
INTRODUCTION
Melanoma is a frequently fatal malignancy of the neural crest-derived melanocytes, the pigment producing cells in skin, uveal tract and mucosal membranes. The cause of death from melanoma is metastasis [1]. While new treatments targeting specific melanoma driver mutations such as BRAF V600E have a significant impact, the benefit of treatment is often short lived and the disease becomes rapidly fatal once resistance develops [2, 3]. Accumulating evidence indicates that the acquisition of invasive and metastatic characteristics by melanoma cells involves the reactivation of a developmental epithelial-to-mesenchymal transition (EMT)-like program [4-6]. The complex mechanisms involved in this process in melanoma remain largely unknown.
EMT describes a reprogramming of epithelial cells that leads to the loss of epithelial characteristics, notably polarity and cell adhesion, and the acquisition of a mesenchymal phenotype with increased invasive abilities. It occurs during normal development as part of processes such as gastrulation and neural crest cell migration [7]. During cancer progression, this phenotype is associated with tumor invasion, metastatic dissemination, and acquisition of resistance to drug treatment [8, 9]. Cadherin switching is a hallmark of EMT, leading to the down-regulation and replacement of the cell surface adhesion molecule E-cadherin by N-cadherin, enabling motility [10-12].
In various cancers, EMT leads to the generation of cancer cells possessing stem cell attributes of tumor-initiation and resistance to chemotherapy [8]. In melanoma, a sub-population of slow-cycling cells (defined by label-retention) which exhibit efficient tumor-initiating capacity, have properties associated with stemness, and are resistant to various anticancer drugs has been identified [13].
Thrombospondins comprise a family of homologous proteins that regulate cellular phenotype and extracellular structure during tissue genesis and remodelling. Thrombospondin 1 (THBS1) was the first member to be identified, and has been shown to modulate tumor progression and metastasis [14, 15]. Whereas, the role of THBS1 in angiogenesis in melanoma is well documented, its role in tumor metastasis is only just emerging [16, 17]. THBS1 has been identified as a major physiological activator of transforming growth factor (TGF) -beta, a potent elicitor of EMT [18, 19]. However, a role of THBS1 in mediating EMT in melanoma remains to be elucidated. This study therefore aimed to reveal the functional roles of THBS1 during melanoma progression by assessing THBS1 expression and its effects on the functional characteristics of melanoma cells, particularly those associated with mesenchymal transformation.
RESULTS
Melanoma cells exhibiting a mesenchymal phenotype express high levels of Thrombospondin 1 associated with TGF-beta signaling
At the molecular level, EMT in melanoma cells is characterized by a series of coordinated changes including down-regulation of the adherens junction molecule E-cadherin and upregulation of N-cadherin [5, 20]. These changes in EMT markers are often associated with functional change toward an invasive phenotype [21]. We evaluated the expression of classical EMT genes, E- and N-cadherins using quantitative real-time RT-PCR (qRT-PCR) in a panel of 54 human melanoma cell lines that were derived from resected melanoma metastases [22]. Expression patterns of these two molecules in the cell lines varied from high N-cadherin with no E-cadherin expression, high E-cadherin with no or low N-cadherin, to intermediate levels of both (Figure 1A).
We divided the lines into those expressing E-cadherin and those lacking E-cadherin expression, and their gene expression patterns were compared using previously generated whole genome microarray expression profiling data [22]. 634 probes representing 552 genes were differentially expressed between the two classes of cell lines (Supplementary Table S1). As expected E-cadherin expression was higher in the lines identified as E-cadherin expressing by qRT-PCR (13.5 fold), and N-cadherin expression was higher in the lines lacking E-cadherin expression by qRT-PCR. A principal components analysis based on the differential expression gene list largely segregated the two classes of cell lines, although with some overlap between lines with intermediate levels of E- and N-cadherin, perhaps representing a mixed phenotype (Supplementary Figure S1). A gene-set enrichment analysis (GSEA) of the cell lines revealed gene sets associated with TGF-beta signaling [23], cell migration [24, 25], ECM modulation [26] and EMT [27-29]. Significantly enriched gene sets can be found in Supplementary Table S2. Based on the GSEA results, and the evidence of opposing E- and N-cadherin expression, we therefore labelled the cell lines mesenchymal- and epithelial-like.
We chose to focus on Thrombospondin 1 (THBS1), which showed 19 fold higher expression in the mesenchymal-like cells. qRT-PCR confirmed THBS1 mRNA expression levels were higher in mesenchymal-like cells (Figure 1B). A subset of high and low THBS1 expressing cell lines, as determined by qRT-PCR, was subjected to solid-phase ELISA. This detected little or no THBS1 secretion in conditioned medium from epithelial-like cells, whereas mesenchymal-like cells secreted significant amounts of THBS1 into the medium (Figure 1C).
As THBS1 is a known activator of TGF-beta [18] and TGF-beta has a pivotal function in the progression of EMT [30, 31], we examined the level of TGF-beta secretion in a subset of high and low THBS1 secreting melanoma cell lines. THBS1 high cell lines secreted high TGF-beta1 in contrast to THBS1 low cell lines that secreted no TGF-beta1 into the medium (Supplementary Figure S2A). To extend these findings, we analyzed a cutaneous melanoma dataset available from The Cancer Genome Atlas (TCGA) (http://www.cbioportal.org) [32, 33]. Mutual exclusivity data from 376 melanoma patients revealed that both THBS1 and TGF-beta1 are co-expressed (Odds ratio =3.4, p=0.036, Fisher’s exact test). TGF-beta1 treatment in two epithelial-like melanoma cell lines induced THBS1 expression in a time-dependent manner (Supplementary Figure S2B). These data indicate that THBS1 expression is associated with TGF-beta signaling in melanoma cells.
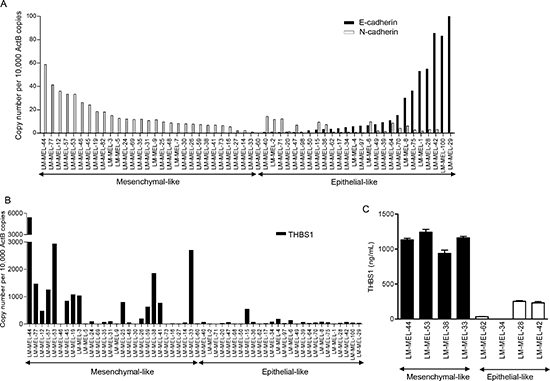
Figure 1: Classification of a panel of melanoma cells lines based on gene expression. qRT-PCR analysis revealed expression of (A) E- and N-cadherins and (B) THBS1 in a panel of 54 melanoma cell lines. (C) Media from 4 mesenchymal- and 4 epithelial-like melanoma cells were collected and subjected to THBS1 ELISA. ANOVA analysis of the two sets of cell lines was significant (** p<0.005).
