translation initiation factors and the elucidation of the process of initiation as a whole [15, 112]. Diverse chemical modification to cap-analogs and nucleosides have given rise to a myriad of novel structures with even greater inhibitory potentials than the common synthetic cap-analog precursor (m7GpppG) or even ordinary methyl-7-GTP (m7GTP) [113] but their use has been limited to in vitro studies since they are not readily cell membrane permeable nor stable in cell culture [114, 115]. In an effort to circumvent such pitfalls, current research has focused on the use “pronucleotide” modifications (nucleosides with protecting groups that convert to their active metabolic form by native cytosolic enzymes) in the development of cap-analogs with greater therapeutic potential. In a recent paper, Ghosh et al. [116] have reported the synthesis of phosphoramidate derivatives of m7GTP, a “pronucleotide” modification that is relatively non-toxic, water soluble and much more stable in blood plasma. One compound, dubbed 4Ei-1 (Table I), was reportedly capable of inhibiting cap- and eIF4E-dependent reporter constructs both in vitro and in vivo when injected into freshly fertilized zebrafish. Although promising, these results are still somewhat incomplete since they have yet to determine whether 4Ei-1 can natively cross phospholipid membranes or inhibit translation in mammalian experimental systems.
Another nucleoside analog, ribavirin, has recently garnered some attention as a novel anti-eIF4E cancer therapeutic [117, 118]. Ribavirin is a nucleoside analog that displays fairly broad anti-viral properties and is currently FDA approved in the treatment of RSV and HCV. The exact mechanism of action of ribavirin in the cell has remained somewhat obscure, with theories ranging from ribavirin acting as a direct inhibitor of viral transcription or mRNA capping to a more indirect method of inhibition by reducing intracellular GTP pools or by promoting an interferon response [119]. A paper by Kentsis et al. [117] has shown data demonstrating binding of ribavirin to eIF4E in multiple assays, inhibition by ribavirin of eIF4E-dependent functions in vivo, as well being able to slow tumor growth in a xenograft mouse model, suggesting that ribavirin can act like a therapeutically viable cap-analog, although this explanation for its pharmacological action has been somewhat controversial [120, 121]. Still, even though mimicking the cellular mRNA cap as a means of directly preventing eIF4E function may seem to be the most “rational” pharmacological approach, it should be emphasized that cellular physiology surrounding the mRNA cap does not begin nor end with translation. Multiple processes governing gene expression, such as pre-mRNA splicing, nucleo-cytoplasmic transport, or mRNA decay (to name a few) utilize the cap structure as point of regulation. Thus, it should always be kept in mind that cap-analogs might have effects other than inhibiting eIF4E-dependant protein synthesis, and that perhaps other pharmacological strategies might prove more specific in design.
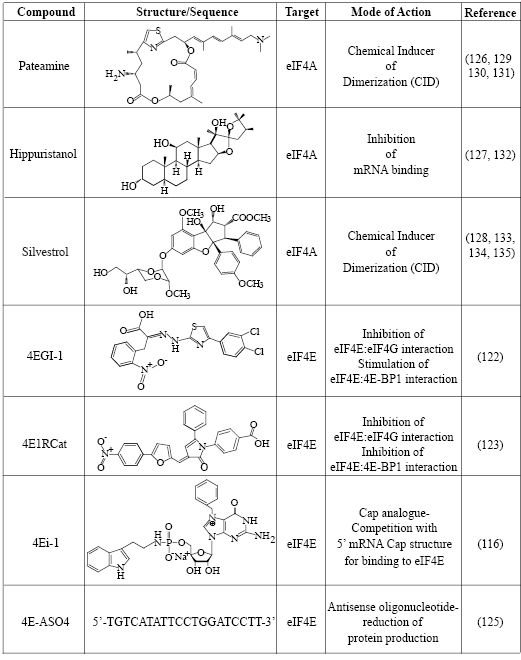
Table I: Summary of Translation Initiation Inhibitors Targeting eIF4F
Targeting the eIF4E:eIF4G Interaction
An alternative approach towards the development of agents that can interfere with eIF4E function would be the discovery and design of small molecule inhibitors that would interfere with eIF4E-eIF4G interaction. As of this writing, there are two small molecules that can compete with and block eIF4G from binding to eIF4E. The first of these to be discovered was from the Wagner lab [122], identified in a screen for small molecules that decreased the specific fluorescence polarization of a labeled peptide fragment encompassing the eIF4G binding domain when titrated with recombinant eIF4E. 4EGI-1 (Table I), as it is called, inhibited cap-dependent translation in in vitro translation extracts, depleted known eIF4E regulated proteins in vivo and elicited apoptosis in several cancer cell lines. NMR spectra further confirmed direct chemical interactions of 4EGI-1 with eIF4E residues. Curiously, 4EGI-1 did not appear to prevent the translational repressor 4E-BP1 from binding to eIF4E (which shares a similar consensus binding motif), but, counterintuitively, seemed to promote the interaction. Whether or not this “gain-of-function” activity contributes to some of the in vitro and in vivo effects observed remains to be tested.
