
INTRODUCTION
Bovine herpesvirus 1 (BoHV-1), an enveloped double-stranded-DNA virus that belongs to the subfamily Alphaherpesvirinae of the Herpesviridae family, causes bovine respiratory disease, vulvovaginitis, abortion, conjunctivitis, balanoposthitis, and severe neonatal disease in cattle [1]. It is divided into three sub-genotypes: BoHV-1.1, BoHV-1.2a, and BoHV-1.2b, which can be differentiated by genomic sequencing and restriction fragment length polymorphisms (RFLPs) [2, 3]. BoHV-1 infects cattle of all ages and breeds worldwide, and it causes severe economic losses to the cattle industry [4]. The morphogenesis and maturation of viral particles is an important stage of viral replication, which contributes to the pathogenesis of herpesviruses. The mature BoHV-1 virion consists of a multilayered architecture that is common in all herpesviruses: a capsid enclosing the double-stranded DNA genome, an envelope, and the tegument. Tegument is a cluster of proteins that occupy the space between the envelope and nucleocapsids. All alpha herpesviruses possess more than 15 tegument proteins [5]. As components of viruses, tegument proteins are delivered into host cells upon viral infection, where some paly roles in the early stage of viral particle production by regulating the expression of viral immediate-early genes, as well as in the late stage of infection via their involvement in viral morphogenesis, assembly, and egress [6, 7]. These proteins play critical roles in each step of envelopment, de-envelopment, and re-envelopment process [5, 8].
UL21 is conserved, multifunctional tegument protein in alphaherpesviruses [9, 10], with sequence similarity ranging between 57 and 94% at the nucleotide level and sequence identity ranging between 27 and 84% at the amino acid level. It has different essential, albeit poorly understood, functions in viral replication and pathogenesis. Previously the crystal structures of UL21 have been reported in HSV-1 [9, 11]. The functions of UL21 have been well-characterized in herpes simplex virus (HSV) and pseudorabies virus (PRV). Previous studies highlighted that a lack of UL21 resulted in decreased viral titers in HSV-1 [12] and PRV [13, 14], and a UL21 deletion mutant caused delay in production of viral mRNA, viral proteins, and virions in HSV-2 and HSV-1 [10, 12, 15]. In HSV-2 ΔUL21-infected cells, nuclear egress was blocked, which reduced viral titer to undetectable levels [10]. In HSV-1 ΔUL21 mutant showed a reduction of UL16 in the tegument of extracellular virions [16], while a PRV ΔUL21 mutant incorporated lower amounts of the tegument proteins UL46 and UL49, as well as a lower level of US3 into mature virions [17]. Biochemical data suggest that UL21 gene expression in PRV facilitates viral DNA processing and is directly linked with capsid maturation [18]. Moreover, UL21 was shown to be a capsid-associated tegument protein in HSV-1 [19] and PRV [18] via an interaction with UL16 [14, 20], which connects the capsid to the envelope to promote secondary envelopment [21, 22]. In HSV-1, UL21 is critical for the function of glycoprotein E by cooperating with the interacting proteins UL11 and UL16. Additionally, in HSV-1, UL21 promotes intracellular viral movement by interacting with microtubules [19]. The cytoplasmic and nuclear localization of UL21 has been observed in HSV-1 [19, 20], particularly to the nuclear rim in HSV-1 and HSV-2 [10, 23].
Herpesvirus assembly and egress is a complex and dynamic process that requires many protein interactions and so are the roles of tegument proteins. As in HSV and PRV, UL16 interacts with UL11 and UL21, and it assists the cellular release of virions via budding and virion morphogenesis [14, 20, 24]. The teguments proteins are compositionally differ among members of the herpesvirus family. The major constituents of the BoHV-1 envelope proteins have been well-characterized [4]; however, the tegument and capsid proteins have not been studied extensively [5]. Nevertheless, the phenotype of UL21 protein in BoHV-1 has not been characterized in cell culture.
The present study was conducted to determine the functions of UL21 in BoHV-1. To this end, we generated a mutant virus with a UL21 deletion that does not express pUL21 as well as its revertant. We linked an epitope tag to the minor capsid protein UL35 in UL21 deletion and intact UL21 to analyze the capsid egress pathway. We found that the ΔUL21 mutant exhibited unenveloped capsid accumulation and that its replication was approximately 1,000-fold lower than that of the wild-type virus. In addition, we found that UL21 formed a complex with UL16. While the UL16 interaction has been reported in other herpesviruses, this is the first report of an interaction between UL21 and UL16 in BoHV-1.
RESULTS
Construction and initial characterization of BoHV-1 mutant viruses
A complete schematic map of the UL21 gene encoding the 575-amino-acid UL21 protein in the BoHV-1 genome is shown in Figure 1A. We constructed a series of recombinant viruses (v), including the UL21 mutant vBoHV-1-ΔUL21 by deleting the entire UL21 coding sequence (Figure 1B), as well as the revertant virus vBoHV-1-ΔUL21R (Figure 1C), the UL21 HA-tagged virus vBoHV-1-UL21-HA (Figure 1D), the UL35 HA-tagged virus vBoHV-1-UL35-HA (Figure 1E), and its UL21 mutant vBoHV-1-ΔUL21-UL35-HA (Figure 1F). Viral genomic DNA was extracted from the vBoHV-1-ΔUL21, vBoHV-1-ΔUL21R, vBoHV-1-UL21-HA, vBoHV-1-UL35-HA, vBoHV-1-ΔUL21-UL35-HA, and wild-type viruses. The UL21 deletion, the reversion of this mutation, and the addition of HA tags to the UL21 or UL35 genes was confirmed by PCR (Figure 2A) and DNA sequencing of the PCR products. The UL21 mRNA level was determined by RT-PCR for all the viruses. A high level of UL21 mRNA expression was observed in viruses with either an intact UL21 gene (wild type BoHV-1) or the revertant UL21 gene (vBoHV-1-ΔUL21R), while UL21 mRNA was barely detected in the ΔUL21 mutant (vBoHV-1-ΔUL21) and the HA-tagged ΔUL21 mutant (vBoHV-1-ΔUL21-UL35-HA) viruses as shown in Figure 2B. In addition, the expression of the HA-tagged UL21 or UL35 in the vBoHV-1-UL21-HA, vBoHV-1-UL35-HA, vBoHV-1-ΔUL21-UL35-HA, and BoHV-1 (wild-type) viruses was detected by Western blotting. As expected, all the HA-tagged proteins were expressed successfully in lysates of infected Mardin-Darby bovine kidney (MDBK) cells, but not in BoHV-1 (wild type) virus and the mock-infected cells (Figure 2C).
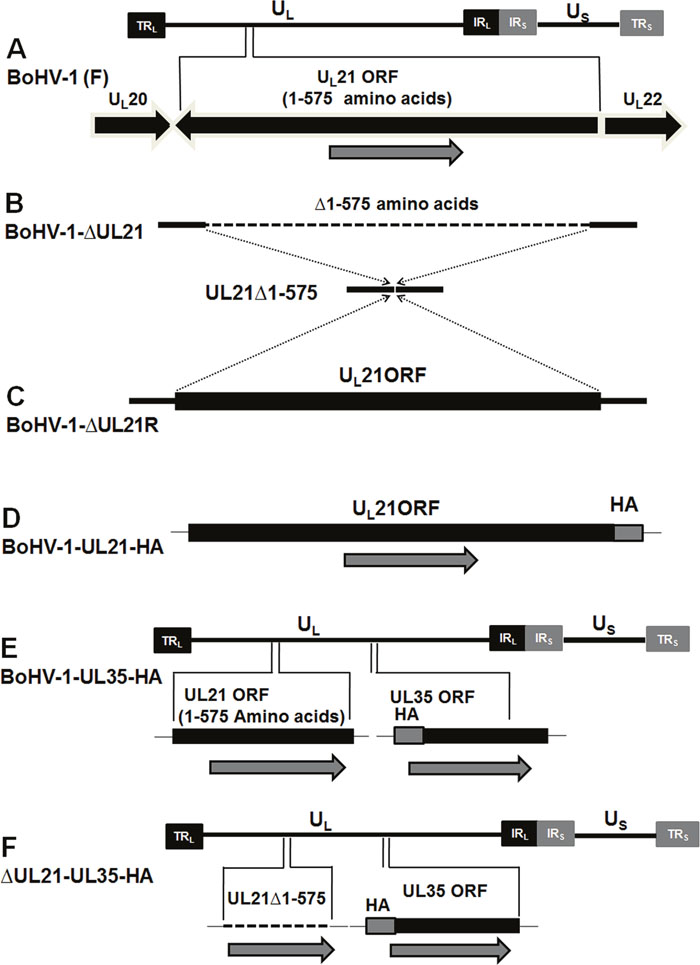
Figure 1: Construction and characterization of the ΔUL21 mutant and revertant viruses. (A) Schematic diagram showing the BoHV-1 genome (~140 kbp) with unique short (US) and unique long (UL) regions, a terminal repeat (TR), and an internal repeat (IR). The region of the genome encoding UL21 (codons 1-575) is located between the UL20 and UL22 genes. (B) Dashed lines show the deletion of the entire UL21 gene from the BoHV-1 genome. (C) Reversion of the UL21 gene by restoring the complete UL21 ORF to its original location in the pBoHV-1-ΔUL21 BAC. (D) The insertion of a sequence encoding an HA tag at the 3′ end of the UL21 gene in the pBoHV-1 BAC. (E) The insertion of a sequence encoding an HA tag at the 3′ end of the UL35 gene in the pBoHV-1 BAC. (F) The addition of a sequence encoding an HA tag at the 3′ end of the UL35 gene in the pBoHV-1-ΔUL21 BAC.