Thrombospondin 1 expression negatively correlates with differentiation markers of melanoma
De-differentiation, which is characterized by the loss of expression of genes involved in pigmentation, is often associated with aggressive phenotype in melanoma [6, 33]. A key determinant of melanoma differentiation sub-population identity is conferred by the expression and activation of microphthalmia-associated transcription factor (MITF). In melanoma, the induction of MITF expression promotes expression of differentiation markers and the inhibition of invasion [35]. Differentiation markers MelanA (MLANA) (fold change: 37), Tyrosinase (TYR) (fold change: 27), and MITF (fold change: 10) were up-regulated in the epithelial-like cells examined. qRT-PCR analysis showed expression of melanoma differentiation markers MLANA, TYR and MITF in the panel of 54 melanoma cell lines. Regression analysis revealed an inverse correlation of THBS1 expression with all three genes tested (p<0.005 in each case) (Supplementary Figure S3). This demonstrates the association between THBS1 expression and a de-differentiated phenotype in melanoma.
Thrombospondin 1 expression is enriched in label-retaining melanoma cells
Others have described label retention as an assay for slow-cycling melanoma cells required for continuous tumor maintenance [13]. To assess whether THBS1 expression is associated with label-retaining cells (LRC) in melanoma, we isolated these cells by using CM-Dil dye, a membrane labeling carbocyanine dye that was distributed equally between daughter cells with each cell division (Figure 2A). After culturing the labelled cells for two weeks, the majority of cells diluted the label to undetectable levels, while a sub-population of cells continued to be brightly labelled (Figure 2B). To ensure that these LRC were not an in vitro artifact of the culture conditions employed, we labelled melanoma cells freshly isolated from the ascites of a patient with advanced metastatic melanoma and immediately injected them subcutaneously into the flank of NOD/SCID mice. After three weeks of tumor growth, a small percentage of LRC could be visualized in a paraffin-embedded xenograft specimen (Figure 2C).
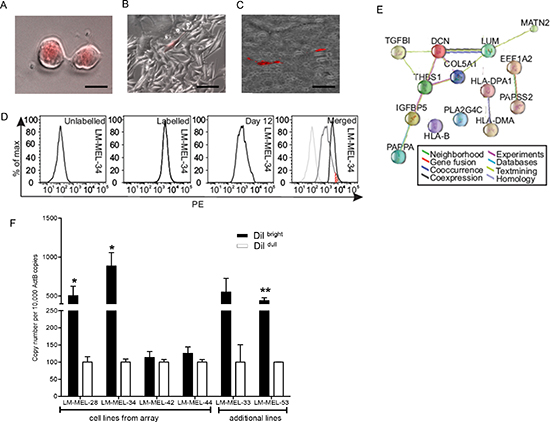
Figure 2: Expression of THBS1 in LRC sub-population within melanoma cell lines. Melanoma cell line, LM-MEL-34 labelled with CM-DiI was imaged after (A) 2 days (scale bar = 50μm) or (B) 14 days (scale bar = 100μm), respectively. (C) Melanoma cells from a patient with malignant melanoma were labelled with CM-Dil and injected subcutaneously into the flank of three animals. After three weeks, tumors were harvested and embedded in paraffin, cut into sections and examined by fluorescent microscopy (scale bar = 500μm). (D) Example of a flow profile of LM-MEL-34 cells unstained, 1 hour after CM-Dil labeling, 12 days after labeling, and merged histogram overlay (from left to right). The red striped area represents the population of cells referred to as LRC. LM-MEL-28, −34, −42 and −44 were sorted into Dilbright and Dildull cells 14 days after labeling with CM-Dil. RNA was subjected to genome-wide gene expression analysis using Illumina HT12 arrays. (E) Upregulated genes were employed to generate an evidence-based protein-interaction network in STRING and demonstrated connections between several ECM-related molecules with higher expression in the Dilbright cells. (F) Dilbright and Dildull cells from the same 4 lines and 2 additional cell lines were separated and qPCR was performed to evaluate expression of THBS1. Values are mean +/− SEM of three experiments in triplicate (* p<0.05, ** p<0.005).
The ‘label-retaining’ phenotype was present in all melanoma cell lines tested (n=10) in vitro and could be visualized by flow cytometry (Figure 2D). Fourteen days after labeling, a small population of cells showed signal intensities close to those measured directly after labeling (Supplementary Figure S4). LRC were therefore FACS-sorted from 4 cell lines based on dye-retention (Dilbright) and subjected to a whole genome expression analysis. Genes significantly up- or down-regulated are listed in Supplementary Table S3. THBS1 was significantly up-regulated in the dye-retention population. Other up-regulated genes were associated with EMT and secretory functions. THBS1 and other proteins enriched in LRCs can be connected to each other based on published associations and analyzed by STRING (Figure 2E). We confirmed significant up-regulation of THBS1 by qRT-PCR performed on Dilbright cells derived from four melanoma cell lines on the array. This was validated independently on two additional cell lines (LM-MEL-33 and LM-MEL-53) (Figure 2F). A gene-set enrichment analysis (GSEA) of this dataset identified gene sets associated with pertinent cell types and biological functions, notably; EMT [27-29], TGF-beta signaling [23], invasion [24, 25], and chemotherapy-resistant cancer cells [36, 37] (Supplementary Table S4).
Thrombospondin 1 expression is associated with increased melanoma cell invasion
Invasion is an important functional change that accompanies EMT [21]. The role of THBS1 in melanoma cell invasion was evaluated using mesenchymal- and epithelial-like melanoma cells in matrigel based transwell invasion assays. THBS1 expressing mesenchymal-like lines were highly invasive. Conversely, epithelial-like cells were less invasive and showed little or no THBS1 expression (Figure 3A).
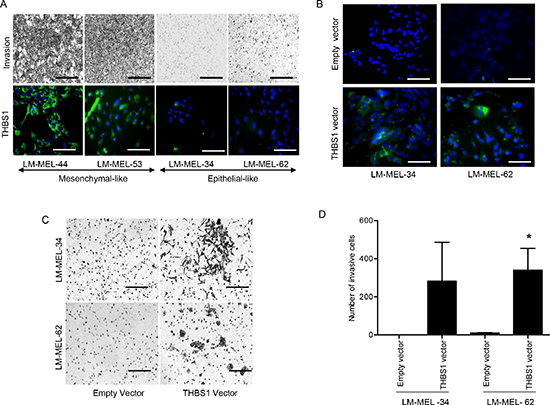
Figure 3: THBS1 expression induces melanoma cell invasion. (A) Mesenchymal- and epithelial-like melanoma cells were subjected to invasion through matrigel-coated Boyden chambers. After 48 hours cells were fixed and images of invasive cells were taken (scale bar = 500μm) or stained using THBS1 antibody in conjunction with an Alexa-488 conjugated secondary antibody and visualized with a fluorescent microscope (scale bar = 100μm). (B) THBS1 protein expression in two clonally derived LM-MEL-34 and LM-MEL-62 cell lines and respective empty vector control was detected using immunostaining (scale bar = 100μm). (C) Invasiveness of epithelial-like cells with empty vector control or THBS-1 expression vector was determined (scale bar = 100μm). (D) The graphs show the total number of invasive cells counted. Values are mean +/− SEM of three experiments in triplicate (* p<0.05).