A second eIF4E-eIF4G inhibitor, found by our group which we named 4E1RCat (Table I), was identified in an ultra high-throughput screen aimed at finding compounds that directly blocked the very same interaction [123]. Here a fluorescence-based assay was used involving recombinant eIF4E and a peptide fragment of eIF4G encompassing the consensus binding motif. Importantly, 4E1RCat blocked the cap-dependent but not HCV IRES-dependent translation of a bicistronic dual-luciferase reporter mRNA with an IC50 of ~25 uM [123]. Like 4EGI-1, 4E1RCat can prevent the association of eIF4G to m7GTP-Sepharose bound eIF4E, but unlike 4EGI-1, it also blocked 4E-BP1 binding with nearly the same efficacy. 4E1RCat was also pharmacologically active in cells and in mice, where it decreased the rate of overall protein synthesis by ~30%. More promisingly, 4E1RCat, like other protein synthesis inhibitors (see below), was able to improve the response to chemotherapy in the Eµ-myc mouse lymphoma model and thus prolong the animal’s tumor-free survival, without any obvious toxicity.
Targeting eIF4E Production
An alternative to using competitive small molecule inhibitors would be the use of bioavailable oligonucleotides to genetically suppress the production of eIF4E protein. Earlier work demonstrated the feasibility of using antisense oligonucleotides (ASO) targeting eIF4E, thus limiting the tumorigenicity of K-Ras transformed cell lines, but was constrained technologically to proof-of-principle in vitro manipulations [124]. More recently, Graff and colleagues at Eli Lilly Research Labs [125] have developed ASOs directed against eIF4E using second-generation backbone antisense modifications that impart improved nuclease resistance and tissue stability to allow for effective systemic therapeutic delivery. In their study, they observed 80% knockdown of eIF4E with their most potent 4E ASO (Table I), which had only a relatively small impact on global protein synthesis (~20% change). Still, known eIF4E-specific pro-growth and pro-survival gene products decreased in a dose-dependent manner. Impressively, administration of 4E ASOs almost completely blocked tumor growth in breast and prostate xenografts. Furthermore the reduction of eIF4E also appeared to prevent endothelial cell tube formation suggesting a possible role for eIF4E in tumor angiogenesis. And from a pharmacological standpoint, reduced eIF4E levels were well tolerated in normal mouse tissues, perhaps reflecting the notion that the sensitivity and tumor selectivity from the specific translational downregulation of oncogenic targets afforded from eIF4E knockdown allows for a greater therapeutic index.
Targeting eIF4A activity
Loss of eIF4E prevents binding of eIF4F to the cap-structure that ultimately reduces its ability to eliminate 5’-end RNA secondary structure and hampers efficient recruitment of the 43S ribosome complexes. A different approach towards limiting translation initiation would be by targeting eIF4A, the source of eIF4F enzymatic activity. Our lab has previously reported on the identification and characterization of three small molecule inhibitors of eIF4A (Table I) [126-128]. Two of these (pateamine A and silvestrol), paradoxically, stimulate eIF4A activity, mostly by forcing the binding of eIF4A to RNA on which much of its enzymatic activity relies [126, 128-131]. The net effect is however an overall reduction in protein synthesis caused by a pronounced block in translation initiation as a result of most of eIF4A being depleted from the eIF4F complex [126, 128, 129]. Hippuristanol, the third eIF4A inhibitor, suppresses eIF4A’s helicase, ATPase and RNA binding properties[127, 132]. All three compounds show high specificity towards eIF4A, making them attractive for studying the biological action of eIF4A in vivo.
Silvestrol has been tested in several different mouse cancer models with encouraging results. The compound does not cause distress, weight loss or liver damage, and does not appear to immunosuppress in the mouse [133, 134]. In some settings, silvestrol alone has therapeutic benefit as chemotherapy: this is the case for xenograft studies of acute lymphoblastic leukemia in SCID mice [134], prostate cancer and breast cancer xenografts in nude mice [133], in the Eµ-Tcl-1, a mouse model of chronic lymphocytic leukemia (CLL), as well as primary human CLL samples [134]. In the latter study, the authors observed that silvestrol was far more toxic towards B-cells than T-cells and that B-cells derived from chronic lymphocytic leukemia patients were more sensitive to the drug than from healthy individuals [134], suggesting preferential targeting of faster growing leukemic cells by silvestrol. Still, in the Eµ-Myc mouse model, a Burkitt’s B-cell lymphoma cancer model, silvestrol alone showed no effect, although this might be explained by the lower doses used [128]. However, when combined with doxorubicin it greatly prolonged the tumor-free survival of tumor-burdened mice [128]. Moreover, this combination therapy was even effective against eIF4E-driven lymphomas, which normally do not respond to conventional monotherapy or combination chemotherapy regime [109]. This is also what is seen in AML cells, where silvestrol increases the cytotoxicity of daunorubicin, etoposide and cytarabine [135].