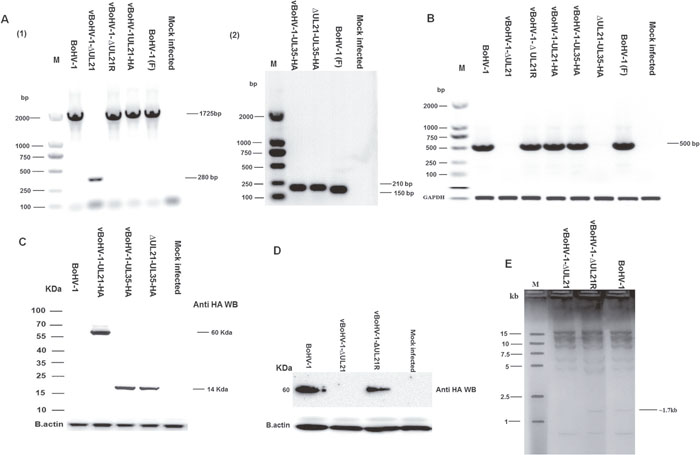
Figure 2: Confirmation of mutant viruses. (A) MDBK cells were infected with each virus at a MOI of 3. PCR were run with the indicated viral DNAs of pUL21 using the UL21 F/R primers (Table 1) and pUL35 using the UL35 F/R primers (Table 1). (B) Expression of pUL21 recombinant viruses was confirmed by RT-PCR with the indicated viral cDNA using primers UL21 F/R (Table 1) via agarose gel electrophoresis. The bovine glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene was used as the internal reference control. DNA ladders are shown in bp to the left of the gel. (C) MDBK cells were infected with indicated viruses at a MOI of 3. Cells were harvested at 18 hpi and separated on a 15% SDS-PAGE and transferred to a PVDF membrane. Then, the membranes were probed sequentially with a mouse anti-HA antibody, followed by HRP-conjugated goat anti-mouse IgG. Beta-actin was used as the internal reference control. (D) MDBK cells were infected at a MOI of 3 with the indicated viruses. At 18 hpi, cell lysates were electrophoresed through 12% polyacrylamide gels and transferred onto PVDF membranes. Membranes were probed with the UL21 antisera. The molecular weights of the protein markers are shown in kDa to the left of the gel. (E) RFLP analysis obtained by restriction enzyme HindIII genomic digestion of UL21 mutant, vBoHV-1-ΔUL21R and BoHV-1 viruses.
To confirm the deletion of UL21 in the vBoHV-1-ΔUL21 virus, Western blotting assay was performed. As shown in Figure 2, panel D, UL21 (approximately 60 kDa) was detected by antiserum against UL21 from lysates of BoHV-1 or vBoHV-1-ΔUL21R- infected MDBK cells, but not from lysates of mock or vBoHV-1-ΔUL21-infected cells.
In addition, a restriction fragment length polymorphism (RFLP) analysis was performed to further confirm the UL21 mutation (deletion). According to the results following the digestion of genomic DNA with HindIII, a fragment of approximately 1.7 kb was missed in the UL21 mutant, compared with the vBoHV-1-ΔUL21R and BoHV-1 (Figure 2E).
Growth defect of the vBoHV-1ΔUL21 mutant
To determine whether the UL21 deletion affects BoHV-1 replication, the growth kinetics of the vBoHV-1ΔUL21 virus was determined in MDBK cells. First, we compared the growth kinetics of the BoHV-1, vBoHV-1-ΔUL21, vBoHV-1-ΔUL21R and vBoHV-1-UL21-HA viruses using plaque assays. The results demonstrated that the replication of the vBoHV-1-ΔUL21 virus was significantly lower than that of the BoHV-1, vBoHV-1-ΔUL21R and vBoHV-1-UL21-HA viruses at 48 hpi. Interestingly, the extracellular titer of the vBoHV-1-ΔUL21 virus was 1,000-folds lower (p < 0.01) than that of the BoHV-1(wild-type), vBoHV-1-ΔUL21R or vBoHV-1-UL21-HA viruses (Figure 3A), while the intracellular titer of the vBoHV-1-ΔUL21 virus was only 100-folds (p < 0.01) lower (Figure 3B) than that of the BoHV-1, vBoHV-1-ΔUL21R and vBoHV-1-UL21-HA viruses. In contrast, the titers of the BoHV-1, vBoHV-1-ΔUL21R and vBoHV-1-UL21-HA viruses were similar (p > 0.05) (Figure 3A and 3B).
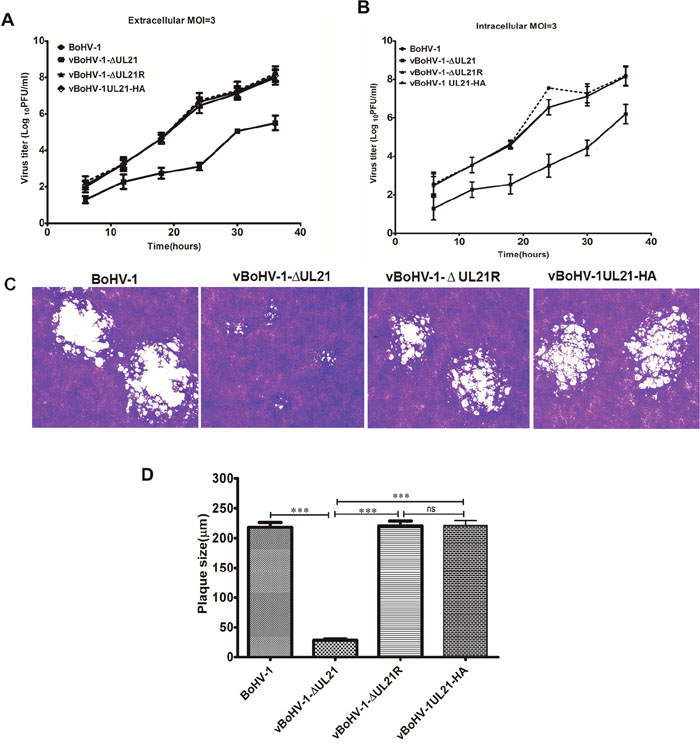
Figure 3: Growth analysis of the BoHV-1 recombinant viruses. (A) Growth of the BoHV-1, vBoHV-1-ΔUL21, vBoHV-1-ΔUL21R and vBoHV-1UL21-HA viruses. For the single-step replication analysis, MDBK cells were infected with the indicated viruses at a MOI of 3, and harvested at 6, 12, 18, 24, 30, and 48 hpi. A t-test of the data revealed very significant differences between the BoHV-1 and vBoHV-1-ΔUL21 viruses in terms of their extracellular (**, p < 0.01) and intracellular (**, p < 0.01) viral titers, while no significant (ns) difference was observed between the BoHV-1, vBoHV-1-ΔUL21R and vBoHV-1UL21-HA viruses (p > 0.05). (B) Plaque size and morphology of the BoHV-1, vBoHV-1-ΔUL21, vBoHV1-ΔUL21R and vBoHV-1UL21-HA viruses. MDBK cells were infected with serial dilutions of each virus in 24-well plates and overlaid with methylcellulose. At 48 hpi, plaques were fixed with formaldehyde and stained with crystal violet. (C) The mean plaque sizes of the aforementioned viruses were determined with an Olympus IX70® Microscope. The difference in the mean plaque size of the vBoHV-1-ΔUL21 virus was shown to be significantly smaller using a t-test (***, p < 0.001). Each independent experiment was performed three times.
To determine the effect of the UL21 deletion on cell-to-cell spreading, the plaque size of the vBoHV-1-ΔUL21 virus was compared to that of the BoHV-1, vBoHV-1-ΔUL21R and vBoHV-1-UL21-HA viruses, and the diameters of 50 plaques for each virus were measured at 48 hpi. The plaques produced by the vBoHV-1-ΔUL21 virus in MDBK cells were 85% smaller (p < 0.01) (Figure 3C) than those produced by the BoHV-1, vBoHV-1-ΔUL21R or vBoHV-1-UL21-HA viruses, while the plaque sizes of the BoHV-1, vBoHV-1-ΔUL21R and vBoHV-1-UL21-HA viruses did not differ significantly (p > 0.05) (Figure 3D). These data suggest that the HA tag did not interfere with UL21 function and furthermore, UL21 is critical for BoHV-1 replication and cell-to-cell spreading.
Localization of BoHV-1 UL21
To gain insight into the function of BoHV-1 UL21 in situ, we investigated the subcellular localization of UL21 using an indirect immunofluorescence assay. MDBK cells were mock infected or infected with the vBoHV-1-UL21-HA virus at a MOI of 3, and fluorescence was observed at 18 hpi using confocal microscopy. At 18 hpi the vBoHV-1-UL21-HA virus reacted with both the UL21 antiserum and the anti-HA-tag antibody, and specific fluorescence was found to localize predominantly in the cytoplasm. Surprisingly, punctate nuclear staining was observed with the UL21 antiserum, whereas nuclear staining with the anti-HA-tag antibody was barely observed. Thus, it is possible that only the UL21 epitopes recognized by the anti-UL21 antiserum were accessible on the surface of UL21 protein in the nucleus, while the HA epitope tag was not surface exposed on the HA-tagged UL21 fusion protein in the nucleus (Figures 4A). As expected, in mock-infected cells, which served as internal controls, no specific fluorescence was observed (Figure 4B).
