
INTRODUCTION
The cyclin/cyclin-dependent kinase (CDK) complexes play an important role in regulating cell cycle progression by phosphorylating specific proteins. Cyclin/CDK activities are modulated by both positive and negative regulators [1, 2]. Cyclin/CDK complexes control phosphorylation of the retinoblastoma protein (Rb) [3]. However, the changes in cyclins and CDKs under the hypoxic tumor microenvironment have not been clearly elucidated. CDK inhibitors (CDKIs) are negative regulators of cyclin/CDK complexes and comprise seven members, including the INK4 and Cip/Kip families. Interestingly, p16INK4a, a cell cycle inhibitor, is involved directly or indirectly in the inhibition of tumor formation. The p16INK4a protein controls apoptosis [4, 5], cellular senescence [6, 7], and invasion through suppressing MMP-2 activity [8]. Additionally, overexpression of p16INK4a decreases expression of the angiogenic factor, vascular endothelial growth factor (VEGF), in glioma cells [9]. However, the mRNA level and protein expression of p16INK4a are reduced in tumors under hypoxic conditions, leading to a normal G1/S arrest [10]. However, the reason for reduced p16INK4a transcript and/or protein levels in tumor hypoxia is unclear.
We performed protein microarray analysis to identify p16INK4a-interacting proteins. Among the interacting proteins, the highest binding affinity was shown by hYSK1. The human analog of YSK1 (hYSK1) is an oxidative stress response kinase induced 3- to 7-fold in response to reactive oxygen intermediates, but is not affected by other environmental stresses, growth factors, or cytokines [11]. hYSK1 targets the Golgi apparatus through the Golgi matrix protein, GM130. Activated hYSK1 phosphorylates 14-3-3ζ, which mediates protein transport, cell adhesion, and polarity for cell migration [12]. A low oxygen (O2) level or hypoxia is a characteristic feature of solid tumors [13]. During the progression of solid tumors towards malignancy, rapidly proliferating cancer cells receive an insufficient supply of growth factors, nutrients, and oxygen from the existing vasculature [14]. This results in an intratumoral hypoxic environment, which induces cell cycle arrest in some cancer cells [10]. In this study, we investigated the role of hYSK1 in regulating p16INK4a during cell cycle progression under hypoxia. The results of the present study revealed that hYSK1 negatively regulates p16INK4a as a representative suppressor of cell cycle progression in both the cytosol and nucleus under tumor hypoxic conditions. Notably hYSK1 interacts with p16INK4a, thereby blocking the nuclear translocation of p16INK4a and subsequently regulates p16INK4a gene expression by activating the transcription factor specificity protein-1 (SP-1) in the nucleus during mild hypoxia. We also found that the hYSK1/CDKI complex increases the promoter activity of matrix metalloproteinase-2 (MMP-2), thereby enhancing the invasiveness and metastatic potential of cancer cells under hypoxic conditions.
RESULTS
p16INK4a interacts with hYSK1
Reciprocal interaction studies form the basis of a powerful test to assess the ability of a protein to retain binding affinity while immobilized on a microarray surface [15]. We used protein arrays comprising 1,800 recombinant human proteins in order to identify the proteins that bind to the CDK inhibitor p16INK4a. We screened p16INK4a interacting proteins using biotin-labeled p16INK4a. Laser scanning and digital image analysis demonstrated that p16INK4a selectively and reproducibly interacted with hYSK1 (Figure 1a). To further confirm that hYSK1 directly interacts with p16INK4a, in vitro binding of hYSK1 and p16INK4a was examined using a glutathione (GST) pull-down assay and co-immunoprecipitation. Full-length p16INK4a proteins were produced in Escherichia coli BL21 as GST-fusion proteins and then purified with GST-Sepharose 4B beads. The 35S-labeled hYSK1 proteins were incubated with either GST or GST-p16INK4a fusion proteins bound to Sepharose beads and precipitated in a pull-down assay. The results indicated that GST-p16INK4a bound to hYSK1, whereas GST alone did not show any binding (Figure 1b). Proteins were extracted from COS-1 cells transiently transfected with p16INK4a, pcDNA3.1-V5-hYSK1, and pcDNA3.1-V5-CDK4, then immunoprecipitated with either p16INK4a or hYSK1 antibody and immunoblotted with anti-V5 or anti-p16INK4a. The results indicated that hYSK1 was co-immunoprecipitated with p16INK4a (Figure 1c, left panel) or reciprocally, p16INK4a was co-immunoprecipitated with hYSK1 (Figure 1c, right panel). The interaction of p16INK4a and CDK4 served as a positive control. These results suggest that p16INK4a directly interacts with hYSK1.
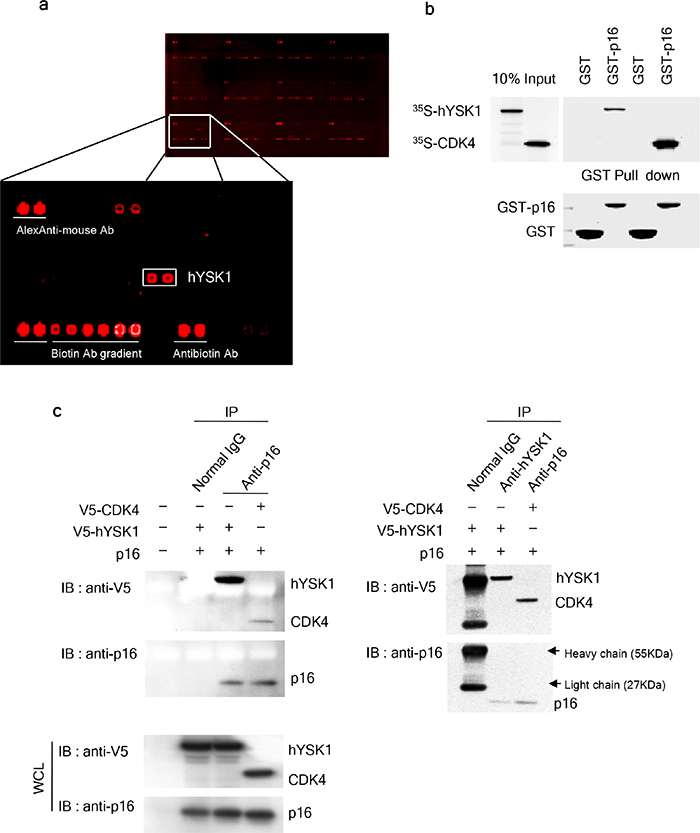
Figure 1: p16INK4a interacts with hYSK1. (a) A human protein microarray was used to screen for novel binding partners of p16INK4a. (b) GST-p16INK4a interacts with in vitro transcribed and translated 35S-met-hYSK1. 35S-met-CDK4, a well-known binding partner of p16INK4a, was used as a positive control. Western blots of in vitro translated and bound proteins are shown (upper panel). Coomassie blue-stained gels show the amount of GST–p16INK4a and GST (as loading control) used in this assay (lower panel). (c) COS-1 cells were transfected with combinations of p16INK4a, pcDNA3.1-V5-hYSK1, and pcDNA3.1-V5-CDK4. Cell lysates were subjected to reciprocal immunoprecipitation and immunoblotting using anti-p16INK4a (left panel) or anti-hYSK1 (right panel).