FUTURE DIRECTIONS
Even though these are still very early days in the development of eIF4F inhibitors as antineoplastics, protein synthesis inhibitors seem poised for a pharmaceutical comeback. Homoharringtonine (HHT) (omacetaxine mepesuccinat), a known inhibitor of peptide-chain elongation, has shown promise in Phase II clinical trials for the treatment of gleevec-resistant chronic myelogenous leukemia (CML) [136] and was approved to be “fast-tracked” by the FDA for clinical development. Homoharringtonine’s anti-neoplastic abilities have been known since the ‘70s [137] and it was considered the premier drug in salvage therapy for CML before getting supplanted by gleevec [138]. Recent data have suggested that part of HHT’s mechanism of action is its ability to downregulate the short-lived anti-apoptotic and pro-oncogenic protein Mcl-1 [139, 140], as would be expected from transient exposure to a general protein synthesis inhibitor and perhaps explaining some of the synergistic effects observed from most inhibitors of translation elongation in the Eµ-Myc model [141]. Other elongation inhibitors have had less success clinically in treatment of cancer due to non-specific toxicity in the patient [142], suggesting a limited therapeutic index for these general protein synthesis inhibitors. Targeting eIF4F might have the benefit of expanding this window of therapeutic success, given the molecular response afforded by blocking translation initiation: one can affect the scope of protein synthesis production (from a more global to more specific mRNA translational inhibition) not only from dosing of a given drug but also by changing the nature of the pharmacological intervention (eIF4A versus eIF4E inhibition). It will be interesting to see whether targeting other regulators of initiation (e.g.-eIF4B. eIF4H, PABP) would yield a similar or even greater therapeutic potential in the treatment of cancer. The next 5 years will indeed be exciting as we monitor the clinical development and progress of translation initiation inhibitors as potential antineoplastics.
ACKNOWLEDGEMENTS
Work in the author’s lab related to the topic presented herein is supported by grants from the Canadian Cancer Society Research Institute (#17099) and the Canadian Institutes of Health Research (CIHR MOP-106530).
Conflict of Interest Statement
The authors declare they have no conflict of interest relevant to the manuscript’s subject.
REFERENCES
1. Felsher DW. Reversibility of oncogene-induced cancer. Curr Opin Genet Dev. 2004; 14:37-42.
2. Solimini NL, Luo J Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007; 130:986-8.
3. Ingolia NT, Ghaemmaghami S, Newman JR Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218-23.
4. Matthews MB, Sonenberg N Hershey JWB (2007) Origins and Principle of Translation Control. In Translational Control in Biology and Medicine, Matthews MB, Sonenberg N, Hershey JWB, eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.), pp. 1-40.
5. Kapp LD Lorsch JR. The molecular mechanics of eukaryotic translation. Annu Rev Biochem. 2004; 73:657-704.
6. Pestova TV, Lorsch JR Hellen CUT (2007) The Mechanism of Translation Initiation in Eukaryotes. In Translational Control in Biology and Medicine, Matthews MB, Sonenberg N, Hershey JWB, eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor) pp. 87-128.
7. Jackson RJ, Hellen CU Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010; 11:113-27.
8. Jang SK, Davies MV, Kaufman RJ Wimmer E. Initiation of protein synthesis by internal entry of ribosomes into the 5’ nontranslated region of encephalomyocarditis virus RNA in vivo. J Virol. 1989; 63:1651-60.
9. Gingras AC, Raught B Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999; 68:913-63.
10. Nielsen PJ Trachsel H. The mouse protein synthesis initiation factor 4A gene family includes two related functional genes which are differentially expressed. EMBO J. 1988; 7:2097-105.
11. Rogers GW, Jr., Komar AA Merrick WC. eIF4A: The godfather of the DEAD box helicases. Prog Nucleic Acid Res Mol Biol. 2002; 72:307-31.
12. Conroy SC, Dever TE, Owens CL Merrick WC. Characterization of the 46,000-dalton subunit of eIF-4F. Arch Biochem Biophys. 1990; 282:363-71.
13. Yoder-Hill J, Pause A, Sonenberg N Merrick WC. The p46 subunit of eukaryotic initiation factor (eIF)-4F exchanges with eIF-4A. J Biol Chem. 1993; 268:5566-73.
14. Ramirez-Valle F, Braunstein S, Zavadil J, Formenti SC Schneider RJ. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol. 2008; 181:293-307.
15. Sonenberg N, Morgan MA, Merrick WC Shatkin AJ. A polypeptide in eukaryotic initiation factors that crosslinks specifically to the 5’-terminal cap in mRNA. Proc Natl Acad Sci USA. 1978; 75:4843-7.