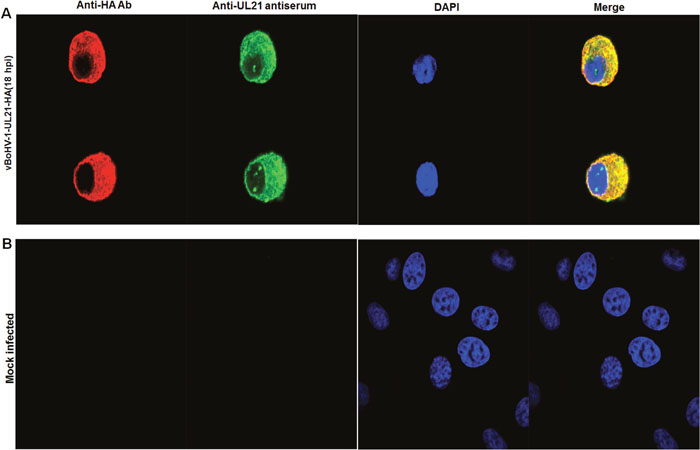
Figure 4: Localization of UL21 using confocal microscopy. (A) MDBK cells were infected with the vBoHV-1-UL21-HA virus at a MOI of 3 for 18 hrs. The infected cells were incubated with primary antibodies (mouse anti-HA) and rabbit polyclonal antisera, and then stained with Cy3-conjugated goat anti-mouse and fluorescein isothiocyanate-conjugated goat anti-rabbit IgG antibodies, Nuclei were stained blue with DAPI. Then, mount with anti-fade mounting medium. Specific capsid fluorescence was visualized under a Zeiss LSM 880 laser-scanning confocal microscope (B) mock-infected cells.
Effect of the UL21 deletion on capsid egress
To investigate the role of UL21 in nucleocapsids egress, an HA tag was attached to the carboxyl-terminus of the minor capsid protein UL35 in the UL21 mutant (vBoHV-1-ΔUL21-UL35-HA) and wild-type (vBoHV-1-UL35-HA) viruses. At 18 hpi, punctate and high-density, concentrated fluorescence was readily detected in the cytoplasm and nuclei of both wild-type and UL21 mutant infected cells (Figure 5A, 5B). While no specific fluorescence was observed in mock-infected cells, which served as internal controls, (Figure 5C). These data suggested that nucleocapsid egress was not impaired by UL21 deletion.
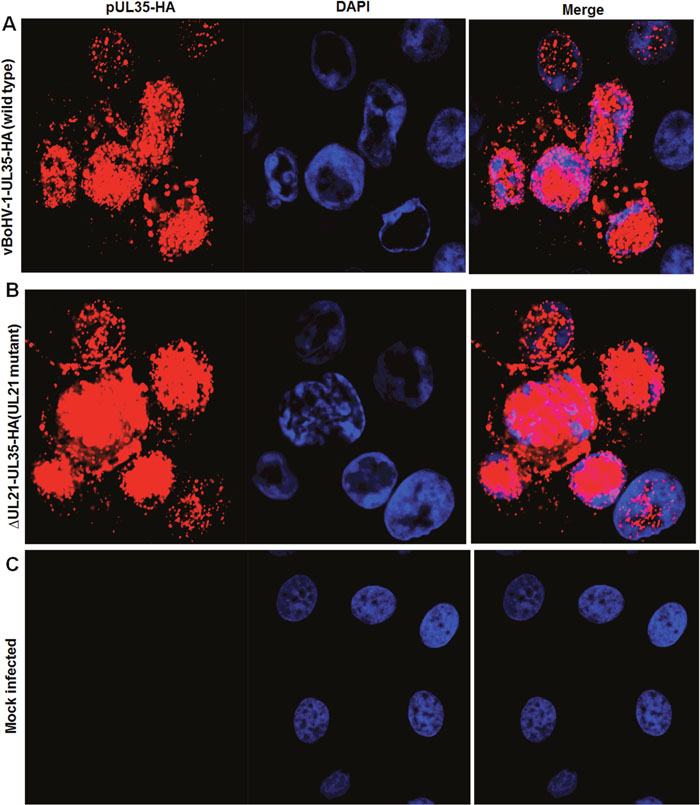
Figure 5: Effect of the UL21 deletion on the capsid egress pathway. MDBK cells were infected with the indicated viruses expressing HA-tagged UL35 at a MOI of 3. At 18 hpi, the cells were fixed and stained with a mouse anti-HA antibody and Cy3-conjugated goat anti-mouse IgG. The nuclei were stained blue with DAPI. Fluorescence was observed under a Zeiss LSM 880 laser-scanning confocal microscope. (A) Cells infected with the vBoHV-1-UL35-HA virus, (B) the vBoHV-1-ΔUL21-UL35-HA virus, as well as (C) mock-infected cells.
To corroborate the fluorescence microscopy data, a TEM analysis was performed. MDBK cells were infected with the BoHV-1, vBoHV-1ΔUL21, or vBoHV-1-ΔUL21R viruses at a MOI of 3. The TEM analysis showed that nucleocapsid formation and movement from the nuclei to the cytoplasm did not appear to differ among the cells infected with the vBoHV-1ΔUL21 (Figure 6A), vBoHV-1-ΔUL21R (Figure 6B), or BoHV-1(Figure 6C) viruses. Furthermore, nucleocapsids quantification showed no significant difference (p > 0.05) in the total number of nucleocapsids in the nuclei infected with BoHV-1, vBoHV-1ΔUL21, or vBoHV-1-ΔUL21R viruses as shown in Figure 8 panel A. These findings showed that primary envelopment and nuclear egress, which was defined by capsid movement from the nuclei to the cytoplasm, remained unaffected, and that the differential distribution of capsids in the cytoplasm of cells infected with the UL21 mutant likely resulted from impairment in secondary envelopment.
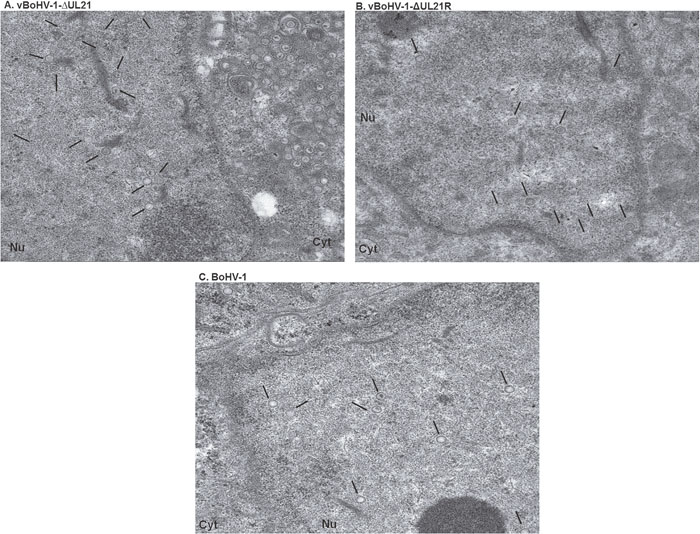
Figure 6: TEM micrographs of nucleocapsids in nuclei. MDBK cells infected with the indicated viruses at a MOI of 3. At 18 hpi, the cells were fixed, sectioned, and analyzed using TEM. Overviews of virus infected cells containing nucleocapsids in the nucleus (A) vBoHV-1-ΔUL21, (B) vBoHV-1-ΔUL21R, (C) BoHV-1, showing no difference. Black arrows point to nucleocapsids.
Effect of the UL21 deletion on secondary envelopment
To analyze in more detail whether the replication defect of the vBoHV-1ΔUL21 virus was associated with capsid assembly and maturation, MDBK cells were infected with the BoHV-1, vBoHV-1ΔUL21, or vBoHV-1-ΔUL21R viruses at a MOI of 3. At 18 hpi, the cells were processed and examined by TEM. The vBoHV-1ΔUL21 virus exhibited a defect in secondary envelopment as evidenced by large numbers of capsid clusters that accumulated in the cytoplasm, which resulted in the failure of the capsids to become fully enveloped. Additionally, in vBoHV-1ΔUL21-infected cells, many cytoplasmic capsids were observed to be partially enveloped or non-enveloped, while very few mature virions were observed in the cytoplasm (Figure 7A and 7B). No difference was observed in the growth between the BoHV-1 and vBoHV-1-ΔUL21R viruses (Figure 7C and 7D), which confirmed that the defect in secondary envelopment and cytoplasmic exit was due to the deletion of the UL21 gene. To quantify the effect of the UL21 deletion, approximately 40 infected cells were counted in more than 25 fields in multiple sections of cells infected with the BoHV-1, vBoHV-1ΔUL21, and vBoHV-1-ΔUL21R viruses. Infected viral particles were categorized as nucleocapsids in the nuclei (A), intracellular mature virions (B), or extracellular enveloped virions (C). Although no significant difference (p > 0.05) was observed in the total number of nucleocapsids in the nuclei (Figure 8 panel A), the number of intracellular enveloped viral particles in the cytoplasm of the vBoHV-1ΔUL21-infected cells was reduced by approximately 80% (Figure 8 panel B), while the number of extracellular mature virions was reduced by approximately 90% (Figure 8 panel C), compared with cells infected with the wild-type or vBoHV-1-ΔUL21R viruses. Therefore, we concluded from the electron microscopic studies that UL21 plays a critical role in secondary envelopment, as well as in the cytoplasmic exit of virions.