Identification of the hYSK1 and p16INK4a binding site
To identify the specific binding regions of hYSK1 and p16INK4a, we generated deletion constructs of p16INK4a and hYSK1 and then examined the binding of different domains of p16INK4a with the full-length hYSK1 produced by in vitro transcription and translation. GST pull-down assay results showed that the D4 domain of p16INK4a, in which 30–60 amino acids were deleted, interacted with the full-length hYSK1 (Figure 2a). We also investigated binding of the full-length p16INK4a to specific regions of hYSK1. Various deleted fragments of hYSK1 were expressed as in vitro transcribed and translated fusion proteins and their possible interaction with the full-length GST-p16INK4a fusion protein was analyzed. GST pull-down assay revealed that the full-length GST-p16INK4a interacted with the deleted fragment (residues 20-40 and 140-200) of hYSK1 (Figure 2b). These results indicated that the amino acids 30-60 of p16INK4a and the amino acids 20-40 and 140-200 of hYSK1 are important binding sites involved in the interaction between hYSK1 and p16INK4a. We also performed a prediction analysis of the p16INK4a and hYSK1 complex using the Fast Fourier transform-based docking algorithm ZDOCK (Figure 2c). The p16INK4a and hYSK1 complex obtained from the protein-protein docking model suggested that p16INK4a bound to the back of the hYSK1 kinase domain, which is located on the opposite side of the ATP binding pocket (Figure 2c). The hydrogen bond networks between the β-strands in the N-terminal domain of hYSK1 and the first, second, and third ankyrin repeats of p16INK4a are expected to be crucial in the p16INK4a and hYSK1 binding (Supplementary Table 1 and Figure 1). For example, the guanidinium side chain of Arg24 in p16INK4a forms a pair of hydrogen bonds with the carboxylate oxygens of Asp24 in hYSK1. Moreover, Ala21, Tyr44, Arg46, Thr79, and Arg87 in p16INK4a form hydrogen bonds with Lys36, Glu17, His41, Lys43, and Glu44 in hYSK1, respectively. In addition, the hydrophobic packing among the Ala21, Tyr44, Val51, and Met53 residues of p16INK4a together with the Leu23, Ile38, Lys43, and Val45 residues of hYSK1 also contribute to the stability of the p16INK4a and hYSK1 complex (Supplementary Figure 1). These hydrogen bonds and hydrophobic interactions might explain the in vitro and in vivo experimental results, which indicated that the segment between Ala20 and Ala60 of p16INK4a and the segment between Phe20 and Ile50 of hYSK1 are the main binding segments of these proteins.
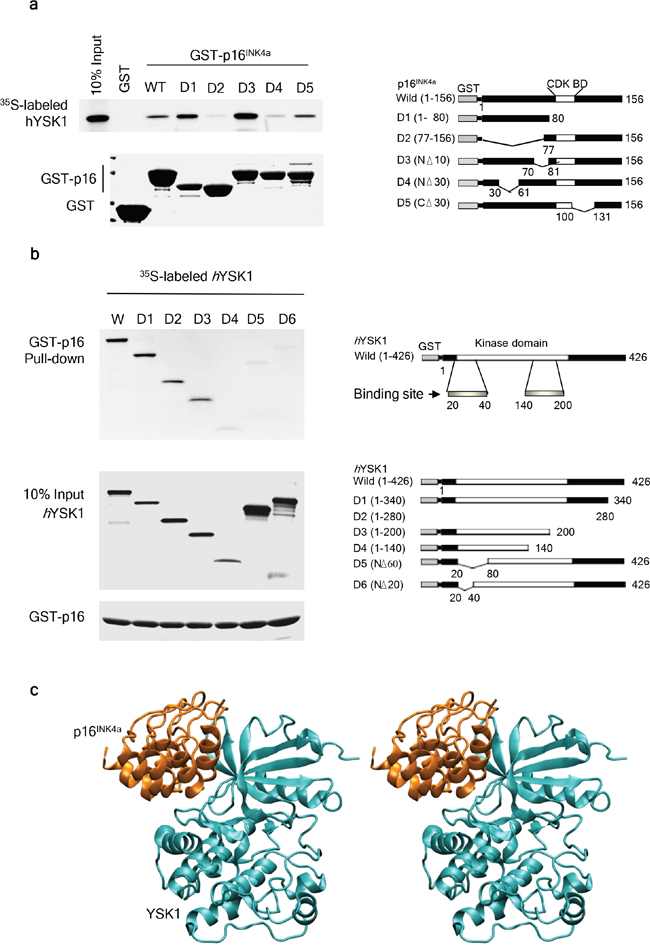
Figure 2: Identification of the binding domains of hYSK1 and p16INK4a. (a) Full length and deletion mutants of p16INK4a were produced in E. coli BL21 as glutathione (GST)-fusion proteins and purified on GST-Sepharose 4B beads. 35S-labeled hYSK1 proteins were incubated with either GST or GST-p16INK4a fusion proteins bound to Sepharose beads and were precipitated by GST pull-down. (b) Full-length and deletion mutants of hYSK1 were produced by in vitro transcription and translation. 35S-labeled hYSK1 proteins were incubated with GST-p16INK4a fusion proteins bound to Sepharose beads and were precipitated by GST pull-down. Proteins bound to the beads were analyzed by autoradiography. Coomassie blue-stained gels were used in the assay as in Figure 1. (c) Predicted model of the p16INK4a and hYSK1 complex generated by computer modeling.
hYSK1 inhibits the expression and translocation of p16INK4a under hypoxic conditions
To examine the role of hYSK1 in the regulation of p16INK4a, we detected the effect of the ectopic expression or knockdown of hYSK1 on the expression level of p16INK4a. The amount-dependent over-expression of hYSK1 decreased p16INK4a expression in SK-MEL-28 melanoma cells (Figure 3a). Transient transfection of SK-MEL-28 cells with short-hairpin RNA designed against hYSK1 (shRNA-hYSK1) completely suppressed hYSK1 expression and restored the expression of p16INK4a (Figure 3b). These results suggest that hYSK1 expression might play an important role in the regulation of p16INK4a. In order to determine whether endogenous hYSK1 and p16INK4a also interacted in cells, we performed immunoprecipitation using a p16INK4a antibody followed by immunoblot analysis with a hYSK1 antibody as described in the Methods section. The p16INK4a protein strongly interacted with hYSK1 at 12 h after exposure to hypoxia (1% O2; Figure 3c). SK-MEL-28 cells were transiently transfected with shRNA-control or shRNA-hYSK1 and endogenous expression of hYSK1 under hypoxic (1% O2) conditions was then assessed after incubation for 36 h. Results showed that the shRNA-hYSK1 completely inhibited hYSK1 expression (Figure 3d). Upon exposure to hypoxia (1% O2), p16INK4a expression was increased in a time-dependent manner in shRNA-hYSK1-transfected SK-MEL-28 cells compared to cells transfected with the shRNA-control (Figure 3d). To determine whether nuclear translocation of the active p16INK4a protein was affected by hYSK1 expression, the distribution of exogenous and endogenous p16INK4a and hYSK1 upon exposure to hypoxia (1% O2) was investigated. The expression of hYSK1 was very low in the cytoplasm and nucleus of HT-1080 and SK-MEL-28 cells during normoxia, but its expression was increased upon exposure to hypoxic conditions (Figure 3e, 3f). Interestingly, the nuclear expression of p16INK4a was decreased in a time-dependent manner, whereas its expression in the cytoplasm was retained under hypoxia following hYSK1 expression (Figure 3e, 3f). If hYSK1 is a negative regulator of p16INK4a expression and translocation in hypoxic conditions, hYSK1 knockdown might be expected to increase the hypoxia-induced p16INK4a protein expression and translocation. Thus, we knocked down hYSK1 expression in HT-1080 and SK-MEL-28 cells to assess whether the expression and nuclear translocation of the p16INK4a protein are inhibited. The expression of p16INK4a was substantially increased after exposure to hypoxia in shRNA-hYSK1-transfected cells compared to that in cells expressing shRNA-control (Figure 3e, 3f).