16. Shatkin AJ, Darzynkiewicz E, Furuichi Y, Kroath H, Morgan MA, Tahara SM Yamakawa M. 5’-Terminal caps, cap-binding proteins and eukaryotic mRNA function. Biochem Soc Symp. 1982; 47:129-43.
17. Marcotrigiano J, Gingras AC, Sonenberg N Burley SK. Cocrystal structure of the messenger RNA 5’ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell. 1997; 89:951-61.
18. Matsuo H, Li H, McGuire AM, Fletcher CM, Gingras AC, Sonenberg N Wagner G. Structure of translation factor eIF4E bound to m7GDP and interaction with 4E-binding protein. Nat Struct Biol. 1997; 4:717-24.
19. Pestova TV Kolupaeva VG. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002; 16:2906-22.
20. Yanagiya A, Svitkin YV, Shibata S, Mikami S, Imataka H Sonenberg N. Requirement of RNA binding of mammalian eukaryotic translation initiation factor 4GI (eIF4GI) for efficient interaction of eIF4E with the mRNA cap. Mol Cell Biol. 2009; 29:1661-9.
21. Park EH, Walker SE, Lee JM, Rothenburg S, Lorsch JR Hinnebusch AG. Multiple elements in the eIF4G1 N-terminus promote assembly of eIF4G1*PABP mRNPs in vivo. EMBO J. 2011; 30:302-16.
22. von Der Haar T, Ball PD McCarthy JE. Stabilization of eukaryotic initiation factor 4E binding to the mRNA 5’-Cap by domains of eIF4G. J Biol Chem. 2000; 275:30551-5.
23. Sonenberg N. ATP/Mg++-dependent cross-linking of cap binding proteins to the 5’ end of eukaryotic mRNA. Nucleic Acids Res. 1981; 9:1643-56.
24. Rogers GW, Jr., Richter NJ, Lima WF Merrick WC. Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J Biol Chem. 2001; 276:30914-22.
25. Lamphear BJ, Kirchweger R, Skern T Rhoads RE. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J Biol Chem. 1995; 270:21975-83.
26. Jacobson A (1996) Poly(A) metabolism and translation: the closed loop model. In In Translational Control in Biology and Medicine, Matthews MB, Sonenberg N, Hershey JWB, eds. (Cold Spring Harbor Press, Cold Spring Harbor), pp. 451-80.
27. Kahvejian A, Svitkin YV, Sukarieh R, M’Boutchou M-N Sonenberg N. Mammalian poly(A)-binding protien is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes & Dev. 2005; 19:104-13.
28. Tarun SZ, Jr. Sachs AB. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J. 1996; 15:7168-77.
29. Lazaris-Karatzas A, Montine KS Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5’ cap. Nature. 1990; 345:544-7.
30. Schneider R Sonenberg N (2007) Translational control in cancer development and progression. In Translational Control in Biology and Medicine, Matthews MB, Sonenberg N Hershey JWB, eds. (Cold SPring Harbor Press, Cold Spring Harbor), pp. 401-31.
31. Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004; 428:332-7.
32. Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C Pandolfi PP. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004; 10:484-6.
33. Petroulakis E, et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell. 2009; 16:439-46.
34. Liang Z, Lei T, LuYing Z YuPing G. The expression of proto-oncogene eIF4E in laryngeal squamous cell carcinoma. Laryngoscope. 2003; 113:1238-43.
35. Kerekatte V, Smiley K, Hu B, Smith A, Gelder F De Benedetti A. The proto-oncogene/translation factor eIF4E: a survey of its expression in breast carcinomas. Int J Cancer. 1995; 64:27-31.
36. Li BD, Liu L, Dawson M De Benedetti A. Overexpression of eukaryotic initiation factor 4E (eIF4E) in breast carcinoma. Cancer. 1997; 79:2385-90.
37. Li BD, Gruner JS, Abreo F, Johnson LW, Yu H, Nawas S, McDonald JC DeBenedetti A. Prospective study of eukaryotic initiation factor 4E protein elevation and breast cancer outcome. Ann Surg. 2002; 235:732-8.
38. Nathan CO, Liu L, Li BD, Abreo FW, Nandy I De Benedetti A. Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene. 1997; 15:579-84.
39. Sorrells DL, Jr., Ghali GE, De Benedetti A, Nathan CA Li BD. Progressive amplification and overexpression of the eukaryotic initiation factor 4E gene in different zones of head and neck cancers. J Oral Maxillofac Surg. 1999; 57:294-9.
40. Haydon MS, Googe JD, Sorrells DS, Ghali GE Li BD. Progression of eIF4e gene amplification and overexpression in benign and malignant tumors of the head and neck. Cancer. 2000; 88:2803-10.
41. Nathan CO, Franklin S, Abreo FW, Nassar R, De Benedetti A Glass J. Analysis of surgical margins with the molecular marker eIF4E: a prognostic factor in patients with head and neck cancer. J Clin Oncol. 1999; 17:2909-14.