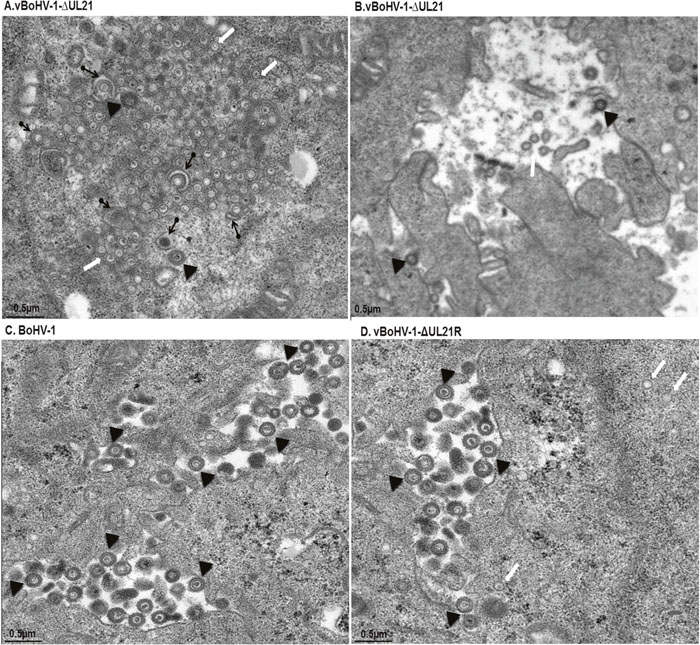
Figure 7: The vBoHV-1-ΔUL21 virus has a defect in secondary envelopment. MDBK cells were infected with the vBoHV-1-ΔUL21 (A and B), BoHV-1 (C), or vBoHV-1-ΔUL21R viruses (D) at a MOI of 3. At 18 hpi, the cells were fixed, sectioned, and analyzed by TEM. Cytoplasmic capsid clusters without secondary envelopment were observed in vBoHV-1-ΔUL21-infected cells (A). In the vBoHV-1-ΔUL21-infected cells, mature, virions were rarely seen in the cytoplasm and outer cell surface (B), compared with BoHV-1- (C) and vBoHV-1-ΔUL21R-infected cells (D). White arrows point to aggregates of unenveloped cytoplasmic capsids, while arrowheads point to enveloped virions and black arrows with rounded tails point to partially enveloped virions.
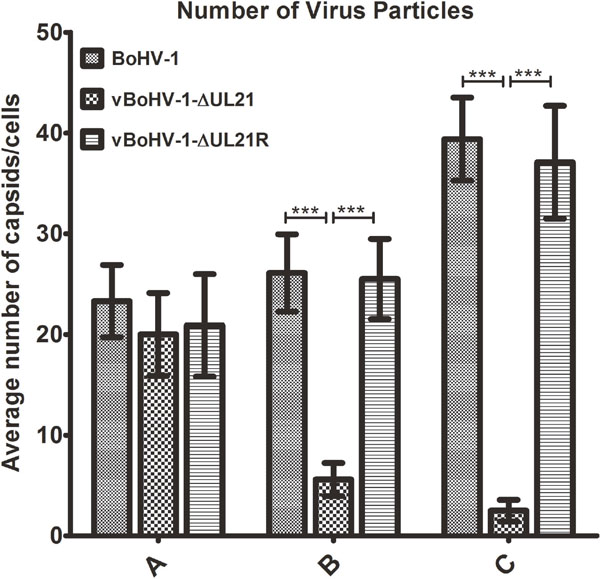
Figure 8: Quantitation of the virion distribution within cells using TEM. MDBK cells were infected with the BoHV-1, vBoHV-1-ΔUL21, or vBoHV-1-ΔUL21R viruses at a MOI of 3. At 18 hpi, the number of virus particles in each well was calculated for 40 infected cells for each individual sample. *** indicates a statistically significant difference (p < 0.001). Bars indicate the mean values for each sample resulting from three independent experiments and error bars indicate the standard deviations. (A) Nucleocapsids in the nuclei. (B) Intracellular mature virions in the cytoplasm. (C) Mature virions on the cell surface.
Identification of the BoHV-1 UL21-interacting protein UL16
To investigate the UL21 interactive proteins, MDBK cells were infected with the vBoHV-1-UL21-HA or BoHV-1 viruses. Cells were lysed at 18 hpi, and incubated with anti-HA-tag monoclonal antibody-conjugated magnetic agarose beads. Following sodium dodecyl sulfate-polyacylamide gel electrophoresis (SDS-PAGE) and silver staining, in addition to the 60-kDa UL21 protein, one differential band corresponding to a 36-kDa protein was detected consistently (Figure 9A). To identify the protein(s), this band was subjected to a Liquid chromatography-mass spectrometry (LC/MS) analysis. The results of the mass spectrometry analysis revealed that most of the data corresponded to peptides from UL16 (32.65% coverage with a 36,422.51 Da molecular weight. The results also indicated the presence of peptides from UL21 (26.9% coverage with a 60,261.18 Da molecular weight) (Figure 9B). The molecular weight is the estimated molecular weight of the peptides, which can be calculated based on their mass-to-charge (m/z) values. Additionally, the observed UL21-HA band exhibited the correct molecular weight of 60 kDa, as the approximate size of UL21 is 59 kDa and the size of the HA tag is approximately 1 kDa which proves the correct insertion and function of HA-tag (Supplementary Materials 1 and 2).
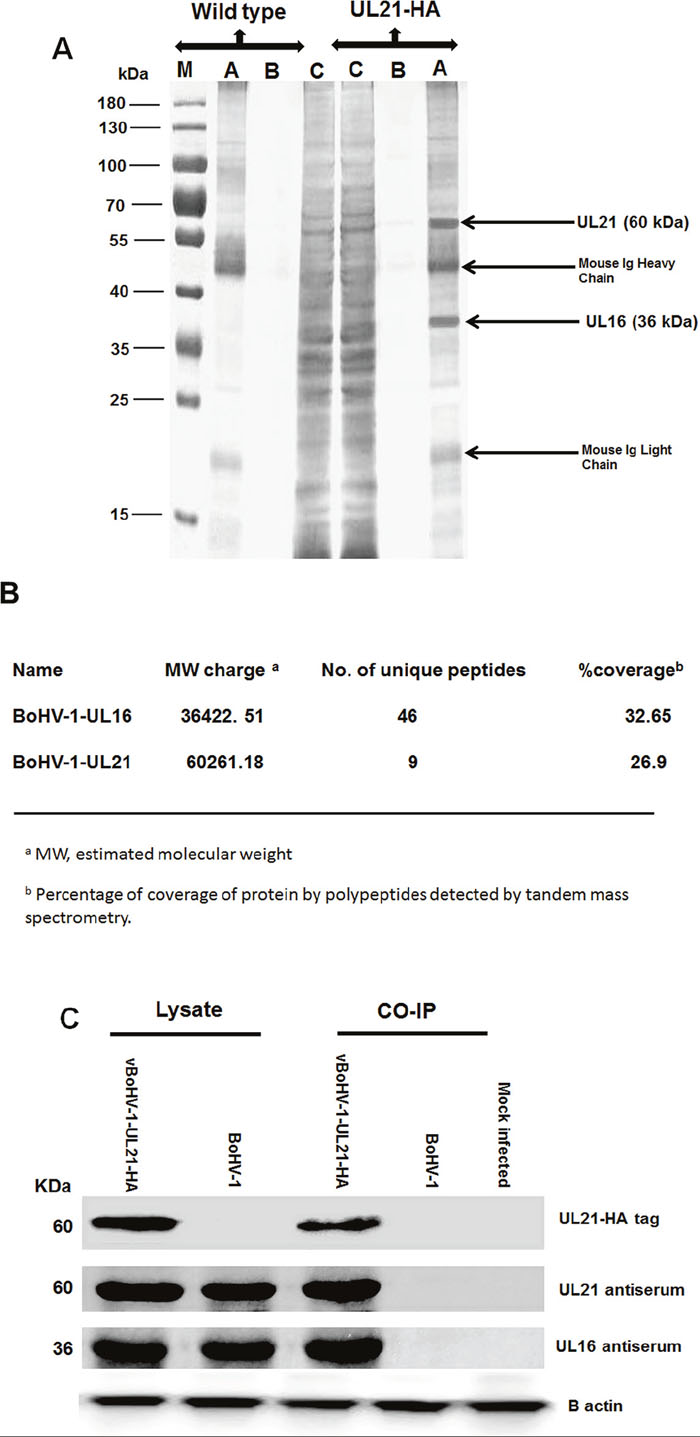
Figure 9: Characterization of the UL21 protein by mass spectrometry. (A) MDBK cells were infected with the vBoHV-1-UL21-HA or BoHV-1 viruses at a MOI of 3. At 18 hpi, cells were harvested and immunoprecipitated with anti-HA antibody conjugated magnetic agarose beads, and the precipitated proteins were loaded onto a 12% SDS-polyacrylamide gel and silver stained. Lane A, the bead eluate; lane B, the third wash; and lane C, the flow-through. Protein molecular weight markers in kDa are indicated to the left of the gel. (B) Mass spectrometry analysis of immunoprecipitated proteins along with the interacting proteins that correspond to the proteins. The expected 60-kDa UL21-HA band, as were 36-kDa band, which were confirmed to be UL16 by LC-MS (C) MDBK cells were infected with the vBoHV-1-UL21-HA or BoHV-1 viruses, and proteins were precipitated using anti-HA antibody-conjugated magnetic agarose beads. The beads were electrophoresed and transferred onto PVDF membranes. Then, the membranes were probed with a mouse anti-HA-tag antibody and with the antisera indicated on the right side of the figure. Beta-actin was used as the internal reference control. The migration positions of proteins are shown in kDa to the left of the gel.