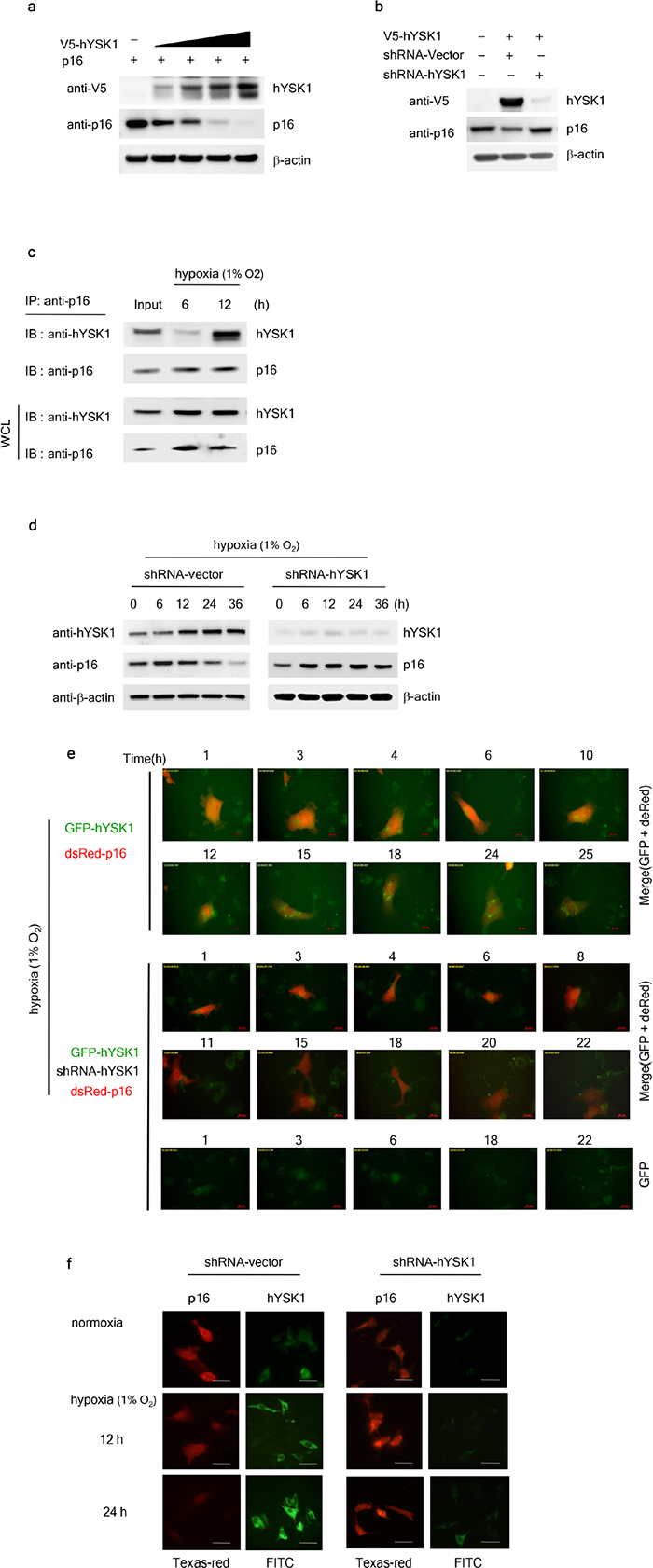
Figure 3: hYSK1 regulates expression and translocation of p16INK4a. (a) The effect of hYSK1 on p16INK4a expression was determined by transfection of pcDNA3.1-V5-hYSK1 in SK-MEL-28 cells. (b) Knocking down hYSK1 abundancerestored the expression of p16INK4a. (c) Immunoprecipitation was performed using anti-p16INK4a in cells incubated under hypoxic (1% O2) conditions. p16INK4a was immunoprecipitated followed by western blotting with anti-hYSK1 and anti-p16. (d) Relative expression of hYSK1 and p16INK4a was assessed under hypoxic (1% O2) conditions by western blot analysis in SK-MEL-28 cells transfected with shRNA-control or shRNA-hYSK1 plasmid. (e) The effect of hYSK1 on subcellular distribution of p16INK4a in HT-1080 cells transfected with a GFP-hYSK1/pDsRed-p16INK4a or GFP-hYSK1/shRNA-hYSK1/pDsRed-p16INK4a plasmid was examined. Transfected cells were incubated under hypoxic (1% O2) conditions were examined by live cell imaging. (f) Time-dependent fluorescence localization of p16INK4a (red) and hYSK1 (green) was visualized at 12 or 24 h after transfection of shRNA-control or shRNA-hYSK1 in SK-MEL-28 cells both in normoxic and hypoxic (1% O2) conditions.
To analyze the effect of hypoxia-induced hYSK1 activation on the expression of p16INK4a, we examined the effect of hYSK1 on p16INK4a expression under normoxic or hypoxic (1% O2) conditions. The expression of p16INK4a was downregulated with the ectopic expression of hYSK1 during both normoxia and hypoxia (Figure 4a). However, p16INK4a expression was substantially elevated under hypoxic conditions in cells transfected with p16INK4a alone and was downregulated in the presence of hYSK1. To investigate whether active hYSK1 can affect the nuclear localization of p16INK4a, the distribution of p16INK4a and hYSK1 upon exposure to hypoxia was examined. Under normoxic conditions, hYSK1 was distributed both in the cytoplasm and nucleus in SK-MEL-28 cells, whereas p16INK4a was localized in the cytoplasm (Figure 4b). Under hypoxic conditions, overexpressed p16INK4a proteins were translocated from the cytoplasm to the nucleus. When hYSK1 and p16INK4a were co-expressed in cells followed by exposure to hypoxia, hYSK1 attenuated the constitutive nuclear accumulation of p16INK4a (Figure 4c). Hypoxia induced hYSK1 expression both in the cytoplasm and nucleus in a time-dependent manner and p16INK4a was largely retained in the cytoplasm corresponding to a time-dependent decrease of its nuclear expression (Figure 4c). If hYSK1 functions as a critical regulator of the cell cycle and transactivation following p16INK4a translocation during early hypoxia, hYSK1 might be expected to regulate the interaction of p16INK4a with CDK4 for cell cycle arrest in hypoxia. Thus, to confirm the binding of p16INK4a and CDK4, which might be influenced by hYSK1, COS-7 cells were transfected with p16INK4a and CDK4 and/or hYSK1. Transfection was followed by co-immunoprecipitation with antibodies against CDK4, hYSK1, or p16INK4a and subsequent immunoblot analysis (Figure 4d). Overexpression of p16INK4a in the presence of either V5-CDK4 or V5-hYSK1 and subsequent immunoprecipitation with p16INK4a revealed that p16INK4a interacted with either CDK4 or hYSK1. However, in COS-7 cells transfected with p16INK4a along with both V5-CDK4 and V5-hYSK1 and immunoprecipitated with anti-p16INK4a, p16INK4a exhibited a stronger interaction with hYSK1 than with CDK4 (Figure 4d). These results indicate that hYSK1 negatively regulates the expression and nuclear localization of p16INK4a by interfering with the interaction between p16INK4a and CDK4. Therefore, we suggest that hYSK1 plays an important role as a p16INK4a inhibitor ex vivo.