42. Franklin S, Pho T, Abreo FW, Nassar R, De Benedetti A, Stucker FJ Nathan CO. Detection of the proto-oncogene eIF4E in larynx and hypopharynx cancers. Arch Otolaryngol Head Neck Surg. 1999; 125:177-82.
43. Nathan CO, Sanders K, Abreo FW, Nassar R Glass J. Correlation of p53 and the proto-oncogene eIF4E in larynx cancers: prognostic implications. Cancer Res. 2000; 60:3599-604.
44. Seki N, Takasu T, Mandai K, Nakata M, Saeki H, Heike Y, Takata I, Segawa Y, Hanafusa T Eguchi K. Expression of eukaryotic initiation factor 4E in atypical adenomatous hyperplasia and adenocarcinoma of the human peripheral lung. Clin Cancer Res. 2002; 8:3046-53.
45. Wang S, Lloyd RV, Hutzler MJ, Rosenwald IB, Safran MS, Patwardhan NA Khan A. Expression of eukaryotic translation initiation factors 4E and 2alpha correlates with the progression of thyroid carcinoma. Thyroid. 2001; 11:1101-7.
46. Rosenwald IB, Hutzler MJ, Wang S, Savas L Fraire AE. Expression of eukaryotic translation initiation factors 4E and 2alpha is increased frequently in bronchioloalveolar but not in squamous cell carcinomas of the lung. Cancer. 2001; 92:2164-71.
47. Wang S, Rosenwald IB, Hutzler MJ, Pihan GA, Savas L, Chen JJ Woda BA. Expression of the eukaryotic translation initiation factors 4E and 2alpha in non-Hodgkin’s lymphomas. Am J Pathol. 1999; 155:247-55.
48. Rosenwald IB, Chen JJ, Wang S, Savas L, London IM Pullman J. Upregulation of protein synthesis initiation factor eIF-4E is an early event during colon carcinogenesis. Oncogene. 1999; 18:2507-17.
49. Shuda M, Kondoh N, Tanaka K, Ryo A, Wakatsuki T, Hada A, Goseki N, Igari T, Hatsuse K, Aihara T, Horiuchi S, Shichita M, Yamamoto N Yamamoto M. Enhanced expression of translation factor mRNAs in hepatocellular carcinoma. Anticancer Res. 2000; 20:2489-94.
50. Martin ME, Perez MI, Redondo C, Alvarez MI, Salinas M Fando JL. 4E binding protein 1 expression is inversely correlated to the progression of gastrointestinal cancers. Int J Biochem Cell Biol. 2000; 32:633-42.
51. Fukuchi-Shimogori T, Ishii I, Kashiwagi K, Mashiba H, Ekimoto H Igarashi K. Malignant transformation by overproduction of translation initiation factor eIF4G. Cancer Res. 1997; 57:5041-4.
52. Bauer C, Brass N, Diesinger I, Kayser K, Grasser FA Meese E. Overexpression of the eukaryotic translation initiation factor 4G (eIF4G-1) in squamous cell lung carcinoma. Int J Cancer. 2002; 98:181-5.
53. Bauer C, Diesinger I, Brass N, Steinhart H, Iro H Meese EU. Translation initiation factor eIF-4G is immunogenic, overexpressed, and amplified in patients with squamous cell lung carcinoma. Cancer. 2001; 92:822-9.
54. Silvera D, Arju R, Darvishian F, Levine PH, Zolfaghari L, Goldberg J, Hochman T, Formenti SC Schneider RJ. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat Cell Biol. 2009; 11:903-8.
55. Eberle J, Krasagakis K Orfanos CE. Translation initiation factor eIF-4A1 mRNA is consistently overexpressed in human melanoma cells in vitro. Int J Cancer. 1997; 71:396-401.
56. Shuda M, Kondoh N, Tanaka K, Ryo A, Wakatsuki T, Hada A, Goseki N, Igari T, Hatsuse K, Aihara T, Horiuchi S, Shichita M, Yamamoto N Yamamoto M. Enhanced expression of translation factor mRNAs in hepatocellular carcinoma. Anticancer Res. 2000; 20:2489-94.
57. Rosenwald IB. The role of translation in neoplastic transformation from a pathologist’s point of view. Oncogene. 2004; 23:3230-47.
58. Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Teruya-Feldstein J, Pelletier J Lowe SW. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007; 21:3232-7.
59. Graff JR Zimmer SG. Translational control and metastatic progression: Enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin. Exp. Met. 2003; 20:265-73.
60. Graff JR, Konicek BW, Carter JH Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008; 68:631-4.
61. Lawson TG, Ray BK, Dodds JT, Grifo JA, Abramson RD, Merrick WC, Betsch DF, Weith HL Thach RE. Influence of 5’ proximal secondary structure on the translational efficiency of eukaryotic mRNAs and on their interaction with initiation factors. J Biol Chem. 1986; 261:13979-89.