To confirm that UL16 form complexes with UL21, co-immunoprecipitation assays were performed with either BoHV-1 or vBoHV-1-UL21-HA-infected cell lysates. Polyclonal antisera against GST-UL16 and GST-UL21 interacted with the immunoprecipitated lysates of the vBoHV1-UL21-HA virus, and 36-kDa (UL16) and 60 kDa (UL21) bands were detected in the lysates of vBoHV1-UL21-HA- and BoHV-1-infected cells (Figure 9C). However, no specific proteins were co-immunoprecipitated in lysates of BoHV-1-infected and mock-infected cells. The co-immunoprecipitation and immunoblotting analyses confirmed the mass spectrometry findings that UL21 interacts with UL16.
Observations of complexes of UL21 with UL16
To confirm the interaction between UL21 and UL16, we performed a co- immunofluorescence assay by using the vBoHV-1-UL21-HA virus. MDBK cells were mock-infected or infected with the vBoHV1-UL21-HA virus at a MOI of 3, fixed at 12 and 24 hpi, permeabilized, and stained with polyclonal rabbit antisera against UL16 and UL21, as well as an anti-HA-tag monoclonal antibody. Confocal microscopy images showed that in the vBoHV1-UL21-HA-infected cells, UL21 co-localized completely with UL16 (Figure 10A and 10B). These data, combined with the co-immunoprecipitation results displayed in Figure 9C, indicated that UL21 interacted with UL16.
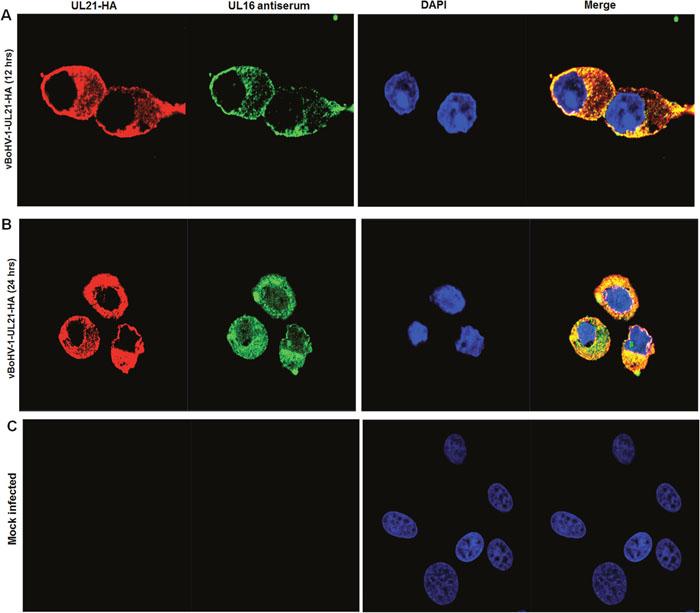
Figure 10: Interaction of UL21 with UL16. MDBK cells were infected with the vBoHV-1-UL21-HA virus at a MOI of 3, fixed for immunofluorescence and stained with a mouse anti-HA antibody or rabbit polyclonal antisera against UL16 and Cy3-conjugated goat anti-mouse IgG or fluorescein isothiocyanate-conjugated goat anti-rabbit IgG antibodies. DNA was stained blue with DAPI. Fluorescence was observed under a Zeiss LSM 880 laser-scanning confocal microscope. (A) The UL21 interaction with UL16 at 12 hpi (B) 24 hpi (C) Mock-infected cells.
DISCUSSION
The functions of UL21 in the life cycle of HSV-1, HSV-2, and PRV have been investigated extensively [10, 12–14, 23], whereas the functions of this conserved protein in BoHV-1 have not been well defined. In the present study, we constructed a UL21 deletion mutant using a BAC system, which revealed the functions of UL21 in BoHV-1 secondary envelopment and cytoplasm exit.
BoHV-1 UL21 is required for efficient virus growth
Other studies have demonstrated that deleting UL21 in HSV-1 [12, 23] and PRV [13, 14] viruses reduced both extracellular and intracellular virus yield and plaque size. The current study confirmed that UL21 in BoHV-1 plays a similar important role in viral growth in cell culture. The deletion of UL21 led to reduced virus titers which were 1,000 and 100-fold lower in the extracellular and intracellular environments at 48 hpi, respectively, compared with that of the wild-type virus, while the plaque size of the vBoHV-1-ΔUL21 virus was 85% smaller than that of the wild-type, vBoHV-1-ΔUL21R, and vBoHV-1-UL21-HA viruses. However, the extent to which UL21 mediates viral growth may be species-dependent. For example, unlike BoHV-1, an HSV-2 UL21 mutant was unable to replicate until the UL21 gene was restored to its original location [10]. These findings demonstrate that the UL21 tegument protein is indispensable for efficient virus replication in cell culture, as deleting the UL21 gene resulted in severe growth defects and smaller plaques compared with the BoHV-1, vBoHV-1-ΔUL21R and vBoHV-1-UL21-HA viruses.
BoHV-1 UL21 is required for secondary envelopment of capsids
In cell culture, we first discovered that in vBoHV-1-ΔUL21-infected cells, the extracellular viral titer was lower than the intracellular viral titer. This showed that deleting UL21 might impair the cytoplasmic exit of BoHV-1. By labeling UL21 with an HA-tag, which was subsequently probed by an anti-HA-tag antibody and anti-UL21 serum, an immunofluorescence analysis showed that UL21 localized predominantly to the cytoplasm, and it only exhibited a minor punctate nuclear staining. Our findings that UL21 localizes to the cytoplasm and nucleus agree with previous studies of HSV-1, PRV [18–21, 23], and HSV-2 [10].
To investigate the potential role of UL21 in viral capsid assembly and movement, HA-tagged viruses were studied with confocal microscopy and TEM. Both methods demonstrated that deleting UL21 did not impair capsid movement from the nuclei to the cytoplasm, but it did affect cytoplasmic maturation and viral particle exit to the extracellular space. A similar association with nucleocapsids egress has been observed in HSV-1 and PRV UL21 [12, 14, 18, 23, 25]. However, in a previous study, HSV-2 UL21 was reported to be involved in nuclear egress [10]. Further observations highlighted that the UL21 deletion caused an accumulation of partially enveloped or non-enveloped virions in the cytoplasm, as well as a decrease in the number of mature virions. This phenomenon demonstrated that the vBoHV-1-ΔUL21 virus exhibited a severe defect in secondary envelopment and, therefore, that UL21 is critical for BoHV-1 secondary envelopment. These results explain the present findings of differential titers between extracellular and intracellular viruses, because significantly fewer viruses matured and exited from the cytoplasm of the vBoHV-1-ΔUL21 virus. Similar findings were reported previously, indicating that UL21 is involved in capsid maturation [18]. While some studies showed that in the absence of UL21, capsid maturation remained unaffected in the PRV [14]. Therefore, although the general function of UL21 in mediating viral morphogenesis of herpesviruses is conserved, its precise function varies in different viruses.
BoHV-1-UL21 interacts with UL16
UL16 is conserved tegument proteins in Alphaherpesvirinae [8]. In contrast to UL21, UL16 has already been studied in some detail in PRV and HSV. In HSV-1 and PRV, deletion of UL16 orthologs caused 10-fold reduction in virus propagation and defects in secondary envelopment [14, 26, 27]. Previously, in PRV the simultaneous absence of UL21 and UL16 led to capsid aggregations in the cytoplasm [14]. Currently in HSV-2 Δ16 mutant resulted in 200-950 fold reduction in virus propagation and suggested that UL16 and UL21 proteins may function together in nuclear egress [28]. UL16 has the capacity to forms complexes with several viral proteins, including VP22, UL11, gE and UL21 [14, 17, 20, 21, 26]. So our recent studies strongly support this interaction between UL21 and UL16.
In the present study, mass spectrometry analysis of proteins co-immunoprecipitated by HA-tagged UL21 showed that UL21 interacted UL16. Our immunoprecipitations observations reveal that the anti-UL16 serum efficiently co-precipitated the UL21 protein as previously reported by coimmunoprecipitation assays in PRV and HSV [14, 20]. Furthermore, an immunofluorescence microscopy analysis showed that UL21 protein co-localized with UL16 which was also observed in HSV-1 [21].