To confirm the role of THBS1 in invasion, a THBS1 expression construct was generated and transfected into epithelial-like cells LM-MEL-34 and LM-MEL-62. A stable clone overexpressing THBS1 was established for each line. Overexpression of THBS1 protein in these epithelial-like melanoma cells was confirmed by immuno-staining (Figure 3B), and greatly enhanced their invasive ability (Figure 3C&D).
In vitro and in vivo silencing of Thrombospondin 1 inhibits invasion of melanoma cells
RNA interference in vitro with two different siRNAs was used to evaluate the role of THBS1 in invasion. siRNA treatment silenced THBS1 expression at mRNA level over 80% in LM-MEL-44, −53, −33 and −38 (Figure 4A). Immuno-staining confirmed reduction in THBS1 protein expression following knockdown with siRNA (Supplementary Figure S5A). ELISA also confirmed reduced secretion of THBS1 following knockdown (Supplementary Figure S5B).
Knockdown of THBS1 in more epithelial-like melanoma cells expressing THBS1 and some epithelial markers, showed significant increase in E-cadherin levels (Supplementary Figure S5C&D). This restoration in E-cadherin levels was associated with a parallel increase in expression of MLANA, TYR and MITF (Supplementary Figure S5E, F&G). Analysis of the cutaneous melanoma dataset consisting of 376 melanoma patients, available from TCGA revealed an increase in mRNA expression of THBS1 in 3% of patients. This altered subset of patients showed significantly low E-cadherin protein expression compared with patients with unaltered THBS1 expression (Supplementary Figure S5H). These data confirm the inverse correlation between THBS1 expression and E-cadherin expression that was observed in our melanoma cell line studies in a large clinical tumor dataset.
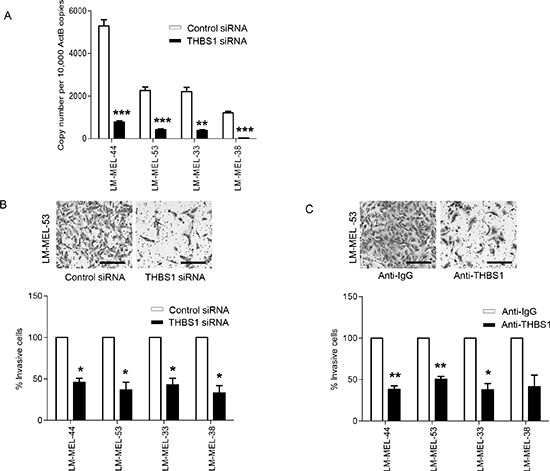
Figure 4: Targeting THBS1 results in loss of invasive potential of melanoma cells in vitro. (A) Melanoma cells were plated out and transfected with either 20nM control siRNA or THBS1 specific siRNA. After 72 hours RNA was extracted and THBS1 qRT-PCR was performed. (B) Melanoma cells were transfected as described and representative images of invasion in LM-MEL-53 are shown after 24 hours (scale bar = 100μm). The graphs show the total number of invasive cells counted and cell numbers from control were set to 100% and compared to THBS1 siRNA transfected cells. (C) Invasiveness of melanoma cells after treatment with control anti-IgG or anti-THBS1 antibodies was tested, images captured (scale bar = 100μm) and quantified as above. Values are mean +/− SEM of four independent experiments in triplicate (* p<0.05, ** p<0.005, *** p<0.0005).
Decreased invasion was observed in all cell lines transfected with THBS1 siRNA, as compared with the control siRNA treated cells (Figure 4B). To further confirm the role of THBS1 in melanoma invasion, melanoma cells were treated with an anti-THBS1 antibody and subjected to invasion assays. Antibody treatment decreased the number of invaded cells in all four cell lines tested (Figure 4C). Thus, loss of THBS1 contributed to the restoration of E-cadherin levels and inhibition of invasion in melanoma cells.
We assessed THBS1 function in an in vivo model of melanoma cell plasticity and invasion, the chick embryo. The model involves the injection of melanoma cells into the neural tube of developing chicken embryos where the cells acquire a motile phenotype and follow the migratory path of their ancestral cell types into the surrounding tissues. It therefore enables assessment of functions that are relevant to the metastatic process such as invasion, cellular polarity and cellular positioning [38, 39]. Melanoma cells were transfected with siRNA targeting THBS1, cultured as a hanging drop for 24 hours and introduced into the trunk neural tube of a developing chick embryo. THBS1 siRNA treated cells demonstrated a significant reduction in emigration from the neural tube in vivo into the surrounding tissue (Figure 5A&B). Cross-sections of chick embryos confirmed that numerous control siRNA treated cells migrate out of the neural tube in contrast to THBS1 siRNA treated cells that predominantly remain at the site of injection (Figure 5C). These results suggest a role for THBS1 in melanoma invasion in vivo in response to physiological environmental signals.
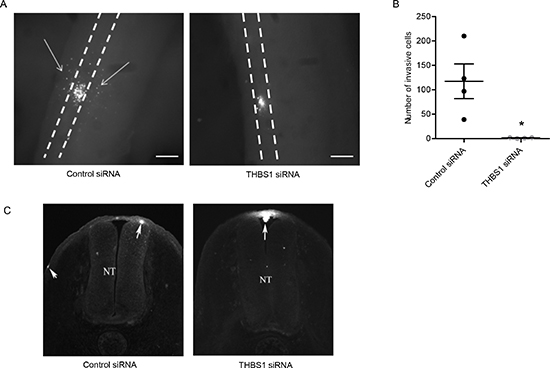
Figure 5: RNAi mediated loss of THBS1 blocks motility in in vivo chick model. Melanoma cells were treated with CM-DiO, transfected with the indicated constructs and cultured as hanging drops to encourage aggregate formation. Similar sized aggregates were introduced into the neural tube of developing chicken and re-incubated within the egg for 2 days. (A) Embryos were harvested and fluorescence pictures from whole-mounts taken (scale bar = 50μm). White dotted line shows the outline of the neural tube and the white arrows indicate melanoma cells that migrated out of the neural tube and into the surrounding tissue. (B) Analysis of cell numbers infiltrating the surrounding tissue from several independent experiments (n=4 for control siRNA and THBS1 siRNA) (* p<0.05). (C) Images from cross-sections of embryos. White arrows indicate melanoma cells and NT denotes neural tube, dorsal is to the top.