62. Pelletier J Sonenberg N. Insertion mutagenesis to increase secondary structure within the 5’ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell. 1985; 40:515-26.
63. Pelletier J Sonenberg N. Photochemical cross-linking of cap binding proteins to eucaryotic mRNAs: effect of mRNA 5’ secondary structure. Mol Cell Biol. 1985; 5:3222-30.
64. Clemens MJ Bommer UA. Translational control: the cancer connection. Int J Biochem Cell Biol. 1999; 31:1-23.
65. Watkins SJ Norbury CJ. Translation initiation and its deregulation during tumorigenesis. Br J Cancer. 2002; 86:1023-7.
66. Hershey JWB Miyamoto S (2000) Translational Control and Cancer. In Translational Control of Gene Expression, Sonenberg, N, Hershey, JWB, Mathews, MB, eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor) pp 637-54.
67. Averous J Proud CG. When translation meets transformation: the mTOR story. Oncogene. 2006; 25:6423-35.
68. De Benedetti A Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004; 23:3189-99.
69. Mamane Y, Petroulakis E, Martineau Y, Sato TA, Larsson O, Rajasekhar VK Sonenberg N. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS ONE. 2007; 2:e242.
70. Rajasekhar VK Holland EC. Postgenomic global analysis of translational control induced by oncogenic signaling. Oncogene. 2004; 23:3248-64.
71. De Benedetti A, Joshi-Barve S, Rinker-Schaeffer C Rhoads RE. Expression of antisense RNA against initiation factor eIF-4E mRNA in HeLa cells results in lengthened cell division times, diminished translation rates, and reduced levels of both eIF-4E and the p220 component of eIF-4F. Mol Cell Biol. 1991; 11:5435-45.
72. Graff JR, Boghaert ER, De Benedetti A, Tudor DL, Zimmer CC, Chan SK Zimmer SG. Reduction of translation initiation factor 4E decreases the malignancy of ras-transformed cloned rat embryo fibroblasts. Int J Cancer. 1995; 60:255-63.
73. Herbert TP, Fahraeus R, Prescott A, Lane DP Proud CG. Rapid induction of apoptosis mediated by peptides that bind initiation factor eIF4E. Curr Biol. 2000; 10:793-6.
74. Zoncu R, Efeyan A Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12:21-35.
75. Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC, Jr. Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5’-cap function. Nature. 1994; 371:762-7.
76. Mader S, Lee H, Pause A Sonenberg N. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4 gamma and the translational repressors 4E-binding proteins. Mol Cell Biol. 1995; 15:4990-7.
77. Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, Kozma SC, Thomas G Sonenberg N. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010; 328:1172-6.
78. Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006; 314:467-71.
79. Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N Hershey JW. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 2004.
80. Cmarik JL, Min H, Hegamyer G, Zhan S, Kulesz-Martin M, Yoshinaga H, Matsuhashi S Colburn NH. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci U S A. 1999; 96:14037-42.
81. Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, Lockett SJ, Sonenberg N Colburn NH. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol. 2003; 23:26-37.
82. Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M Sonenberg N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006; 25:2781-91.
83. Duncan R, Milburn SC Hershey JW. Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. J Biol Chem. 1987; 262:380-8.
84. Tarun SZ, Jr. Sachs AB. A common function for mRNA 5’ and 3’ ends in translation initiation in yeast. Genes Dev. 1995; 9:2997-3007.
85. Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes & Dev. 1991; 5:2108-16.
86. Preiss T Hentze MW. Dual function of the messenger RNA cap structure in poly(A)-tail-promoted translation in yeast. Nature. 1998; 392:516-20.
87. Iizuka N, Najita L, Franzusoff A Sarnow P. Cap-dependent and cap-independent translation by internal initiation of mRNAs in cell extracts prepared from Saccharomyces cerevisiae. Mol Cell Biol. 1994; 14:7322-30.
88. Michel YM, Poncet D, Piron M, Kean KM Borman AM. Cap-poly(A) synergy in mammalian cell-free extracts. Investigation of the requirements for poly(A)-mediated stimulation of translation initiation. J. Biol. Chem. 2000; 275:32268-76.
89. Svitkin YV, Ovchinnikov LP, Dreyfuss G Sonenberg N. General RNA binding proteins render translation cap dependent. EMBO J. 1996; 15:7147-55.
90. Lang V, Zanchin NI, Lunsdorf H, Tuite M McCarthy JE. Initiation factor eIF-4E of Saccharomyces cerevisiae. Distribution within the cell, binding to mRNA, and consequences of its overproduction. J Biol Chem. 1994; 269:6117-23.
91. Vasilescu S, Ptushkina M, Linz B, Muller PP McCarthy JE. Mutants of eukaryotic initiation factor eIF-4E with altered mRNA cap binding specificity reprogram mRNA selection by ribosomes in Saccharomyces cerevisiae. J Biol Chem. 1996; 271:7030-7.