Unlike HSV-2, where UL21 deletion prevented virus propagation and blocked nuclear egress [10], the absence of UL21 in BoHV-1 did not affect nuclear egress. In HSV-1 [12, 15, 23], and PRV [13, 14], where UL21 deletion mutants caused comparatively modest defects in virus growth while the UL21 was found to be critical for BoHV-1 replication. In PRV UL21 mutant affected capsid maturation [18] and led to formation of DNA-deprived capsids [25], while in our study the UL21 mutant affected the secondary envelopment. These results suggested that although the UL21 is conserved among alphaherpesviruses, its precise function varies in different viruses.
Collectively, our results demonstrate that BoHV-1 UL21 plays critical roles in viral secondary envelopment and cell-to-cell spreading in tissue culture. Furthermore, we demonstrate that UL16 forms a complex with UL21 in BoHV-1. However, further research should be conducted to clarify whether the interaction of UL16 with UL21 correlates with UL21 function and BoHV-1 lacking UL16 would have a similar phenotype as BoHV-1 lacking UL21.
MATERIALS AND METHODS
Ethical statement regarding animal experiments
Experimental protocols involving animals were approved by the China Hubei Province Science and Technology Department, which is responsible for animal ethics (permit no. HZAURAB-2015-006), in accordance with the recommendations of the China Regulations for the Administration of Affairs Concerning Experimental Animals (1988) and the Hubei Regulations for the Administration of Affairs Concerning Experimental Animals (2005). The animal use in this study was supervised by the Ethical Committee for Experimental Animals of Huazhong Agricultural University, Wuhan, China.
Cells and viruses
BoHV-1 mutant viruses (designated vBoHV-1) were derived from the BoHV-1 strain IBRV HB06 (which was isolated by our laboratory and stored as no. V201024 at the China Center for Type Culture Collection at Wuhan University, and used as the wild-type strain in the present study). BoHV-1 and mutant viruses were propagated and titrated in the Madin-Darby bovine kidney (MDBK) cell line as described previously [29]. Viral proteins were obtained from lysates of infected MDBK cells. Plasmid transfections of MDBK cells were conducted via the calcium chloride transfection method [30].
Construction of UL21 recombinant viruses
The UL21 gene of BoHV-1 (GenBank accession no. AJ004801.1) encodes UL21, which comprises 575 amino acids. The procedures used to construct a UL21 deletion mutant with a BoHV-1 bacterial artificial chromosome (BoHV-1 BAC) were described previously [31]. Briefly, the primers ΔUL21 F/R (Table 1), with 40-bp upper and lower homolog arms of the deleted region and sequences specific to the flanking sequences of a kanamycin resistance gene cassette, were used to polymerase chain reaction (PCR)-amplify the kanamycin resistance gene from the pEPkan-S plasmid, which was kindly provided by Dr. Nikolaus Osterrieder [32]. Then, the PCR products containing the upper and lower homologous arms of the deleted region and the positive selection marker of the 3′ I-SceI-aphAI cassette, which includes AphAI conferring kanamycin resistance and an adjoining I-SceI site, were purified using the Cycle Pure Kit (Omega Bio-Tek, Norcross, GA, USA) and confirmed by DNA sequencing. Subsequently, these gel-purified, DpnI-digested, linear PCR amplicons were electroporated (1.8 kV, 25 μF capacitance, and 200 Ω resistance) into Escherichia coli GS1783 competent cells that contained a BoHV-1 BAC-positive clone (pBoHV-1 BAC). Chloramphenicol-resistant and kanamycin-sensitive colonies were identified and confirmed by PCR using the primers UL21-KAN-F/R (Table 1) and DNA sequencing, and they were named pBoHV-1-ΔUL21 BAC. Recombinant viruses were rescued by transfecting the respective BAC DNA into MDBK cells, and they were named vBoHV-1-ΔUL21 (the ΔUL21 mutant).
Table 1: List of primers used in this study
Primer name |
Sequence (5’ to 3’) |
|---|---|
ΔUL21-F |
TTGTTCCCCCCGTCGGCCCGCTCGCGCGGCCGCCGCGGCGGCTCAGCGTGCCACAAGGGCTAGGGATAACAGGGTAATCGATTT |
ΔUL21-R |
TTATTGATTGCCGCGTCGCCGCCCTTGTGGCACGCTGAGCCGCCGCGGCCGCGCGAGGCCAGTGTTACAACCAATTAACC |
UL21-HA- F |
GCGAAGGGGAGGGAGAAGGGGAAGCTGGAGCCGGCACTGAACCCTACCCATACGACGTCCCAGACTACGCTTAGGCTCAGCGTGCCACATAGGGATAACAGGGTAATCGATTT |
UL21-HA -R |
TATTATTGATTGCCGCGTCGCCGCCCTTGTGGCACGCTGAGCCTAAGCGTAGTCTGGGACGTCGTATGGGTAGCCAGTGTTACAACCAATTAACC |
UL21-REPAIR-F |
TAAGGGTATATTATTGATTGCCGCGTCGCCGCCCTTGTGGCACGCTGAGCCTAGGGTTCAGTGCCGGCTC |
UL21-REPAIR-R |
TTTTGCGTCATTGTTCCCCCCGTCGGCCCGCTCGCGCGGCCGCCGCGGCGATGGAGCTCGCCTACCACAG |
UL21-KAN-F |
ACGCACCGCAGCAGCTCATC |
UL21-KAN-R |
CGCCAATAAAGACGCCTCCAA |
UL35-HA- F |
ATACGAGGAGGACGCTGGCGCGGCGGCGTCTGCCGCGCGCACGTACCCATACGACGTCCCAGACTACGCTTGACGCTTTTTTCGGTAGGGATAACAGGGTAATCGATTT |
UL35-HA -R |
ATTATTGCGATCGCACAATTGCGCCGATCCGAAAAAAGCGTCAAGCGTAGTCTGGGACGTCGTATGGGTAGCCAGTGTTACAACCAATTAACC |
UL35- F |
ATGTTTGCGACCTACGACCACG |
UL35- R |
CGCACAATTGCGCCGATCC |
cDNA UL21- F |
CGTGTACGCGCCCAACCGGGAGTGCTT |
cDNA UL21- R |
TGAACCCGTAGTCCGCCACGGATTCG |
GAPDH- F |
GGCCTGAACCACGAGAAGTATAA |
GAPDH- R |
CCCTCCACGATGCCAAAGT |
UL21-F |
GCGGATCCATGGAGCTCGCCTACC |
UL21-R |
CGAAGCTTCTAGGGTTCAGTGCCGG |
UL16-F |
GCGGATCC ATGGCCGAGGACCCCGCCGCTGCGG |
UL16-R |
CGAAGCTT TCAGCCCCACGGCGAGCCGCGCGCT |
Reversion of pBoHV-1-ΔUL21
A repair virus (vBoHV-1-ΔUL21R) was constructed by restoring the entire UL21 open reading frame (ORF) to its original location using the pBoHV-1-ΔUL21 BAC (Figure 1C). The UL21 coding sequence with flanking upstream and downstream sequences was amplified using the primers UL21-Repair F/R (Table 1). All the λ red recombination steps were performed according to a previous protocol [31], and positive clones were identified by PCR using the primers UL21-KAN-F/R (Table 1) and DNA sequencing. The UL21 revertant virus vBoHV-1-ΔUL21R was rescued by transfecting the respective pBoHV-1-ΔUL21R BAC DNA into MDBK cells.
Construction of hemagglutinin (HA)-tagged recombinant viruses
To investigate the effect of the UL21 deletion, an HA tag was fused in frame with the sequence encoding the minor viral capsid protein UL35 (VP26) of the pBoHV-1 BAC (Figure 1E) and the pBoHV-1-ΔUL21 BAC (Figure 1F) using the UL35-HA F/R primers (Table 1). The HA tag was also fused in frame with the sequence encoding the carboxyl-terminus of UL21 in the pBoHV-1 BAC (Figure 1D) using the primers UL21-HA F/R (Table 1) via two-step, λ-red mediated mutagenesis [33]. All the two-step, λ-red recombination events were performed according to previously described methods [31], and positive clones were identified by PCR using the primers UL21-KAN-F/R, UL35- F/R (Table 1) and DNA sequencing. Recombinant viruses were rescued by transfecting the respective BAC DNA into MDBK cells, and the resultant mutants were designated as the vBoHV-1-UL35-HA, vBoHV-1-ΔUL21-UL35-HA, and vBoHV-1-UL21-HA viruses.
RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR)
Freshly grown MDBK cells were infected with the indicated viruses at a MOI of 3 plaque-forming units (PFU) per cell. Total RNA was extracted from the cells using TRIzol reagent (Invitrogen, Auckland, New Zealand), and reverse transcription of cDNA was performed using the PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, Otsu, Shiga, Japan) following the manufacturer’s instructions. The cDNA was quantified by RT-PCR with the specific primers cDNA-UL21 F/R (Table 1). The PCR products were sequenced by the Sangon Company (Wuhan, China).
RFLPs
To further confirm the deletion in the recombinant mutants, genomic DNA was extracted from the mutant virus and the wild-type strain, and it was analyzed for RFLPs using the restriction enzyme HindIII (Takara, Dalian, China) as described previously [34]. After enzymatic digestion, the resulting DNA fragments were separated on a 0.7 % agarose gel at room temperature for approximately 15 h under a constant voltage of 20 V.