Thrombospondin 1 is upregulated in a subset of drug resistant melanoma cell lines
Drug resistance can be associated with the EMT process [4]. To examine whether THBS1 expression was associated with the acquisition of drug resistance, six melanoma cell lines were treated with chemotherapy agents. qRT-PCR showed induction of THBS1 expression in melanoma cells treated with cytotoxic chemotherapy agents, such as taxol, cisplatin and 5-flurouracil after 72 hours (Supplementary Figure S6A). Next, we assessed whether THBS1 might play a role in therapies specifically targeting activated BRAF. To relate our results to findings from studies with BRAF V600E-specific inhibitors, we clustered gene expression microarray data from a publication examining mechanisms of resistance to the activated BRAF-inhibitor PLX4032 by analysing the gene set that we identified to be up-regulated in the mesenchymal-like cells (Figure 2E & Supplementary Figure S6B) [40]. Data from two of PLX4032 resistant cell lines (M229 and M238) clustered together and demonstrated up-regulation of mesenchymal-associated genes including THBS1 in comparison to the parental cell lines (Supplementary Figure S6B). Both lines have previously been described as undergoing a change in morphology characteristic of EMT during the acquisition of resistance, whereas the cell line that did not up-regulate the LRC genes (M249) acquired resistance via a genetic mutation of NRAS and did not undergo morphological change [40]. These data show that in some cases BRAF-resistance is accompanied by an induction of THBS1 expression and acquisition of a mesenchymal gene expression pattern.
We next established two PLX4720 resistant cell lines from the V600E BRAF mutated parental lines by continuous culture in the presence of 5μM or 1μM of the drug over 10 weeks (LM-MEL-28 R1 and LM-MEL-64 R3). qRT-PCR and ELISA revealed that the expression and secretion of THBS1 was higher in LM-MEL-28R1 and LM-MEL-64R3, when compared with the drug sensitive parental cell lines (Figure 6A&B). Furthermore, depletion of THBS1 with siRNA treatment resulted in significant restoration of drug sensitivity in the resistant cell lines (Figure 6C&D). These data show that inhibition of THBS1 can impact on sensitivity to drugs that act via an independent signaling pathway. This raises the possibility that the THBS1/TGF-beta axis may enable drug resistance though a mechanism mediated by cellular plasticity and unrelated to the commonly described alternative MAPK signaling mechanisms.
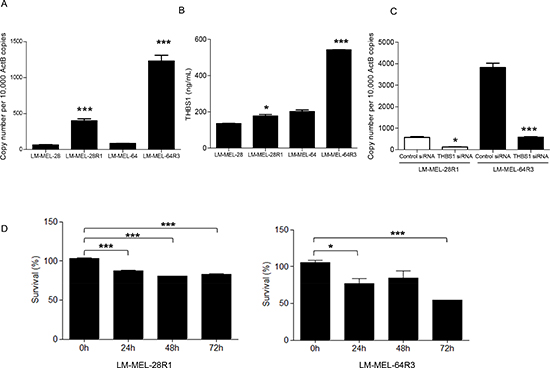
Figure 6: Melanoma cells surviving treatment with BRAF V600E inhibitor up-regulate THBS1. THBS1 expression by qRT-PCR (A) and secretion by ELISA (B) in PLX4720 resistant LM-MEL-28R1 and LM-MEL-64R3 melanoma cell lines relative to their sensitive parental lines was determined. (C) LM-MEL-28R1 and LM-MEL-64R3 melanoma cell lines were treated with control or THBS1 siRNA and knockdown was evaluated with qRT-PCR and (D) percentage survival was quantified from absorbance obtained from MTS measurement. Bars are mean values+/− SEM from three independent experiments in triplicate (* p<0.05, ** p<0.005, *** p<0.0005).
Thrombospondin 1 is expressed in metastatic melanoma patient tumors
THBS1 protein expression patterns in melanoma tumors were determined by immunohistochemical staining of a tissue microarrays (TMA) comprising of tumors from 103 patients with stage III and IV metastatic melanoma. THBS1 expression was detected in 61% of metastatic melanoma patient tumors (Figure 7A&B). The subcellular location was identified as predominantly cytoplasmic.
DISCUSSION
This study examined the roles of THBS1, a gene which is highly expressed in mesenchymal-like but not or at lower levels in epithelial-like melanoma cell lines. THBS1 was associated with increased invasion and drug resistance. THBS1 expression was also increased in label-retaining melanoma cell sub-populations. Inhibition, either with neutralizing antibody or RNAi, reduced invasion of melanoma cells both in vitro and in vivo. RNAi mediated loss of THBS1 also restored drug sensitivity in BRAF V600E mutated cells that developed drug resistance following exposure to a BRAF inhibitor.
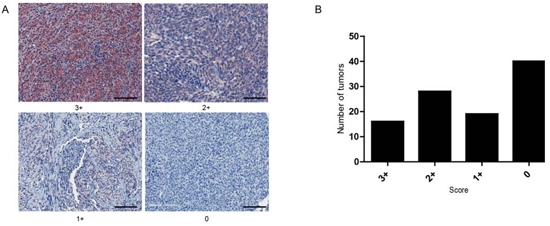
Figure 7: THBS1 immunostaining in melanoma tumor tissue. (A) Localization of THBS1 in metastatic melanoma tumor biopsies. Representative tumors show score staining intensities (scale bar = 50μm). (B) Graph shows number of tumors scored.
Phenotypes similar to the epithelial- and mesenchymal-like melanoma cells described here have been demonstrated by Hoek et al., who classified melanoma cell lines based on invasion, high proliferation rate, and susceptibility to growth inhibition by TGF-beta treatment. Cell lines unaffected by TGF-beta were in general invasive and lost expression of MITF and associated differentiation markers. Switching between these proliferative and invasive phonotypes was proposed as a mechanism for tumor progression [41]. As these sub-populations conform to the EMT dichotomy, the Hoek phenotype switching model can be seen as a variant of EMT occurring in a non-epithelial cancer. Induction of EMT is often associated with wide-spread transcriptional changes via the activation of specific transcription factors and epigenetic regulatory mechanisms [9]. Recent research showed that a reversible EMT-TF (transcription factor) reprogramming involving upregulation of ZEB1/TWIST1 paralleled by downregulation of SNAIL2/ZEB2 occurs in human melanoma. This EMT-TF reprogramming is followed by the reduction in the expression of MITF [42] which may lead to the establishment of functional divergent melanoma cell populations based on the “rheostat model” [43]. In our system, THBS1 is highly expressed in mesenchymal-like cells that express low MITF and display an invasive phenotype. This contrasts with earlier studies where low levels of THBS1 expression have been associated with increased recurrence rates and decreased overall survival rates in several human cancers [44].
Importantly, THBS1 is a multifunctional protein with different functional and structural domains and has a variety of biological activities [15, 44]. Due to its several binding partners, the sum of downstream signals derived from the molecules occupied by the full-length protein may result in different consequences for the cell depending on the availability of THBS1 interacting proteins [45]. THBS1 has been previously described as an inhibitor of angiogenesis and tumourigenesis [17, 46] and mimetics of THBS1 have been trialled as anti-angiogenic agents [45]. It is conceivable that targeting angiogenic factors with non-redundant roles can achieve not only preventing tumor angiogenesis and metastasis, but also hindering the direct growth and invasion of tumor cells. Different approaches have been proposed to exploit protein-protein interactions in order to develop novel inhibitor peptide sequences or non-peptide, small molecule mimetics of THBS1 domains that block angiogenesis [45].