92. Andreev DE, Dmitriev SE, Terenin IM, Prassolov VS, Merrick WC Shatsky IN. Differential contribution of the m7G-cap to the 5’ end-dependent translation initiation of mammalian mRNAs. Nucleic Acids Res. 2009; 37:6135-47.
93. De Gregorio E, Preiss T Hentze MW. Translational activation of uncapped mRNAs by the central part of human eIF4G is 5’ end-dependent. RNA. 1998; 4:828-36.
94. De Gregorio E, Preiss T Hentze MW. Translation driven by an eIF4G core domain in vivo. EMBO J. 1999; 18:4865-74.
95. Jefferies HB, Reinhard C, Kozma SC Thomas G. Rapamycin selectively represses translation of the “polypyrimidine tract” mRNA family. Proc Natl Acad Sci U S A. 1994; 91:4441-5.
96. Terada N, Patel HR, Takase K, Kohno K, Nairn AC Gelfand EW. Rapamycin selectively inhibits translation of mRNAs encoding elongation factors and ribosomal proteins. Proc Natl Acad Sci U S A. 1994; 91:11477-81.
97. Mendez R, Myers MG, Jr., White MF Rhoads RE. Stimulation of protein synthesis, eukaryotic translation initiation factor 4E phosphorylation, and PHAS-I phosphorylation by insulin requires insulin receptor substrate 1 and phosphatidylinositol 3-kinase. Mol Cell Biol. 1996; 16:2857-64.
98. Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, Lowe SW Pelletier J. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A. 2008; 105:10853-8.
99. Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007; 318:1108-13.
100. Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001; 98:10314-9.
101. Podsypanina K, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/- mice. Proc Natl Acad Sci U S A. 2001; 98:10320-5.
102. Hudes G, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007; 356:2271-81.
103. Lane HA Breuleux M. Optimal targeting of the mTORC1 kinase in human cancer. Curr Opin Cell Biol. 2009; 21:219-29.
104. Jaeschke A, Hartkamp J, Saitoh M, Roworth W, Nobukuni T, Hodges A, Sampson J, Thomas G Lamb R. Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol. 2002; 159:217-24.
105. Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005; 19:1773-8.
106. Shah OJ, Wang Z Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004; 14:1650-6.
107. Thoreen CC Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009; 5:725-6.
108. Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009; 7:e38.
109. Wendel HG, Malina A, Zhao Z, Zender L, Kogan SC, Cordon-Cardo C, Pelletier J Lowe SW. Determinants of sensitivity and resistance to rapamycin-chemotherapy drug combinations in vivo. Cancer Res. 2006; 66:7639-46.
110. Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM Ruggero D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010; 17:249-61.
111. She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, Solit DB Rosen N. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010; 18:39-51.
112. Hickey ED, Weber LA, Baglioni C, Kim CH Sarma RH. A relation between inhibition of protein synthesis and conformation of 5’-phosphorylated 7-methylguanosine derivatives. J Mol Biol. 1977; 109:173-83.
113. Cai A, Jankowska-Anyszka M, Centers A, Chlebicka L, Stepinski J, Stolarski R, Darzynkiewicz E Rhoads RE. Quantitative assessment of mRNA cap analogues as inhibitors of in vitro translation. Biochemistry. 1999; 38:8538-47.
114. Jemielity J, Kowalska J, Rydzik AM Darzynkiewicz E. Synthetic mRNA cap analogs with a modified triphosphate bridge – synthesis, applications and prospects. New Journal of Chemistry. 2010:829-44.
115. Wagner CR, Iyer VV McIntee EJ. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med Res Rev. 2000; 20:417-51.
116. Ghosh B, Benyumov AO, Ghosh P, Jia Y, Avdulov S, Dahlberg PS, Peterson M, Smith K, Polunovsky VA, Bitterman PB Wagner CR. Nontoxic chemical interdiction of the epithelial-to-mesenchymal transition by targeting cap-dependent translation. ACS Chem Biol. 2009; 4:367-77.
117. Kentsis A, Topisirovic I, Culjkovic B, Shao L Borden KL. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc Natl Acad Sci U S A. 2004; 101:18105-10.
118. Assouline S, Culjkovic B, Cocolakis E, Rousseau C, Beslu N, Amri A, Caplan S, Leber B, Roy DC, Miller WH, Jr. Borden KL. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): a proof-of-principle clinical trial with ribavirin. Blood. 2009; 114:257-60.
119. Tam RC, Lau JY Hong Z. Mechanisms of action of ribavirin in antiviral therapies. Antivir Chem Chemother. 2001; 12:261-72.
120. Yan Y, Svitkin Y, Lee JM, Bisaillon M Pelletier J. Ribavirin is not a functional mimic of the 7-methyl guanosine mRNA cap. RNA. 2005; 11:1238-44.