Growth curve analysis
Virus replication kinetics was determined as follows. MDBK cells were infected with the vBoHV-1-ΔUL21, vBoHV-1-ΔUL21R, or wild-type BoHV-1 viruses at a MOI of 3 and incubated at 37°C for 2 h. Then, the inoculum was removed and infected cell monolayers were rinsed with low pH (3.0) citrate-buffered saline (40 mM citric acid, 135 mM NaCl, 10 mM KCl) for 1 min. The cells were washed twice with 1× phosphate-buffered saline (PBS) (136.9 mM NaCl, 2.7 mM KCl, 7.0 mM Na3PO4, and 0.9 mM Na3PO4 at pH 7.4), and fresh medium was added. Intracellular and extracellular viruses were harvested at 6, 12, 18, 24, 30, and 48 h. After three freeze-thaw cycles, 1000 RPM for 5 min centrifugation was used to clear the infected cell debris. The titers of the extracellular and intracellular viruses were determined by a plaque assay.
The plaque assay was performed in MDBK cells grown in 24-well plates. Serial dilutions of the viruses were prepared in Dulbecco's modified Eagle medium (Hyclone, Logan, UT, USA) supplemented with 1% fetal bovine serum (Gibco, Grand Island, NY, USA) and a 2% penicillin and streptomycin solution (Hyclone, Vienna, Austria) and inoculated into each well. After incubation for 2 h, the viral inoculum was removed, and the cells were overlaid with a 1:1 solution of carboxymethyl cellulose (3.2%) and Dulbecco's modified Eagle medium supplemented with 2% fetal bovine serum and 1% penicillin and streptomycin solution, and they were incubated for 48 h at 37°C in a 5% CO2 atmosphere. The cells were fixed at 48 h post-infection (hpi) with 4% paraformaldehyde and stained with crystal violet blue for 30 min. Plaque size was determined using an Olympus (Tokyo, Japan) IX70® microscope.
To measure plaque size, confluent MDBK cells grown in a six-well culture plate (Corning, New York, NY, USA) were infected with 200 PFU of the indicated viruses in each well. For each virus, 50 plaques were measured microscopically in two independent experiments.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting
Approximately 8.8 ×108 MDBK cells in a 100-mm petri dish were infected with the indicated viruses at a MOI of 3 for 18 h, and then the infected cells were washed and collected in 1× cold PBS with a cell scraper. Cells were collected and pelleted by centrifugation at 160 × g for 10 min at 4°C and lysed with lysis buffer (1% Triton X-100, 1 mM ethylenediaminetetraacetic acid, 50 mM Tris, and 150 mM NaCl) containing 1× protease inhibitor cocktail (Sigma-Aldrich, Shanghai, China). The proteins were separated by 15% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Hong Kong, China). The membranes were blocked overnight in 5% skim milk containing 0.1% Tween 20 in 1× PBS. Then, the membranes were probed with a mouse anti-HA-tag antibody (1:1,000 dilution; Beyotime, Haimen, China) or an antibody against β-actin as an internal reference (1:1,000 dilution; Beyotime). Goat anti-mouse antibodies conjugated with horse radish peroxidase (HRP) (1:5,000 dilution) (Southern Biotech, Birmingham, MI, USA) were overlaid for 90 min at room temperature. Then, the antibodies were removed, and the membranes were washed with Tris-buffered saline containing 0.1% Tween-20. The bound IgG was detected subsequently using the Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA).
Silver staining
Silver staining was conducted as described previously [35]. Following electrophoresis, gels were fixed for 1 h in fixative solution (10% acetic acid, 40% ethanol, and 50% H2O), rinsed with distilled water overnight, and incubated in a 0.02% thiosulfate sodium solution for 1 min. After rinsing three times with water for 2 min, the gels were incubated in a 0.1% silver nitrate solution for 20 min at 4°C. The bands in the gel were developed in a 0.05% formaldehyde and 3% sodium carbonate solution, and the reaction was stopped by adding a 5% acetic acid solution for 5 min.
Mass spectrometry
For the mass spectrometry analysis, a confluent monolayer of MDBK cells were was grown in 100-mm culture dishes and infected with the vBoHV1-UL21-HA and wild-type BoHV-1 viruses at a MOI of 3. At 18 hpi, the cells were lysed using lysis buffer. The cell lysates were added to magnetic agarose beads conjugated with an anti-HA-tag monoclonal antibody (MBL, Nagoya, Japan) and incubated for 5 to 6 h at 4°C with gentle rotation. Then, the agarose beads were washed five times, and protein complexes were eluted by heating them in 50 μl of 2× reducing SDS-PAGE sample buffer. The bound proteins were separated by SDS-PAGE and silver stained, and the expected band was excised and placed into a sterile 1.5-ml microcentrifuge tube for further processing via trypsin digestion for Liquid chromatography mass spectrometry (LC-MS), which was performed by Shanghai Applied Protein Technology, Shanghai, China.
Immunofluorescence confocal microscopy
MDBK cells grown on glass coverslips were infected with the indicated viruses. At different time points, the cells were fixed in 4% paraformaldehyde at 37 C for 20 min, permeabilized with 0.2% Triton X-100, and blocked with 1% bovine serum albumen (Biosharp, Hefei, China) and 0.1% Tween 20 in PBS. The cells were incubated with mouse anti-HA antibodies (1:5,000 dilution; Beyotime) and then stained with Cy3-conjugated goat anti-mouse antibodies (1:1,000 dilution; Beyotime). Nuclei were stained blue with 4, 6-diamidino-2-phenylindole (DAPI) (Beyotime). After each step, the cells were washed three times with 1× PBS. Then, anti-fade mounting medium was used to mount the coverslips onto slides. The cells were examined, and images were captured with a Zeiss LSM 880 laser-scanning confocal microscope (Carl Zeiss, Jena, Germany).
Transmission electron microscopy (TEM)
TEM was performed as described previously [31, 36]. MDBK cells were infected with the indicated viruses at a MOI of 3 PFU per cell. At 18 hpi, the cells were pelleted by centrifugation at 500 × g for 10 min at 4°C in cold PBS and fixed with 5% glutaraldehyde in 0.1 M sodium cacodylate buffer at 4°C for 2 h, and then they were post-fixed with 1% osmium tetroxide, followed by washing three times with PBS. The fixed cells were dehydrated with uranyl acetate and a graded ethanol series (35%, 50%, 70%, 90%, and 100%), polymerized using three changes of propylene oxide, embedded, sectioned, and examined by a transmission electron microscope (Hitachi 7000FA®, Tokyo, Japan).
Protein expression and production of polyclonal antisera
Polyclonal antisera were produced according to a previously described method [37], with a slight modification. The entire coding sequences of the UL16 and UL21 were cloned by PCR amplification using the UL16 F/R, UL21F/R primer sets (Table 1), respectively, with KOD-Plus (Toyobo, Osaka, Japan). The PCR products were cloned into the prokaryotic expression vector pGEX-KG using the GST Gene Fusion System (Novagen, Darmstadt, Germany) via the BamHI and HindIII restriction sites (Takara, Dalian, China). The cloned products were confirmed by DNA sequencing and named pGEX-16 and pGEX-21. The expression plasmids were transformed into E. coli strain BL21 (DE3) (TransGen Biotech, Beijing, China). Expression of all the glutathione S-transferase (GST) fusion proteins was induced using different concentrations of isopropyl-β-D-thiogalactopyranoside (UL16, 0.8 mM for 16 h at 37°C; UL21, 0.8 mM for 4 h at 37°C). The bacterial cell pellets were collected by centrifugation (10,000 RPM/10 min), resuspended, and lysed by sonication. Then, the GST fusion proteins were separated from the lysate using Glutathione Sepharose 4B (GE Healthcare, Uppsala, Sweden). Subsequently, the purified proteins were used to immunize rabbits as described previously [38]. All the resulting polyclonal antibodies were used at a dilution of 1:3,000 (in 1% nonfat milk dissolved in 20 mM Tris, pH 7.6, 135 mM NaCl, and 0.1% Tween-20) for an immunoblotting assay, except that the rabbit polyclonal antiserum was diluted 1:100 for an immunofluorescence assay.
Identification of UL21-interacting proteins
Co-immunoprecipitation assay for UL21-HA from infected MDBK cells
MDBK cells were seeded into 100-mm culture dishes and infected with the vBoHV-1-UL21-HA or wild-type BoHV-1 viruses at a MOI of 3, or mock infected as negative controls. At 18 hpi, the cells were lysed using lysis buffer. The cell lysates were added to magnetic agarose beads conjugated with an anti-HA-tag monoclonal antibody (MBL) and incubated for 5 to 6 h at 4°C with gentle rotation. Then, the agarose beads were washed five times, and protein complexes were eluted by heating the agarose in 50μl of 2× reducing SDS-PAGE sample buffer. For western blotting, the bound proteins were separated by 12% SDS-PAGE (Bio-Rad, Hercules, CA, USA) and transferred to a PVDV membrane (Millipore) that was blocked with 5% milk in PBS containing 1% Tween 20, and probed with polyclonal rabbit antisera against UL16 and UL21 (1:3,000 dilutions) and a mouse anti-HA-tag antibody (1:1,000 dilution, Beyotime) or with a β-actin antibody as an internal reference (1:1,000; Beyotime). Binding of the primary antibodies was detected using goat anti-rabbit and goat anti-mouse HRP-conjugated antibodies (1:5,000 dilutions; Southern Biotech, Birmingham, AL, USA). A chemiluminescent substrate (Thermo Fisher Scientific) was used to detect the proteins, and images were obtained using film.