For instance, a THBS1 derived anti-angiogenic peptide ABT-510 has been tested clinically and failed to show antitumor effects in melanoma and sarcoma patients in phase II clinical trials [47, 48]. Most notably, we show a role for THBS1 in TGF-beta regulation indicating therapeutic potential beyond inhibiting angiogenesis. This may provide opportunities for therapeutic targeting despite the clinical failure of the THBS1-derived anti-angiogenic peptide. As with a wide range of molecules in cancer, an anti-angiogenic molecule could well lead to hypoxia in tumors and as a consequence the induction of an aggressive phenotype resulting ultimately in de-differentiation and metastasis [49-52].
THBS1 can be epigenetically silenced in melanomas, marking it as a potential tumor-suppressor gene [46]. Contrasting, THBS1 has been described as an immune modulator, both through the activation of regulatory T cells, and by facilitating the activation of TGF-beta, an immunosuppressive cytokine. A recent study showed that the transcription factor SNAIL induces EMT in melanoma and leads to an increase in THBS1 expression and results in immunosuppression and enhanced metastasis [16]. Similarly, we demonstrated that in vivo THBS1 knockdown abrogates the invasive/plastic potential of melanoma cells within the chicken neural tube. The avian embryo has emerged as a useful site for analysing melanoma cell behaviour within a developmental microenvironment since it provides imaging and surgical accessibility to manipulate and monitor the transplanted tumor [53-55]. Additionally, the biology of cells within the neural crest is well understood and since melanocytes are neural-crest-derived cells, it is likely that the environmental signals in this model are pertinent to melanoma cell motility. Thus the interaction between melanoma and motile embryonic neural crest cells provide a functional context, and thereby a valuable model for interrogating the molecular regulation of migrating melanoma [38, 39].
Although we have clearly demonstrated that THBS1 mediates enhanced melanoma invasion in vitro and in vivo, previous studies that evaluated the role of THBS1 in cancer cell invasion have yielded mixed results. Although THBS1 expression was inversely correlated with cell invasion in thyroid cancer in one study [56], other studies reported that THBS1 promotes cell invasion in breast cancer, thyroid cancer, colon cancer and prostate tumors [57-59]. In addition, knock-out of THBS1 in an animal model of breast cancer led to growth of the primary tumor but a decrease in the number of metastases [61]. Thus, the pathological roles and clinical significance of THBS1 may depend on the nature of the model and on the stage of tumor progression. To better evaluate role of THBS1 in human melanoma, we determined its expression in metastatic melanoma tumor biopsies. THBS1 protein was located in tumor cells and was undetectable in stroma. This contrasts with gastric cancer, where THBS1 localized to stroma [62], indicating apparent tissue-specific differences depending on cancer type.
The mechanism whereby THBS1 promotes melanoma invasion remains poorly understood, however we demonstrate that this is likely linked to TGF-beta activation. TGF-beta 1 is secreted in a biologically inactive latent form and activation of this latent TGF-beta 1 is required for biological activity [19]. THBS1 has been identified as a major physiological activator of TGF-beta 1 both in vivo and in vitro [18, 63]. The interaction between tripolymer THBS1 and latent TGF-beta 1 causes a conformational change in the latent complex and exposes epitopes that are critical for binding to the cellular receptor. Our data show that THBS1 and TGF-beta 1 is expressed and secreted by mesenchymal-like melanoma cells and further, that THBS1 is inducible in epithelial-like cells upon exposure to TGF-beta 1. The interplay between these molecules supports a role for positive feedback within the tumor and extends the observations of others that TGF-beta 1 induces EMT in melanoma [21, 31] and that THBS1 is a TGF-beta 1 activator [18]. We propose that THBS1 promotes EMT in melanoma through activation of latent TGF-beta 1 during the progression of melanoma.
Finally we demonstrate a striking association between the expression of THBS1 and drug resistance, not only to cytotoxic drugs, but also in melanoma cells resistant to BRAF inhibition. The list of genetic and non-genetic changes that are associated with resistance to the targeted MAPK inhibitors continues to grow and our data adds to this with the clear demonstration that THBS1 can play a functional role in BRAF inhibitor resistance. Although a direct mechanism cannot readily be implied from an understanding of its various biological specificities, an indirect role can readily be explained via TGF-beta-induced EMT, or through association with the LRC phenotype. Slow cycling cells have recently been associated with BRAF inhibitor-resistance and melanoma invasiveness [13]. In our studies THBS1 expression in LRC was associated with an EMT-like phenotype and potentially implicates TGF-beta pathway activation in LRC to mediate tumor progression. These findings contrast with those of Roesch et al., who did not identify EMT possibly explained by differences in the experimental systems utilized. Nonetheless it is clear that further studies of LRC, THBS1 and the role of TGF-beta signaling will be critical to better understand heterogeneity, plasticity and the mediators of phenotype switching as contributors to treatment failure. As a central player in these processes, THBS1 well may serve as a target in strategies to better treat malignant melanoma.
MATERIAL AND METHODS
Cell culture
Melanoma cell lines were established from resected melanoma metastases by mechanical dissociation of tissue with subsequent overnight digestion in media containing collagenase IV at 37°C. Established cell lines were Mycoplasma-tested using the MycoAlert test (Lonza Rockland, Inc., USA). All tissue donors provided written informed consent for tissue collection and research, which was covered by protocols approved by the Austin Health Human Research Ethics Committee, Melbourne, Australia (approval number H2012/04446). All cell lines were matched with their donors by HLA-typing. Cells were cultured in RPMI1640 supplemented with 10% fetal calf serum (FCS) as described previously [22].
qRT-PCR
RNA for qPCR was extracted using the RNEasy kit (Qiagen, Germany) or the Acturus® RNA Picopure® kit (Life Technologies, USA). Reverse transcription was carried out using the High Capacity cDNA RT kit (Applied Biosystems, Life Technologies, USA). Following reverse transcription, qRT-PCR was performed using SYBR Green (Qiagen, Germany). Beta-Actin (ActB) was used as internal control. Following primers were used: ActB (forward) 5’-ctg gaa cgg tga agg tga ca-3’ and (reverse) 5’-cgg cca cat tgt gaa ctt tg-3’, E-cadherin (forward) 5’-gcc gag agc tac acg ttc a-3’ and (reverse) 5’-gac cgg tgc aat ctt caa a-3’, N-cadherin (forward) 5’-ctc cat gtg ccg gat agc-3’ and (reverse) 5’-cga ttt cac cag aag cct cta c -3’, THBS1 (forward) 5’- caa tgc cac agt tcc tga tg-3’ and (reverse) 5’-tgg aga cca gcc atc gtc-3’, TYR (forward) 5’-gct gcc aat ttc agc ttt aga -3’ and (reverse) 5’-ccg cta tcc cag taa gtg ga -3’, MLANA (forward) 5’-gag aaa aac tgt gaa cct gtg gt -3’ and (reverse) 5’-gac tgt tct gca gag agt ttc tca t -3’, MITF (forward) 5’-cat tgt tat gct gga aat gct aga -3’ and (reverse) 5’-tgc taa agt ggt aga aag gta ctg c -3’.