121. Westman B, Beeren L, Grudzien E, Stepinski J, Worch R, Zuberek J, Jemielity J, Stolarski R, Darzynkiewicz E, Rhoads RE Preiss T. The antiviral drug ribavirin does not mimic the 7-methylguanosine moiety of the mRNA cap structure in vitro. RNA. 2005; 11:1505-13.
122. Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA Wagner G. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007; 128:257-67.
123. Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, Fu H, Kozakov D, Vajda S Pelletier J. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A. 2011; 108:1046-51.
124. Rinker-Schaeffer CW, Graff JR, De Benedetti A, Zimmer SG Rhoads RE. Decreasing the level of translation initiation factor 4E with antisense RNA causes reversal of ras-mediated transformation and tumorigenesis of cloned rat embryo fibroblasts. Int J Cancer. 1993; 55:841-7.
125. Graff JR, et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007; 117:2638-48.
126. Bordeleau ME, Matthews J, Wojnar JM, Lindqvist L, Novac O, Jankowsky E, Sonenberg N, Northcote P, Teesdale-Spittle P Pelletier J. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc Natl Acad Sci U S A. 2005; 102:10460-5.
127. Bordeleau M-E, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J Pelletier J. Functional Characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat Chem Biol. 2006; 2:213-20.
128. Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SM, Wendel HG, Brem B, Greger H, Lowe SW, Porco JA, Jr. Pelletier J. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest. 2008; 118:2651-60.
129. Bordeleau ME, Cencic R, Lindqvist L, Oberer M, Northcote P, Wagner G Pelletier J. RNA-mediated sequestration of the RNA helicase eIF4A by Pateamine A inhibits translation initiation. Chem Biol. 2006; 13:1287-95.
130. Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, Romo D Liu JO. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell. 2005; 20:709-22.
131. Low WK, Dang Y, Bhat S, Romo D Liu JO. Substrate-Dependent Targeting of Eukaryotic Translation Initiation Factor 4A by Pateamine A: Negation of Domain-Linker Regulation of Activity. Chem Biol. 2007; 14:715-27.
132. Lindqvist L, Oberer M, Reibarkh M, Cencic R, Bordeleau ME, Vogt E, Marintchev A, Tanaka J, Fagotto F, Altmann M, Wagner G Pelletier J. Selective pharmacological targeting of a DEAD box RNA helicase. PLoS ONE. 2008; 3:e1583.
133. Cencic R, Carrier M, Galicia-Vazquez G, Bordeleau ME, Sukarieh R, Bourdeau A, Brem B, Teodoro JG, Greger H, Tremblay ML, Porco JA, Jr. Pelletier J. Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS ONE. 2009; 4:e5223.
134. Lucas DM, et al. The novel plant-derived agent silvestrol has B-cell selective activity in chronic lymphocytic leukemia and acute lymphoblastic leukemia in vitro and in vivo. Blood. 2009.
135. Cencic R, Carrier M, Trnkus A, Porco JA, Jr., Minden M Pelletier J. Synergistic effect of inhibiting translation initiation in combination with cytotoxic agents in acute myelogenous leukemia cells. Leuk Res. 2009.
136. Lipton JH, Wetzler M, Nicolini F, Baccarani M, Baer MR, Masszi T, Cram D, Benichou A, Nanda N Cortes JE (2010) Safety of omacetaxine mepesuccinate (OM) subcutaneous (SQ) injection for the treatment of chronic myeloid leukemia (CML) patients (pts) resistant or intolerant to tyrosine kinase inhibitors (TKIs): Analysis of two phase II studies. 2010 ASCO Annual Meeting, J Clin Oncol. 2010; abstr 6568.
137. Powell RG, Weisleder D Smith CRJ. Antitumor alkaloids from Cephalotaxus harrintonia: structure and activity. J. Pharm. Sci. 1972; 61:1227-30.
138. Quintas-Cardama A, Kantarjian H Cortes J. Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009. Cancer. 2009; 115:5382-93.
139. Tang R, Faussat AM, Majdak P, Marzac C, Dubrulle S, Marjanovic Z, Legrand O Marie JP. Semisynthetic homoharringtonine induces apoptosis via inhibition of protein synthesis and triggers rapid myeloid cell leukemia-1 down-regulation in myeloid leukemia cells. Mol Cancer Ther. 2006; 5:723-31.
140. Chen R, Guo L, Chen Y, Jiang Y, Wierda WG Plunkett W. Homoharringtonine reduced Mcl-1 expression and induced apoptosis in chronic lymphocytic leukemia. Blood. 2011; 117:156-64.
141. Robert F, Carrier M, Rawe S, Chen S, Lowe S Pelletier J. Altering chemosensitivity by modulating translation elongation. PLoS ONE. 2009; 4:e5428.
142. Pelletier J Peltz SW (2007) Therapeutic Opportunities in Translation. In Translational Control in Biology and Medicine, Matthews MB, Sonenberg N, Hershey JWB, eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.), pp. 855-895.