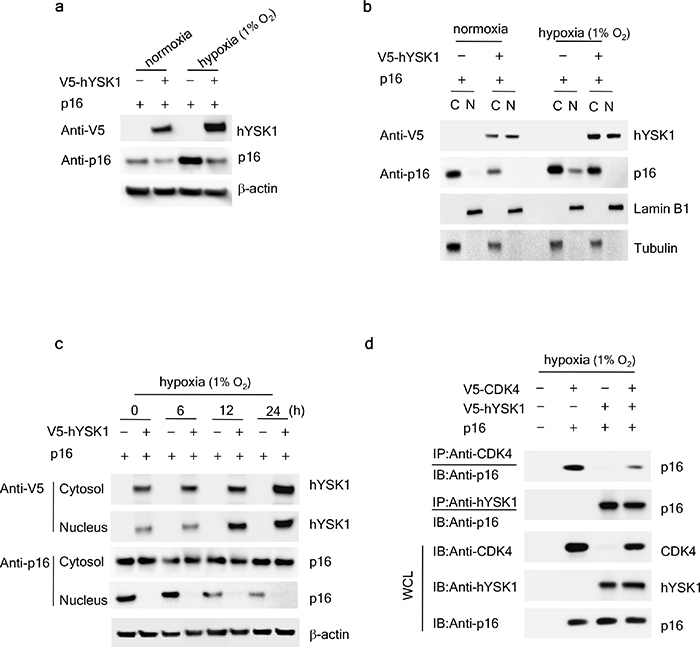
Figure 4: hYSK1 regulates the expression of p16INK4a in the nucleus. (a) The effect of hYSK1 on p16INK4a expression was determined both in normoxic and hypoxic (1% O2) conditions after transfection of pcDNA3.1-V5-hYSK1 and pcDNA3.1-p16INK4a in SK-MEL-28 cells. (b) The cytosolic and nuclear expression of hYSK1 and p16INK4a was examined after transfection of pcDNA3.1-V5-hYSK1 and pcDNA3.1-p16INK4a in SK-MEL-28 cells incubated under normoxic or hypoxic (1% O2) conditions. Lamin B1 and tubulin were used as loading controls for nuclear and cytosol fractions, respectively. (c) The expression of hYSK1 and p16INK4a was examined both in the cytosol and nucleus at various time points under hypoxic conditions (1% O2). (d) Immunoprecipitation was performed using anti-CDK4 or anti-p16INK4a in COS-7 cells exposed to hypoxic (1% O2) conditions. p16INK4a, hYSK1, and CDK4 in the lysates were co-precipitated and detected by western blotting.
hYSK1 inhibits p16INK4a gene transcription and induces MMP-2 promoter activity; role of SP-1
Under hypoxic conditions, gene transcription of p16INK4a was increased until 12 h and then gradually declined until 24 h when cells were transfected with shRNA-control. However, in cells transfected with shRNA-hYSK1, the elevated gene transcription of p16INK4a was sustained until 24 h (Figure 5a). These findings suggested that shRNA-hYSK1-transfected cells exhibited increased p16INK4a gene transcription compared to shRNA-control-transfected cells at each time point.
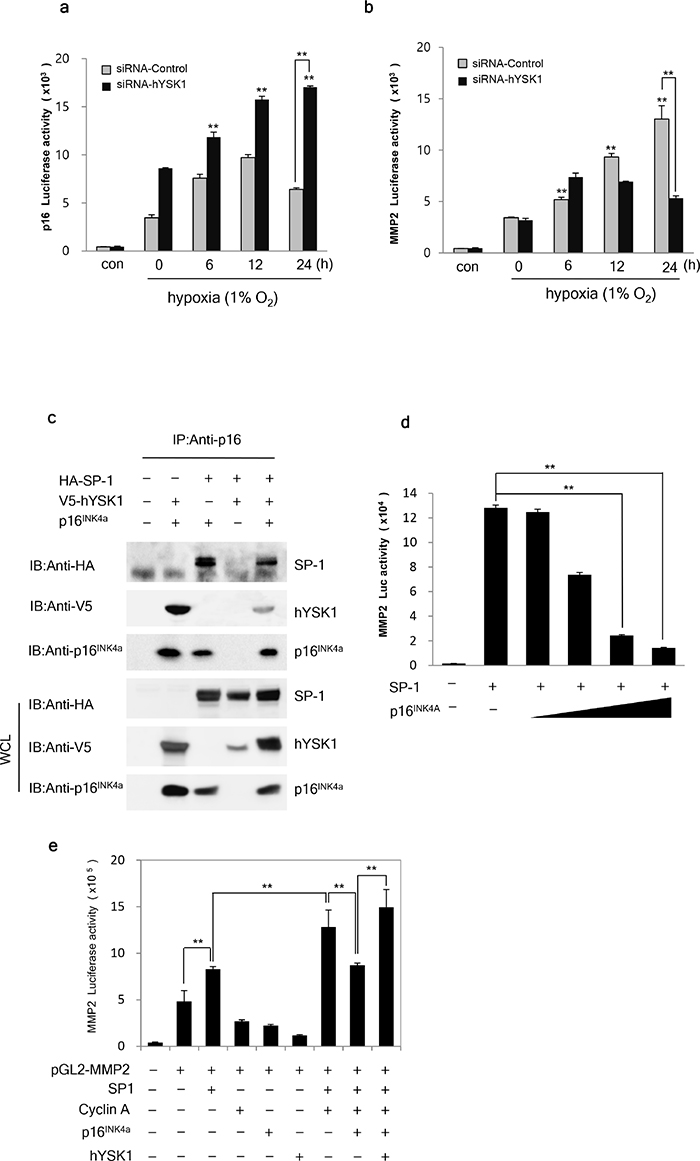
Figure 5: hYSK1 enhances the transcriptional activity of SP-1 transcription factor in the MMP-2 promoter through interfering p16INK4a. (a) A time course study of the silencing effect of hYSK1 on p16INK4a luciferase activity in the SK-MEL-28 cell line under hypoxic conditions. (b) Time-dependent analysis of the silencing effect of hYSK1 on MMP-2 luciferase activity in the HT1080 cell line was examined under hypoxic conditions. (c) The interaction of hYSK1, SP-1 and p16INK4a was detected by immunoprecipitation with a p16INK4a antibody. (d) The effect of p16INK4a on SP-1 activation of the transcription of MMP-2. (e) The SP-1 transcriptional activation of MMP-2 luciferase activity was assessed by various combinations of transfected SP-1, cyclin A, p16INK4a or hYSK1. The expression of hYSK1, p16INK4a and SP-1 were detected by western blot in SK-MEL-28 cells.