Gene-expression arrays and data analysis
RNA was analysed on Illumina HT-12 v3 arrays at the Australia Genome Research Facility (AGRF, Australia). Raw data were read in to the R environment for statistical computing (http://www.r-project.org/) using the limma package [64], background was corrected using the normexp function, and log2 transformed and quantile normalized. Differential expression for the cell line panel was determined using the ANOVA tool in Partek Genomics Suite (Partek GS). Differential expression for the smaller label-retaining data set employed the RankProd package for differential expression analysis, using a percentage of false positive (PFP – approximation of false discovery rate (FDR)) cut-off of 5% to determine statistical significance [65]. Gene-set enrichment analysis (GSEA) settings were; gene set permutation, a FDR of 5%, and the MSigDB 4.0 database (category C2). Lists of differentially expressed genes were used to create network diagrams in STRING [66], with connections between molecules based on different levels of molecular interaction evidence, such as known physical interaction, correlated gene expression, and literature mining. Microarray data from Nazarian R et al. were obtained from NCBI GEO (accession GSE24862). Affymetrix CEL files were normalized by RMA [67] in Partek GS. Principal components analysis and hierarchical clustering of all datasets were performed in Partek GS, with hierarchical clustering using average linkage and Pearson’s dissimilarity as a distance metric.
Solid phase ELISAs
THBS1 and TGF-beta 1 protein amount in cell line supernatants was measured by Quantikine human Thrombospondin 1 (R&D Systems, USA) or TGF-beta1 immunoassay kit (Life Technologies, USA) as per the manufacturer’s instructions.
TGF-beta 1 treatment
LM-MEL-42 and LM-MEL-34 was treated with 5ng/ml TGF-beta 1 (Pepro Tech Inc.,USA). RNA was extracted 24, 48 and 72 hours following treatment.
VybrantDye labeling
LM-MEL-44, LM-MEL-28, LM-MEL-33, LM-MEL-34, LM-MEL-42 and LM-MEL53 were stained with 5μl of VybrantDye CM-Dil (Life technologies, USA) for 25min at 37°C/5% CO2.
Flow cytometry and sorting
Melanoma cell lines were stained with Pacific-Blue (Life Technologies, USA) according to the manufacturer’s protocol and flow cytometry was performed using a FACSCanto II. Cell sorting utilized a FACSAria III (all Becton, Dickinson and Company, USA).
Animal experiments
Ethical approval was provided by the Austin Health Animal Ethics Committee. Ascites cells from the peritoneal fluid of a patient with advanced stage IV melanoma were confirmed as melanoma by a pathologist in the Austin Hospital. Red blood cells and debris were removed with Ficoll-Plaque centrifugation. Cells were stained with CM-Dil, resuspended in PBS and injected subcutaneously in mice (n=3). Tumor growth was monitored three times a week, and mice were sacrificed after three weeks. Tumors were removed and paraffin-embedded.
Invasion assay
Invasion assays were performed in Boyden chamber inserts with Matrigel coating (Becton, Dickinson and Company, USA). Insert membranes were stained with 4’, 6-diamidino-2-phenylindole (DAPI) or a 0.1% crystal violet solution (Sigma, USA). Cells were photographed with a monochromatic Olympus camera. The total number of invaded cells was counted from three representative fields of view per chamber, at 10× magnification.
THBS1 over-expression construct
The plasmid encoding THBS1 was obtained from Sino Biological Inc. Cells were transfected using Lipofectamine 2000 (Life Technologies, USA). Stably transfected cells were selected for and maintained in media containing 0.6mg/ml G418, with colonies isolated using cloning cylinders (Merck, USA).
THBS1 knockdown and immunofluorescence
For transient siRNA transfection, cells at 30% confluence were transfected using a control siRNA and two different Silencer select siRNAs targeting THBS1 (s14100 and s14098) at 20nM final concentration (Ambion, USA) with Lipofectamine RNAiMAX according to the manufacturer’s protocol (Invitrogen, USA). Cells were incubated with siRNA complex for 48 hours and then fixed with 4% paraformaldehyde, stained with anti-THBS1 antibody (A6.1, NB100-2059, Novus Biologicals, USA) was applied at 2.5μg/mL concentration overnight at 4°C and with Alexa flour 488 conjugated secondary antibody for 45 minutes at room temperature (Molecular probes, USA). Cells were counter stained with DAPI for 10 minutes.
In vivo chick embryo model
Melanoma cells were treated with THBS1-specific siRNAs or scrambled control siRNA as described and labelled with CM-DiO or Dil as per manufacturer’s instructions (Invitrogen, USA). Cells were grown overnight in a hanging-drop fashion to allow the formation of aggregates. Fertile chicken eggs were incubated at 38°C for 48 hours prior to transplantation. Cell aggregates consisting of 50-400 cells were harvested and carefully injected with a glass pipette into the trunk neural tube lumen of developing chicken embryos. The eggs were then sealed with adhesive tape and re-incubated for 2 days. After incubation, embryos were removed from the eggs and fixed with 4% paraformaldehyde and whole mounts or cross sections were analyzed for the localization of melanoma cells using Lumar V12 Zeiss microscope.
Treatment with chemotherapy agents
Melanoma cells were treated with cytotoxic chemotherapy agents, such as Taxol (40nM), 5-Flourouracil (150μM) or Cisplatin (1μM) or DMSO (all Sigma Aldrich Pty Ltd., USA) for 72 hours.
Generation of PLX4720 resistant cell lines
LM-MEL-28 and LM-MEL-64 were grown in media with 5μM or 1μM PLX4720 (Scientifix Pty Ltd, Australia) for 10 weeks; media was exchanged every third day. Parental cell lines treated with DMSO served as control. Cells for RNA extraction were harvested at 80% confluency.
Proliferation assays
10,000 cells per well were plated out in 96 well plates and treated as indicated. Relative cell numbers were measured using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Corporation, USA).
Immunohistochemistry and pathological evaluation
Paraffin embedded tissue slides were deparaffinised and rehydrated, endogenous peroxidise activity was blocked with 3% Hydrogen peroxide, antigen retrieval was performed in 10mmol/L citrate buffer, and nonspecific binding was blocked with blocking reagent. THBS1 antibody (A6.1, NB100-2059, Novus Biologicals, USA) was applied at 6μg/mL concentration and incubated overnight at 4°C, followed by 60 minute incubation with secondary anti-mouse antibody HRP (Dako). The chromogen used was 3-amino-9-ethylcarbazole (AEC). Human placenta was used as the positive control for THBS1 and a negative control, for which the primary antibody was substituted with the same concentration of mouse IgG. Slides were scanned using a ScanScope XT (Aperio) and immunohistochemical reactivity was evaluated by two independent investigators. The expression of THBS1 was categorized into four grades. They were arbitrarily scored as 3, strong staining; 2, moderate staining; 1, weak staining and 0, no staining.