Immunofluorescence assay
MDBK cells grown on 25-mm glass coverslips were infected with the vBoHV1-UL21-HA virus or mock infected. At 18 hpi, the cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 1% bovine serum albumen (Biosharp) and 0.1% Tween 20 in PBS. To detect the interactions of UL16 with UL21, the cells were incubated with mouse anti-HA-tag antibodies (1:5,000 dilutions) or rabbit polyclonal antisera against UL16 (1:100 dilutions) as indicated. Then, they were stained with Cy3-conjugated goat anti-mouse antibodies (1:1000 dilutions; Beyotime) or fluorescein isothiocyanate-conjugated goat anti-rabbit IgG antibodies (1:1000 dilutions; Beyotime). After each step, the cells were washed three times with 1× PBS. The nuclei were visualized by DAPI staining (Beyotime). Images were captured using a Zeiss LSM 880 laser-scanning confocal microscope.
Statistical analysis
All experiments were performed independently at least three times. The data were expressed as means ± standard deviations, and the data from the various groups were compared using a t-test and two-way analysis of variance using GraphPad Prism software (GraphPad, La Jolla, CA, USA). Differences were considered to be statistically significant at p < 0.05 (*), and very significant at p < 0.01 (**) or p < 0.001 (***).
CONFLICTS OF INTEREST
The authors declare that they have no potential conflicts of interest.
FUNDING
This work was funded by the National Key Research and Development Program of China (grant No. 2016YFD0500906), the Special Fund for China Agriculture Research System (Beef/Yak Cattle) (grant No. CARS-38), the Fund for National Distinguished Scholars in Agricultural Research and Technical Innovative Teams.
REFERENCES
1. Muylkens B, Thiry J, Kirten P, Schynts F, Thiry E. Bovine herpesvirus 1 infection and infectious bovine rhinotracheitis. Vet Res. 2007; 38: 181-209.
2. d'Offay JM, Fulton RW, Eberle R. Complete genome sequence of the NVSL BoHV-1.1 Cooper reference strain. Arch Virol. 2013; 158: 1109-13.
3. Fulton RW, d'Offay JM, Eberle R, Moeller RB, Campen HV, O'Toole D, Chase C, Miller MM, Sprowls R, Nydam DV. Bovine herpesvirus-1: evaluation of genetic diversity of subtypes derived from field strains of varied clinical syndromes and their relationship to vaccine strains. Vaccine. 2015; 33: 549-58.
4. Jones C, Chowdhury S. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex and development of improved vaccines. Anim Health Res Rev. 2007; 8: 187-205.
5. Mettenleiter TC. Herpesvirus assembly and egress. J Virol. 2002; 76: 1537-47.
6. Lobanov VA, Maher-Sturgess SL, Snider MG, Lawman Z, Babiuk LA, van Drunen Littel-van den Hurk S. A UL47 gene deletion mutant of bovine herpesvirus type 1 exhibits impaired growth in cell culture and lack of virulence in cattle. J Virol. 2010; 84: 445-58.
7. Fuchs W, Granzow H, Klupp BG, Kopp M, Mettenleiter TC. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J Virol. 2002; 76: 6729-42.
8. Mettenleiter TC. Budding events in herpesvirus morphogenesis. Virus Res. 2004; 106: 167-80.
9. Metrick CM, Chadha P, Heldwein EE. The unusual fold of herpes simplex virus 1 UL21, a multifunctional tegument protein. J Virol. 2015; 89: 2979-84.
10. Le Sage V, Jung M, Alter JD, Wills EG, Johnston SM, Kawaguchi Y, Baines JD, Banfield BW. The herpes simplex virus 2 UL21 protein is essential for virus propagation. J Virol. 2013; 87: 5904-15.
11. Metrick CM, Heldwein EE. Novel structure and unexpected RNA-binding ability of the C-terminal domain of herpes simplex virus 1 tegument protein UL21. J Virol. 2016; 90: 5759-69.
12. Mbong EF, Woodley L, Frost E, Baines JD, Duffy C. Deletion of UL21 causes a delay in the early stages of the herpes simplex virus 1 replication cycle. J Virol. 2012; 86: 7003-7.
13. Klopfleisch R, Klupp BG, Fuchs W, Kopp M, Teifke JP, Mettenleiter TC. Influence of pseudorabies virus proteins on neuroinvasion and neurovirulence in mice. J Virol. 2006; 80: 5571-6.
14. Klupp BG, Bottcher S, Granzow H, Kopp M, Mettenleiter TC. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J Virol. 2005; 79: 1510-22.
15. Muto Y, Goshima F, Ushijima Y, Kimura H, Nishiyama Y. Generation and characterization of UL21-null herpes simplex virus type 1. Front Microbiol. 2012; 3: 394.
16. Meckes DG Jr, Marsh JA, Wills JW. Complex mechanisms for the packaging of the UL16 tegument protein into herpes simplex virus. Virology. 2010; 398: 208-13.
17. Michael K, Klupp BG, Karger A, Mettenleiter TC. Efficient incorporation of tegument proteins pUL46, pUL49, and pUS3 into pseudorabies virus particles depends on the presence of pUL21. J Virol. 2007; 81: 1048-51.
18. de Wind N, Wagenaar F, Pol J, Kimman T, Berns A. The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J Virol. 1992; 66: 7096-103.
19. Takakuwa H, Goshima F, Koshizuka T, Murata T, Daikoku T, Nishiyama Y. Herpes simplex virus encodes a virion-associated protein which promotes long cellular processes in over-expressing cells. Genes Cells. 2001; 6: 955-66.
20. Harper AL, Meckes DG Jr, Marsh JA, Ward MD, Yeh PC, Baird NL, Wilson CB, Semmes OJ, Wills JW. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J Virol. 2010; 84: 2963-71.
21. Han J, Chadha P, Starkey JL, Wills JW. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc Natl Acad Sci U S A. 2012; 109: 19798-803.
22. Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol. 2011; 9: 382-94.
23. Baines JD, Koyama AH, Huang T, Roizman B. The UL21 gene products of herpes simplex virus 1 are dispensable for growth in cultured cells. J Virol. 1994; 68: 2929-36.
24. Guo H, Shen S, Wang L, Deng H. Role of tegument proteins in herpesvirus assembly and egress. Protein Cell. 2010; 1: 987-98.
25. Wagenaar F, Pol JM, de Wind N, Kimman TG. Deletion of the UL21 gene in Pseudorabies virus results in the formation of DNA-deprived capsids: an electron microscopy study. Vet Res. 2001; 32: 47-54.
26. Starkey JL, Han J, Chadha P, Marsh JA, Wills JW. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J Virol. 2014; 88: 110-9.
27. Baines JD, Roizman B. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J Virol. 1991; 65: 938-44.
28. Gao J, Hay TJ, Banfield BW. The product of the herpes simplex virus 2 UL16 gene is critical for the egress of capsids from the nuclei of infected cells. J Virol. 2017; 91.
29. Zhang M, Fu S, Deng M, Xie Q, Xu H, Liu Z, Hu C, Chen H, Guo A. Attenuation of bovine herpesvirus type 1 by deletion of its glycoprotein G and tk genes and protection against virulent viral challenge. Vaccine. 2011; 29: 8943-50.
30. Chowdhury SI, Batterson W. Transinhibition of herpes simplex virus replication by an inducible cell-resident gene encoding a dysfunctional VP19c capsid protein. Virus Res. 1994; 33: 67-87.
31. Raza S, Deng M, Shahin F, Yang K, Hu C, Chen Y, Chen H, Guo A. A bovine herpesvirus 1 pUL51 deletion mutant shows impaired viral growth in vitro and reduced virulence in rabbits. Oncotarget. 2016; 7: 12235-53. https://doi.org/10.18632/oncotarget.7771.
32. Tischer BK, von Einem J, Kaufer B, Osterrieder N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques. 2006; 40: 191-7.
33. Tischer BK, Smith GA, Osterrieder N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol. 2010; 634: 421-30.
34. Luppi MM, Luiz AP, Coelho FM, Ecco R, da Fonseca FG, Resende M. Genotypic characterization of psittacid herpesvirus isolates from Brazil. Braz J Microbiol. 2016; 47: 217-24.
35. Loret S, Guay G, Lippe R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol. 2008; 82: 8605-18.
36. Zhang K, Brownlie R, Snider M, van Drunen Littel-van den Hurk S. Phosphorylation of bovine herpesvirus 1 VP8 plays a role in viral DNA encapsidation and is essential for its cytoplasmic localization and optimal virion incorporation. J Virol. 2016; 90: 4427-40.
37. Lam N, Letchworth GJ. Bovine herpesvirus 1 U(L)3.5 interacts with bovine herpesvirus 1 alpha-transinducing factor. J Virol. 2000; 74: 2876-84.
38. Klupp BG, Granzow H, Mettenleiter TC. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J Virol. 2000; 74: 10063-73.