Furthermore, unlike the MMP-2 activity under normoxic conditions, the MMP-2 gene transcription in shRNA-hYSK1-transfected cells was decreased from 12 h upon exposure to hypoxic conditions (Figure 5b). Moreover, COS-7 cells were transfected with p16INK4a, V5-hYSK1, or HA-SP-1 and its cell lysates were immunoprecipitated with a p16INK4a antibody. The results showed that p16INK4a interacted with both hYSK1 and SP-1, forming a complex comprised of p16INK4a, hYSK1, and SP-1 (Figure 5c). Ectopic expression of increasing amounts of p16INK4a in HT-1080 cells, which have a homozygous deletion of p16INK4a, resulted in decreased SP-1-dependent MMP-2 expression (Figure 5d). Combinatorial transfection of the MMP-2 reporter, SP-1, p16INK4a, cyclin A, or hYSK1 gene constructs resulted in increased MMP-2 transcription in the presence of SP-1 and cyclin A. Its effect was significantly abolished by transfection with p16INK4a and was restored in the presence of hYSK1 (Figure 5e). These results indicate that hYSK1 controls the expression of p16INK4a and MMP-2 by regulation of SP-1 activity through masking p16INK4a under hypoxic conditions.
hYSK1 enhances proliferation and migration of cancer cells by downregulating p16INK4a
Next, we examined the physiological consequences of p16INK4a regulation by hYSK1 in cancer cells. A2058 or SK-MEL-28 invasive melanoma cells transiently transfected with either siRNA-control or siRNA-hYSK1 revealed that hYSK1 silencing reduced cell proliferation (Figure 6a). Moreover, migration of HT-1080 and SK-MEL-28 cells stably transfected with shRNA-hYSK1 was dramatically reduced under hypoxic conditions compared to cells transfected with the shRNA-control (Figure 6b, 6c). To determine whether hYSK1 could activate cell migration through downregulation of p16INK4a, we performed co-transfection with various combinations of hYSK1, p16INK4a, or siRNA-hYSK1 in HT-1080 cells. The results indicated that the migration index was increased by the addition of 2% serum under hypoxic (1% O2) conditions and that cell migration was further enhanced in hYSK1-transfected cells but was decreased in p16INK4a- and hYSK1/siRNA-hYSK1-transfected HT-1080 cells (Figure 6d, 6e). The decrease in migration index mediated by p16INK4a was recovered by transfection with hYSK1. The hYSK1-induced recovery of p16INK4a-repressed cell migration was abrogated by transfection with siRNA-hYSK1. The cell migration enhanced by hYSK1 and its modulation by p16INK4a was also examined using a wound healing assay with cells transfected using different combinations of hYSK1, p16INK4a, and siRNA-hYSK1 (Supplementary Figure 2). We present a model (Figure 6d) depicting the interaction between hYSK1 and p16INK4a and its consequences on cell proliferation and migration.
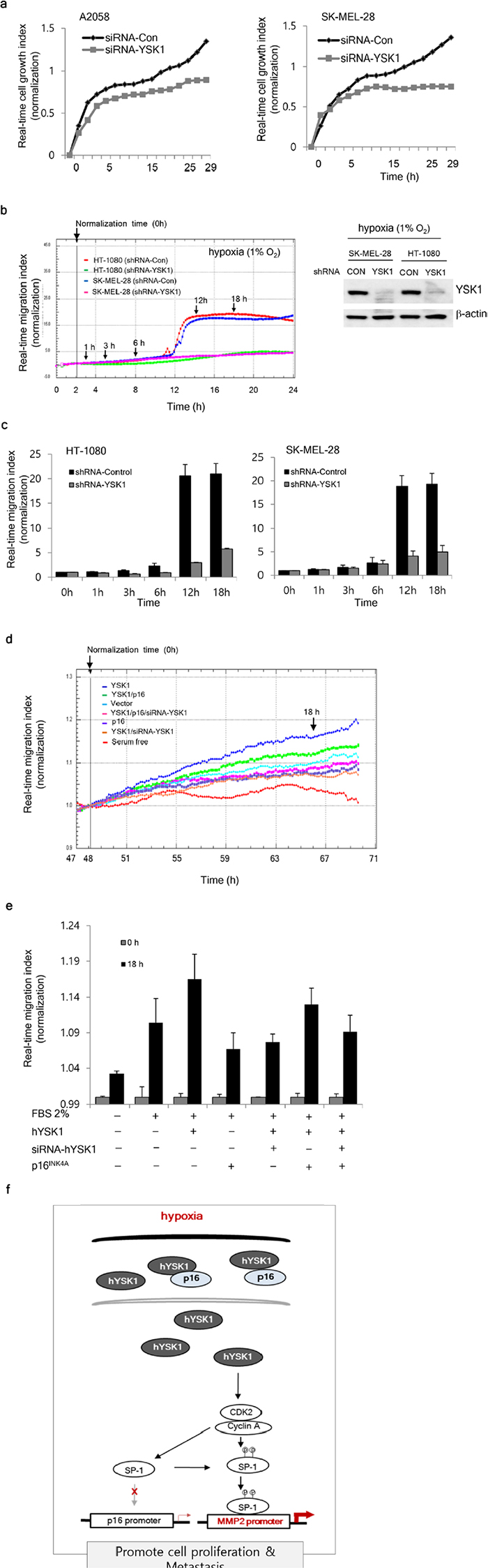
Figure 6: Physiologic effect of the interaction of hYSK1 and p16INK4a in cancer cells. (a) Growth of A5028 and SK-MEL-28 cells transfected with si-hYSK1 was retarded compared to cells transfected with the si-control plasmid. (b, c) Under hypoxic conditions, the migration of HT-1080 and SK-MEL-28 cells was decreased in cells transfected with shRNA-hYSK1 compared with cells transfected with si-control. (d, e) The effect of various transfection combinations on HT-1080 cancer cell migration. (f) A representative scheme of the regulation of p16INK4a by hYSK1 in a hypoxic tumor. The increased expression of hYSK1 in hypoxic conditions retains cytosolic p16INK4a and activates SP-1 transcriptional activity of the MMP-2 promoter. Thereby, cell proliferation and migration is promoted.