Statistical analysis
All statistical comparisons of data sets were performed using Student’s two-tailed t-test in Prism software version 5.00 (GraphPad Software Inc).
ACKNOWLEDGEMENTS
We would like to thank Dr. Robert Weinberg (MIT Ludwig Center for Molecular Oncology) for helpful comments.
We would like to acknowledge the Melanoma Research Alliance (MRA) and the Victorian State Government Operational Infrastructure Support Program for partial funding of this project. AB and AJ were supported by Cure Cancer Australia Foundation Fellowships. JC was supported by Australian National Health and Medical Research Council Practitioner Fellowship. The establishment of an expression profile across 58 melanoma cell lines was funded by a grant from the Austin Hospital Research Foundation to AB.
Disclosure of Potential Conflicts of Interest:
No potential conflicts of interest are disclosed
REFERENCES
1. Miller AJ and Mihm MC. Melanoma. N Engl J Med. 2006; 355:1. 51–65.
2. Lito P, Rosen N and Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013; 19:11. 1401–1409.
3. Villanueva J, Vultur A and Herlyn M. Resistance to BRAF inhibitors: unraveling mechanisms and future treatment options. Cancer Res. 2011; 71:23. 7137–7140.
4. Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J, Hutchinson P, Tse G, Lachuer J, Puisieux A, Pringle JH, Ansieau S and Tulchinsky E. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell. 2013; 24:4. 466–480.
5. Kim JE, Leung E, Baguley BC and Finlay GJ. Heterogeneity of expression of epithelial-mesenchymal transition markers in melanocytes and melanoma cell lines. Front Genet. 2013; 4:97
6. Wehbe M, Soudja SM, Mas A, Chasson L, Guinamard R, de Tenbossche CP, Verdeil G, Van den Eynde B and Schmitt-Verhulst AM. Epithelial-mesenchymal-transition-like and TGFbeta pathways associated with autochthonous inflammatory melanoma development in mice. PLoS One. 2012; 7:11. e49419
7. Duband JL. Diversity in the molecular and cellular strategies of epithelium-to-mesenchyme transitions: Insights from the neural crest. Cell Adh Migr. 2010; 4:3. 458–482.
8. Scheel C and Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012; 22:5-6. 396–403.
9. Bedi U, Mishra VK, Wasilewski D, Scheel C and Johnsen SA. Epigenetic plasticity: a central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget. 2014; 5:8. 2016–2029.
10. Kalluri R and Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:6. 1420–1428.
11. Polyak K and Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009; 9:4. 265–273.
12. Singh A and Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010; 29:34. 4741–4751.
13. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M and Hoth M, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013; 23:6. 811–825.
14. Carlson CB, Lawler J and Mosher DF. Structures of thrombospondins. Cell Mol Life Sci. 2008; 65:5. 672–686.
15. Miyata Y and Sakai H. Thrombospondin-1 in urological cancer: pathological role, clinical significance, and therapeutic prospects. Int J Mol Sci. 2013; 14:6. 12249–12272.
16. Kudo-Saito C, Shirako H, Takeuchi T and Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009; 15:3. 195–206.
17. Tsuchida R, Osawa T, Wang F, Nishii R, Das B, Tsuchida S, Muramatsu M, Takahashi T, Inoue T, Wada Y, Minami T, Yuasa Y and Shibuya M. BMP4/Thrombospondin-1 loop paracrinically inhibits tumor angiogenesis and suppresses the growth of solid tumors. Oncogene. 2013;
18. Bige N, Shweke N, Benhassine S, Jouanneau C, Vandermeersch S, Dussaule JC, Chatziantoniou C, Ronco P and Boffa JJ. Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction. Kidney Int. 2012; 81:12. 1226–1238.
19. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012; 13:10. 616–630.
20. Mikesh LM, Kumar M, Erdag G, Hogan KT, Molhoek KR, Mayo MW and Slingluff CL. Evaluation of molecular markers of mesenchymal phenotype in melanoma. Melanoma Res. 2010; 20:6. 485–495.
21. Liu F, Gomez Garcia AM and Meyskens FL. NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase-2 and epithelial-mesenchymal transition in melanoma cells. J Invest Dermatol. 2012; 132:8. 2033–2041.
22. Behren A, Anaka M, Lo PH, Vella LJ, Davis ID, Catimel J, Cardwell T, Gedye C, Hudson C, Stan R and Cebon J. The Ludwig institute for cancer research Melbourne melanoma cell line panel. Pigment cell & melanoma research. 2013; 26:4. 597–600.
23. Verrecchia F, Chu ML and Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. The Journal of biological chemistry. 2001; 276:20. 17058–17062.
24. Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, Walter M, Fehm T, Solomayer E, Riess O, Wallwiener D, Kurek R and Neubauer HJ. Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer research. 2006; 66:10. 5278–5286.
25. Wu Y, Siadaty MS, Berens ME, Hampton GM and Theodorescu D. Overlapping gene expression profiles of cell migration and tumor invasion in human bladder cancer identify metallothionein 1E and nicotinamide N-methyltransferase as novel regulators of cell migration. Oncogene. 2008; 27:52. 6679–6689.
26. Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, Kanapin A, Lewis S, Mahajan S, May B, Schmidt E and Vastrik I, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic acids research. 2009; 37:(Database issue). D619–622.
27. Anastassiou D, Rumjantseva V, Cheng W, Huang J, Canoll PD, Yamashiro DJ and Kandel JJ. Human cancer cells express Slug-based epithelial-mesenchymal transition gene expression signature obtained in vivo. BMC cancer. 2011; 11:529
28. Gotzmann J, Fischer AN, Zojer M, Mikula M, Proell V, Huber H, Jechlinger M, Waerner T, Weith A, Beug H and Mikulits W. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene. 2006; 25:22. 3170–3185.
29. Jechlinger M, Grunert S, Tamir IH, Janda E, Ludemann S, Waerner T, Seither P, Weith A, Beug H and Kraut N. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003; 22:46. 7155–7169.
30. Moustakas A and Heldin P. TGFbeta and matrix-regulated epithelial to mesenchymal transition. Biochimica et biophysica acta. 2014;
31. Perrot CY, Gilbert C, Marsaud V, Postigo A, Javelaud D and Mauviel A. GLI2 cooperates with ZEB1 for transcriptional repression of CDH1 expression in human melanoma cells. Pigment cell & melanoma research. 2013; 26:6. 861–873.
32. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C and Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signalling. 2013; 6(269):pl1
33. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C and Schultz N. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discovery. 2013; 2(5):401-405
34. Salti GI, Manougian T, Farolan M, Shilkaitis A, Majumdar D and Das Gupta TK. Micropthalmia transcription factor: a new prognostic marker in intermediate-thickness cutaneous malignant melanoma. Cancer research. 2000; 60:18. 5012–5016.