DISCUSSION
p16INK4a, a tumor suppressor, is silenced by methylation in various cancers. Mutational inactivation of p16INK4a has been shown to promote tumorigenesis [16]. The tumor suppressor function of p16INK4a has been linked to its inhibition of CDK that leads to disassembly of the cyclin D1/CDK complex, thereby resulting in reduced cell proliferation [17, 18]. The growing tumor maintains an intratumoral hypoxic environment that stimulates the malignant progression of preneoplastic cells [19]. Expression of p16INK4a is decreased in cancer cells under severe hypoxia [12]. Moreover, ectopic expression of p16INK4a was reported to inhibit hypoxia-induced migration of the human breast cancer cells, MDA-MB231 [20]. However, the mechanisms underlying the loss of p16INK4a function under hypoxic conditions and its role in the resultant increased proliferation and migration of cancer cells, remain elusive. In the present study, we sought to elucidate the inactivation mechanisms of p16INK4a under tumor hypoxia and examine the effects of loss of p16INK4a function on tumor progression.
Using protein microarray analysis, we identified hYSK1, a protein that facilitates cell migration [21], as an interacting partner of p16INK4a. Data from GST pull-down and immunoprecipitation assays further confirmed the interaction between hYSK1 and p16INK4a. Our study revealed that the D4 (30–60 amino acid) domain of p16INK4a interacted with the 20–40 amino acid and 140–200 amino acid segments of hYSK1. Computational docking analysis indicated that the possible binding site of p16INK4a would be located at the other side of the ATP binding pocket of hYSK1. We found that ectopic hYSK1 expression dose-dependently diminished the expression of p16INK4a, which was restored by silencing hYSK1. Moreover, the elevated hYSK1 expression in cells exposed to hypoxia showed a concomitant decrease in p16INK4a, which was further restored with hYSK1 knockdown. With hypoxic stimulation, hYSK1 interacted with p16INK4a and retained p16INK4a in the cytoplasm, thereby blocking its nuclear localization. These findings raised the possibility that the binding between hYSK1 and p16INK4a, and the subsequent inhibition of p16INK4a expression might contribute to tumor progression. One of the mechanisms by which p16INK4a inhibits tumor growth under hypoxic conditions is mediated through the interaction of p16INK4a with CDK4 [8] and a subsequent decrease in CDK4 catalytic activity [18]. The finding that the interaction between p16INK4a and CDK4 under hypoxic conditions is reduced in the presence of hYSK1 suggests that hYSK1 impairs the p16INK4a-mediated decrease in CDK4 activity and hence, promotes the growth of hypoxic tumor cells by sequestering p16INK4a in the cytoplasm through protein-protein interaction.
To further elucidate the mechanisms involved in hYSK1 regulation of p16INK4a and its consequences on cell migration, we first found that under hypoxic conditions, hYSK1 silencing increased the promoter activity of p16INK4a, but decreased the promoter activity of MMP-2. The p16INK4a protein was reported to inhibit MMP-2 expression by inactivating SP-1 [22], which is a strong activator of p16INK4a and p21WAF1 [23]. Because transactivation of both p16INK4a and MMP-2 promoters is regulated by SP-1, we focused on the impact of hYSK1 on the regulation of SP-1. However, immunoprecipitation of cells transfected with either V5-hYSK1 or HA-SP-1 with anti-hYSK1 or anti-SP-1 failed to detect either SP-1 or hYSK1, respectively. These findings suggest that hYSK1 does not interact with SP-1 directly. We therefore sought to examine the mechanisms underlying the negative and positive regulation of p16INK4a and MMP-2 promoter activity by hYSK1, respectively. SP-1 plays a key role in the transactivation of MMP-2 [22]. Haidweger et al. reported that SP-1 is phosphorylated by CDK2-cyclin A [5]. p16INK4a negatively regulates MMP-2 expression by downregulating SP-1 function [22]. Thus, our finding that p16INK4a diminished the SP-1 plus cyclin-A-dependent increase in MMP-2 promoter activity, which was restored in the presence of hYSK1, suggests that the hYSK1 negation of p16INK4a enhanced MMP-2 promoter activity. The increased p16INK4a promoter activity and decreased MMP-2 promoter activity in cells transfected with siRNA-hYSK1 under hypoxic conditions confirmed the negative and positive regulation of p16INK4a and MMP-2 promoter activity by hYSK1. These findings were associated with decreased migration of different cancer cells transfected with siRNA-hYSK1.
Thus, our study reports the novel finding that inactivation of p16INK4a is partly mediated through its binding with hYSK1, which sequesters p16INK4a in the cytoplasm. The hYSK1-mediated inactivation of p16INK4a results in elevated MMP-2 promoter activity, which contributes to increased migration of cancer cells under hypoxia. Furthermore, a hYSK1 inhibitor might provide a new therapeutic window for the development of novel anticancer agents for activation of the tumor suppressor, p16INK4a.
MATERIALS AND METHODS
Plasmid construction
pcDNA3.1-v5-hYSK1 and deletion fragments of hYSK1 were generated by PCR using the human cDNA clone-hYSK1 (Origene Technologies, MD, USA) as a template. The PCR product was purified, digested with EcoRI/XhoI, and cloned into the EcoRI/XhoI sites of pcDNA3.1-v5-HisA (Invitrogen, MA, USA). For TOPO-GFP-hYSK1, the PCR product was inserted into the pcDNA3.1-NT-GFP-TOPO vector (Invitrogen, MA, USA). The GST-p16INK4a and GST-deletion fragments of p16INK4a were inserted in-frame into the BamHI/XhoI site of the pGEX-5X-1 vector (Amersham Biosciences, PA, USA). For pDsRed-p16INK4a, the PCR product was cloned into the ApeI site of the pDsRed vector (Clontech Laboratories, CA, USA). HA-SP-1 was cloned into the EcoRI/XhoI site of the pCMV-HA and pCMV-Myc vectors (Clontech Laboratories, CA, USA). The shRNA-hYSK1 plasmid was constructed into the pSilencer 4.1-CMV-hyg vector (Ambion, NY, USA). The pGL2-p16-luc vector was a gift from Dr. Gordon Peters (Cancer Research UK, London) and the pGL2-MMP-2-luc vector was a gift from Dr. Etty N. Benveniste (The University of Alabama at Birmingham, Birmingham, AL). Various expression vectors were amplified in E. coli XL1-blue or BL21 cells and plasmids were purified using a Qiagen midi kit (Qiagen, Hilden, Germany). The DNA sequences of all plasmids were confirmed by sequencing (Dye Terminator ABI Type Seq., Bionex, NJ, USA)
Cell culture and transfection
Human melanoma (SK-MEL-28, A2058), human fibrosarcoma (HT-1080), and COS-7 cells (African green monkey kidney) were purchased from the American Type Culture Collection (ATCC, VA, USA). SK-MEL-28, A2058, and HT-1080 cells were cultured in MEM containing penicillin (100 units/mL), streptomycin (100 μg/mL), sodium pyruvate (1 mM), and 10% fetal bovine serum (FBS) (Invitrogen, Gibco). COS-1 and COS-7 cells were grown in DMEM containing penicillin (100 units/mL), streptomycin (100 μg/mL), and 10% FBS. Cells were maintained at 37°C in a humidified atmosphere of 95% air/5% CO2. The pcDNA3.1-p16INK4a or pcDNA3.1-v5-hisA-hYSK plasmid was transfected using the jetPEI poly transfection reagent (Polyplus) into HT-1080, COS-1, or COS-7 cells to generate p16INK4a- or p16INK4a-hYSK1-expressing cells. For transient gene silencing, si-control and si-hYSK1 (Dharmacon, CO, USA) were transfected into HT-1080, SK-MEL-28, and A2058 cells. shRNA-control and shRNA-hYSK1 were transfected into HT-1080 and SK-MEL-28 cells. For the reporter gene assay, COS-7 cells were seeded in 24-well plates and incubated for 24 h followed by transfection with pcDNA3-p16INK4a, HA-SP-1, cyclin A, pGL2-p16-luc, or pGL2-MMP-2-luc reporter plasmids using the jetPEI poly transfection reagent based on the manufacturer’s instructions. The pRL-TK reporter plasmid was used as an internal control. Cells were harvested after 24 h and disrupted with 5X lysis buffer. Luciferase activity was determined after normalization to pRL-TK activity (Promega, WI, USA). For immunofluorescence microscopy, SK-MEL-28 cells were transfected with shRNA-control or shRNA-hYSK1 and incubated for 24 h. The cells were transferred to a hypoxic chamber and incubated for an additional 12 or 24 h. The cells were then washed with PBS and fixed with methanol for 5 min at room temperature. After blocking with 2% BSA in PBST for 10 min, cells were sequentially incubated with p16INK4a and hYSK1 primary monocloncal antibodies followed by incubation with Texas-red and FITC-conjugated secondary antibodies.
Protein microarray
To evaluate novel protein-protein interactions, a protein microarray was used as a powerful detection method [24]. The p16INK4a protein was biotinylated and probed on a ProtoArray® Protein Microarray (v.1.0) printed on thin nitrocellulose slides (Promega, WI, USA). Briefly, the ProtoArray™ Human Protein Microarray slide was blocked and probed with biotin labeled p16INK4a. Proteins on the array were detected using streptavidin conjugated Alexa Fluor 647 and scanned for analysis (Invitrogen, MA, USA) after drying.
Immunoprecipitation
Transfected COS-7 cells or SK-MEL-28 cells were harvested in NET-NL lysis buffer containing 50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% NP-40, 0.2 mM PMSF, and a mixture of protease inhibitors (Roche Diagnostics, Basel, Switzerland). Cell lysates (200 μg) were clarified by centrifugation before overnight incubation at 4°C with p16INK4 (Pharmingen, CA, USA), CDK4 (Cell Signaling, MA, USA), hYSK1, and SP-1 (Santa Cruz Biotechnology, TX, USA) antibodies in NET-NR buffer [50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% NP-40, 2 μg/mL BSA, 0.2 mM PMSF, and a mixture of protease inhibitors (Roche Diagnostics Basel, Switzerland)]. An aliquot of 50 μL pre-washed protein A/G-agarose beads (Roche Diagnostics; 50% slurry) was then added to the mixture and incubated for 2 h at 4°C. Immunoprecipitates were recovered by centrifugation, washed three times in NET-NW buffer [50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% NP-40, and 0.2 mM PMSF] and resolved by SDS-PAGE and western blotting.
Western blot analysis
Cells were disrupted on ice for 30 min in cell lysis buffer [20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium vanadate, 1 μg/mL leupeptin, and 1 mM phenylmethylsulfonyl fluoride (PMSF)]. After centrifugation at 20,817 g for 15 min, the supernatant fraction was harvested as a total cellular protein extract. Protein concentration was then determined using a protein assay reagent (Bio-Rad labs, CA, USA). The total cellular protein extracts were separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes in 20 mM Tris-HCl (pH 8.0), containing 150 mM glycine and 20% (v/v) methanol. Membranes were blocked with 5% nonfat dry milk in 1x TBS containing 0.05% Tween 20 (TBS-T) and incubated with antibodies against p16, YSK1, CDK4, SP-1, MMP-2, lamin B1, α-tubulin, and β-actin. Blots were washed thrice in 1 x TBS-T buffer followed by incubation with the appropriate HRP-linked IgG. The specific proteins in the blots were visualized using an enhanced chemiluminescence (ECL) detection kit (GE Healthcare Bioscience, PA, USA).
GST pull-down assay
Full length and deletion mutants of hYSK1 or CDK4 were translated in vitro with L-[35S] methionine using the TNT Quick Coupled Transcription/Translation System (Promega, WI, USA). GST fusion proteins were collected on glutathione-Sepharose beads (GE Healthcare Bioscience, PA, USA) and incubated for 4 h at 4°C with 35S-Met-labeled hYSK1, its deletion mutants, or CDK4. The bound proteins were washed thrice and boiled with 2.5X sample buffer for 3 min, centrifuged, and the supernatant fraction was then examined by 15% SDS-PAGE analysis. Binding was detected by autoradiography.
Protein-protein docking
Docking structures of p16INK4a and YSK1 were generated using the rigid-body global search algorithm and further refinement by energy-minimization. Rigid-body docking was performed by the Fast Fourier Transform-Based Docking algorithm ZDOCK [25–27], where p16INK4a and YSK1 were treated as solid objects. Subsequent energy-minimization and molecular dynamics simulations of the p16INK4a-YSK1 complex were performed using the Impact modules from the Schrödinger Software Suite [28, 29]. Details are provided in the Supplementary Materials.
Fluorescence live cell imaging
To track individual hYSK1 and p16INK4a expression, HT-1080 cells were transfected with GFP-TOPO-hYSK1 and pDsRed-p16INK4a or shRNA-hYSK1. The cells were incubated for 12 h in a 5% CO2 incubator and transferred to a hypoxic (1% CO2) chamber. The movement of cells was tracked every 8-10 min for 24 h at 16 fixed regions by an inverted microscope (BX50, Olympus, Tokyo, Japan). Cell movement was analyzed using AVI meta imaging software.
Cell proliferation and migration assay
Proliferation of A2058 and SK-MEL-28 cells transfected with si-control or si-hYSK1, was assessed by determining the cell growth index using the xCELLigence RT-CES system and E plates (Roche Diagnostics Basel, Switzerland), which monitors the cellular events in real time by measuring electrical impedance across inter-digitized gold micro-electrodes integrated on the bottom of the culture plates. The impedance measurement indicates the quantitative value of cell number, viability, and morphology [30]. The cells were suspended in MEM containing 10% FBS and seeded in the E-plate 96 (2,000 cells/well/100 μl). The cells were monitored every 30 min for a period of 29 h. Wound migration of cells was measured using Culture-Inserts (Ibidi GmbH, Martinsried, Germany). The Culture-Inserts were placed in 6-well plates, and HT-1080 cells were seeded at a density of 5×104 cells in each well with the Culture-Inserts. After 24 h of incubation, the Culture-Inserts were removed, and a cell-free gap of 500 μm was created. Phase contrast images of the closed gap were captured at 0 h (control) and 12 h of hypoxic incubation using an inverted microscope (magnification, 10X). For real-time cell migration, we used the xCELLigence RTCA DP system and fibronectin-coated CIM plates (Roche Diagnostics Basel, Switzerland) [31]. Cells were seeded at 10,000 cells per well and the migratory behavior of each cell line was monitored for 18 h. The assay was performed based on the manufacturer’s instructions (Roche Diagnostics Basel, Switzerland).