35. Goding CR. Commentary. A picture of Mitf in melanoma immortality. Oncogene. 2011; 30:20. 2304–2306.
36. Huang F, Reeves K, Han X, Fairchild C, Platero S, Wong TW, Lee F, Shaw P and Clark E. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selection. Cancer research. 2007; 67:5. 2226–2238.
37. Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H, Nagle R and Bearss D. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007; 26:27. 3909–3919.
38. Bailey CM, Morrison JA and Kulesa PM. Melanoma revives an embryonic migration program to promote plasticity and invasion. Pigment cell & melanoma research. 2012; 25:5. 573–583.
39. Busch C, Krochmann J and Drews U. The chick embryo as an experimental system for melanoma cell invasion. PLoS One. 2013; 8:1. e53970
40. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A and Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010; 468:7326. 973–977.
41. Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U, Kobert N, Schaerer L, Hemmi S and Dummer R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008; 68:3. 650–656.
42. Tulchinsky E, Pringle JH, Caramel J and Ansieau S. Plasticity of melanoma and EMT-TF reprogramming. Oncotarget. 2014; 5:1. 1–2.
43. Hoek KS and Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigment cell & melanoma research. 2010; 23:6. 746–759.
44. Sargiannidou I, Zhou J and Tuszynski GP. The role of thrombospondin-1 in tumor progression. Exp Biol Med (Maywood). 2001; 226:8. 726–733.
45. Taraboletti G, Rusnati M, Ragona L and Colombo G. Targeting tumor angiogenesis with TSP-1-based compounds: rational design of antiangiogenic mimetics of endogenous inhibitors. Oncotarget. 2010; 1:7. 662–673.
46. Lindner DJ, Wu Y, Haney R, Jacobs BS, Fruehauf JP, Tuthill R and Borden EC. Thrombospondin-1 expression in melanoma is blocked by methylation and targeted reversal by 5-Aza-deoxycytidine suppresses angiogenesis. Matrix Biol. 2013; 32:2. 123–132.
47. Baker LH, Rowinsky EK, Mendelson D, Humerickhouse RA, Knight RA, Qian J, Carr RA, Gordon GB and Demetri GD. Randomized, phase II study of the thrombospondin-1-mimetic angiogenesis inhibitor ABT-510 in patients with advanced soft tissue sarcoma. J Clin Oncol. 2008; 26:34. 5583–5588.
48. Markovic SN, Suman VJ, Rao RA, Ingle JN, Kaur JS, Erickson LA, Pitot HC, Croghan GA, McWilliams RR, Merchan J, Kottschade LA, Nevala WK, Uhl CB, Allred J and Creagan ET. A phase II study of ABT-510 (thrombospondin-1 analog) for the treatment of metastatic melanoma. Am J Clin Oncol. 2007; 30:3. 303–309.
49. Almendros I, Montserrat JM, Torres M, Dalmases M, Cabanas ML, Campos-Rodriguez F, Navajas D and Farre R. Intermittent hypoxia increases melanoma metastasis to the lung in a mouse model of sleep apnea. Respir Physiol Neurobiol. 2013; 186:3. 303–307.
50. Feige E, Yokoyama S, Levy C, Khaled M, Igras V, Lin RJ, Lee S, Widlund HR, Granter SR, Kung AL and Fisher DE. Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:43. E924–933.
51. Liu S, Kumar SM, Martin JS, Yang R and Xu X. Snail1 mediates hypoxia-induced melanoma progression. Am J Pathol. 2011; 179:6. 3020–3031.
52. Widmer DS, Hoek KS, Cheng PF, Eichhoff OM, Biedermann T, Raaijmakers MI, Hemmi S, Dummer R and Levesque MP. Hypoxia contributes to melanoma heterogeneity by triggering HIF1alpha-dependent phenotype switching. J Invest Dermatol. 2013; 133:10. 2436–2443.
53. Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM and Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007; 7:4. 246–255.
54. Kulesa PM, Kasemeier-Kulesa JC, Teddy JM, Margaryan NV, Seftor EA, Seftor RE and Hendrix MJ. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc Natl Acad Sci U S A. 2006; 103:10. 3752–3757.
55. Schriek G, Oppitz M, Busch C, Just L and Drews U. Human SK-Mel 28 melanoma cells resume neural crest cell migration after transplantation into the chick embryo. Melanoma Res. 2005; 15:4. 225–234.
56. Tanaka K, Sonoo H, Kurebayashi J, Nomura T, Ohkubo S, Yamamoto Y and Yamamoto S. Inhibition of infiltration and angiogenesis by thrombospondin-1 in papillary thyroid carcinoma. Clin Cancer Res. 2002; 8:5. 1125–1131.
57. Duquette M, Sadow PM, Lawler J and Nucera C. Thrombospondin-1 Silencing Down-Regulates Integrin Expression Levels in Human Anaplastic Thyroid Cancer Cells with BRAF(V600E): New Insights in the Host Tissue Adaptation and Homeostasis of Tumor Microenvironment. Front Endocrinol (Lausanne). 2013; 4:189.
58. Firlej V, Mathieu JR, Gilbert C, Lemonnier L, Nakhle J, Gallou-Kabani C, Guarmit B, Morin A, Prevarskaya N, Delongchamps NB and Cabon F. Thrombospondin-1 triggers cell migration and development of advanced prostate tumors. Cancer Res. 2011; 71:24. 7649–7658.
59. Radziwon-Balicka A, Santos-Martinez MJ, Corbalan JJ, O'Sullivan S, Treumann A, Gilmer JF, Radomski MW and Medina C. Mechanisms of platelet-stimulated colon cancer invasion: role of clusterin and thrombospondin 1 in regulation of the P38MAPK-MMP-9 pathway. Carcinogenesis. 2014; 35:2. 324–332.
60. Wang TN, Qian X, Granick MS, Solomon MP, Rothman VL, Berger DH and Tuszynski GP. Thrombospondin-1 (TSP-1) promotes the invasive properties of human breast cancer. J Surg Res. 1996; 63:1. 39–43.
61. Yee KO, Connolly CM, Duquette M, Kazerounian S, Washington R and Lawler J. The effect of thrombospondin-1 on breast cancer metastasis. Breast Cancer Res Treat. 2009; 114:1. 85–96.
62. Lin XD, Chen SQ, Qi YL, Zhu JW, Tang Y and Lin JY. Overexpression of thrombospondin-1 in stromal myofibroblasts is associated with tumor growth and nodal metastasis in gastric carcinoma. J Surg Oncol. 2012; 106:1. 94–100.
63. Uchida H, Kuroki M, Shitama T, Hayashi H and Kuroki M. Activation of TGF-beta1 through up-regulation of TSP-1 by retinoic acid in retinal pigment epithelial cells. Curr Eye Res. 2008; 33:2. 199–203.
64. Ritchie ME, Silver J, Oshlack A, Holmes M, Diyagama D, Holloway A and Smyth GK. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007; 23:20. 2700–2707.
65. Breitling R, Armengaud P, Amtmann A and Herzyk P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS letters. 2004; 573:1-3. 83–92.
66. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:43. 15545–15550.
67. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:2. 249–264.