Statistical analysis
Values were expressed as means ± S.E.M. from at least three independent experiments. Statistical significance was determined by Student’s t-test and a p-value less than 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We wish to thank Dr. Ann M. Bode and Zunnan Huang in The Hormel Institute, University of Minnesota, USA, Dr. Bum Ju Ahn in Seoul National University, South Korea for supporting experiments.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
REFERENCES
1. Osada S, Izawa M, Saito R, Mizuno K, Suzuki A, Hirai S, Ohno S. YSK1, a novel mammalian protein kinase structurally related to Ste20 and SPS1, but is not involved in the known MAPK pathways. Oncogene. 1997; 14:2047-57. https://doi.org/10.1038/sj.onc.1201043.
2. Pombo CM, Bonventre JV, Molnar A, Kyriakis J, Force T. Activation of a human Ste20-like kinase by oxidant stress defines a novel stress response pathway. EMBO J. 1996; 15:4537-46.
3. Krtolica A, Krucher NA, Ludlow JW. Hypoxia-induced pRB hypophosphorylation results from downregulation of CDK and upregulation of PP1 activities. Oncogene. 1998; 17:2295-304. https://doi.org/10.1038/sj.onc.1202159.
4. Fojas de Borja P, Collins NK, Du P, Azizkhan-Clifford J, Mudryj M. Cyclin A-CDK phosphorylates Sp1 and enhances Sp1-mediated transcription. EMBO J. 2001; 20:5737-47. https://doi.org/10.1093/emboj/20.20.5737.
5. Haidweger E, Novy M, Rotheneder H. Modulation of Sp1 activity by a cyclin A/CDK complex. J Mol Biol. 2001; 306:201-12. https://doi.org/10.1006/jmbi.2000.4406.
6. Rocco JW, Sidransky D. p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp Cell Res. 2001; 264:42-55. https://doi.org/10.1006/excr.2000.5149.
7. Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002; 2:38-47. https://doi.org/10.1038/nrc704.
8. Zygmunt A, Tedesco VC, Udho E, Krucher NA. Hypoxia stimulates p16 expression and association with cdk4. Exp Cell Res. 2002; 278:53-60. https://doi.org/S0014482702955643.
9. Hanks SK. Genomic analysis of the eukaryotic protein kinase superfamily: a perspective. Genome Biol. 2003; 4:111.
10. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003; 5:741-7. https://doi.org/10.1038/ncb1024.
11. Alcouffe C, Boutonnat J, Clement-Lacroix J, Mousseau M, Ronot X. Non-toxic and short treatment with gemcitabine inhibits in vitro migration of HT-1080 cells. Anticancer Drugs. 2004; 15:803-7. https://doi.org/00001813-200409000-00010.
12. Box AH, Demetrick DJ. Cell cycle kinase inhibitor expression and hypoxia-induced cell cycle arrest in human cancer cell lines. Carcinogenesis. 2004; 25:2325-35. https://doi.org/10.1093/carcin/bgh274.
13. Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004; 23:1949-56. https://doi.org/10.1038/sj.emboj.7600196.
14. Mellor H. Cell motility: golgi signalling shapes up to ship out. Curr Biol. 2004; 14:R434-5. https://doi.org/10.1016/j.cub.2004.05.038.
15. Merkel JS, Michaud GA, Salcius M, Schweitzer B, Predki PF. Functional protein microarrays: just how functional are they? Curr Opin Biotechnol. 2005; 16:447-52.
16. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993; 366:704-7.
17. Ragione FD, Russo GL, Oliva A, Mercurio C, Mastropietro S, Pietra VD, Zappia V. Biochemical characterization of p16INK4- and p18-containing complexes in human cell lines. J Biol Chem. 1996; 271:15942-9.
18. Martin EA, Robinson PJ, Franklin RA. Oxidative stress regulates the interaction of p16 with Cdk4. Biochem Biophys Res Commun. 2000; 275:764-7. https://doi.org/10.1006/bbrc.2000.3383.
19. Li L, Lu Y. Inhibition of hypoxia-induced cell motility by p16 in MDA-MB-231 breast cancer cells. J Cancer. 1:126-35.
20. Xue L, Wu J, Zheng W, Wang P, Li J, Zhang Z, Tong T. Sp1 is involved in the transcriptional activation of p16(INK4) by p21(Waf1) in HeLa cells. FEBS Lett. 2004; 564:199-204. https://doi.org/10.1016/S0014-5793(04)00352-7.
21. Preisinger C, Short B, De Corte V, Bruyneel E, Haas A, Kopajtich R, Gettemans J, Barr FA. YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3zeta. J Cell Biol. 2004; 164:1009-20. https://doi.org/10.1083/jcb.200310061.
22. Wang CH, Chang HC, Hung WC. p16 inhibits matrix metalloproteinase-2 expression via suppression of Sp1-mediated gene transcription. J Cell Physiol. 2006; 208:246-52. https://doi.org/10.1002/jcp.20660.
23. Pagliuca A, Gallo P, Lania L. Differential role for Sp1/Sp3 transcription factors in the regulation of the promoter activity of multiple cyclin-dependent kinase inhibitor genes. J Cell Biochem. 2000; 76:360-7. https://doi.org/10.1002/(SICI)1097-4644(20000301)76:3<360::AID-JCB3 >3.0.CO;2-1.
24. Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, et al. Global analysis of protein activities using proteome chips. Science. 2001; 293:2101-5.
25. Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003; 52:80-7. https://doi.org/10.1002/prot.10389.
26. Chen R, Weng Z. A novel shape complementarity scoring function for protein-protein docking. Proteins. 2003; 51:397-408. https://doi.org/10.1002/prot.10334.
27. Mintseris J, Pierce B, Wiehe K, Anderson R, Chen R, Weng Z. Integrating statistical pair potentials into protein complex prediction. Proteins. 2007; 69:511-20. https://doi.org/10.1002/prot.21502.
28. Baugh EH, Lyskov S, Weitzner BD, Gray JJ. Real-time PyMOL visualization for Rosetta and PyRosetta. PLoS One. 6:e21931. https://doi.org/10.1371/journal.pone.0021931.
29. Watts KS, Dalal P, Murphy RB, Sherman W, Friesner RA, Shelley JC. ConfGen: a conformational search method for efficient generation of bioactive conformers. J Chem Inf Model. 50:534-46. https://doi.org/10.1021/ci100015j.
30. Moreno S, Ibraghimov-Beskrovnaya O, Bukanov NO. Serum and urinary biomarker signatures for rapid preclinical in vivo assessment of CDK inhibition as a therapeutic approach for PKD. Cell Cycle. 2008; 7:1856-64. https://doi.org/6055.
31. Nogueira E, Fidalgo M, Molnar A, Kyriakis J, Force T, Zalvide J, Pombo CM. SOK1 translocates from the Golgi to the nucleus upon chemical anoxia and induces apoptotic cell death. J Biol Chem. 2008; 283:16248-58. https://doi.org/M70972420010.1074/jbc.M709724200.