
INTRODUCTION
Glioblastoma (GBM) is the most commonly diagnosed primary brain tumor in adults. These tumors are highly aggressive and the prognosis for patients is extremely poor, with median overall survival of 14.6 months and 5-year survival rates less than 10% following standard of care treatment [1, 2]. With near 100% relapse rates and limited treatment options at the time of recurrence, GBM represents one of the biggest therapeutic challenges of our time.
The development of effective treatments for GBM, both primary and recurrent, has been challenged by the intracranial location and infiltrative growth of these tumors, extensive molecular heterogeneity, and associated immunosuppression. Although surgery is a key component of the standard treatment, in many cases complete or even partial resection is deemed unattainable due to the eloquent nature of the involving brain tissue. Furthermore, despite maximal surgical resection, the highly infiltrative nature of GBM ensures local recurrence, in contrast to the metastatic nature of other aggressive tumors. Tumors are further protected by the blood brain barrier (BBB), a semipermeable membrane of endothelial cells connected by tight junctions, which prevents the passage of most conventional drugs to tumor sites [3, 4]. A hallmark adaptation of GBM is the development of a profoundly immunosuppressive tumor microenvironment (TME) that cripples endogenous antitumor immune responses and limits the effectiveness of immunotherapies [5, 6].
The programmed cell death (PD) pathway is an endogenous negative feedback mechanism for T-cell activity that is often exploited by human tumors, including GBM, to suppress the antitumor efficacy of incoming CD8+ cytotoxic T lymphocytes (CTLs) [7, 8]. Immune checkpoint inhibition with monoclonal antibodies targeting the programmed cell death-1 (PD-1) protein or its ligand, PD-L1, has produced significant clinical results in the treatment of several cancers, most notably metastatic melanoma [9, 10] and non-small cell lung cancer (NSCLC) [11, 12], and has shown promise in preclinical studies for the treatment of GBM [13, 14]. CheckMate 143 (NCT02017717) was the first randomized phase III clinical trial of PD pathway inhibition in the setting of GBM, including a comparison of nivolumab (Opdivo) and the anti-vascular endothelial growth factor (VEGF) antibody, bevacizumab (Avastin), in the treatment of recurrent GBM. Early results presented at WFNOS conference 2017 revealed that nivolumab did not prolong overall survival (OS) in these patients, the primary endpoint of the study, and consequently this arm of the trial was closed [15].
Although this failure can be regarded as a setback, it provides an opportunity to reevaluate existing treatment strategies, spurring research that will improve our understanding of GBM, ultimately leading to the development of more effective therapies for patients with this devastating illness. A thorough analysis of the CheckMate 143 trial and factors potentially contributing to its failure is imperative in accomplishing this goal. In this review we discuss the basic science concepts underlying the rationale to target PD-1 pathway in GBM.
CLINICAL MANAGEMENT OF GBM
The current standard of care for newly diagnosed GBM is maximal surgical resection and concurrent radiotherapy (RT) and temozolomide (TMZ) chemotherapy, followed by 6 months of adjuvant TMZ [1]. A recent phase III trial evaluating the addition of tumor treating fields (TTFs) to the TMZ and radiation protocol showed increased OS from 16 months to 21 months in newly diagnosed GBM [16]. However, even with the best available treatment, GBM has a near 100% relapse rate with a median time to recurrence of 7 months [2]. Treatment options at this time are limited; repeat surgery is considered for approximately 25% of patients and re-irradiation is only possible as a palliative option in rare cases [17]. Chemotherapy response rates, including to TMZ, typically do not exceed 10% and none have been shown to prolong OS [18–22]. Although two large randomized trials failed to show increased OS with addition of bevacizumab, an anti-VEGF monoclonal antibody, to the current treatment strategy [23, 24], it is regularly used in the treatment of recurrent GBM due to a demonstrated ability to prolong progression-free survival (PFS), reduce the use of immunosuppressive corticosteroids, and improve patient quality of life, both as a monotherapy and in combination with other cytotoxic agents [25–28]. Overall, the median survival for patients with recurrent disease typically ranges between 6.6 and 9.6 months, with one recent study showing overall survival rates of slightly over one year for patients treated with TTFs [29].
GBM AND IMMUNOTHERAPY
The field of immunotherapy centers on the natural ability of the host immune system to identify and destroy malignant cells, an ability that is often impaired in the setting of cancer, particularly GBM [6]. Immunotherapies are targeted towards activating and enhancing endogenous host immune responses. Among those being investigated are: 1) T-cell based therapies like chimeric antigen receptor (CAR) T-cells and adoptive transfer of immune cells to directly bolster antitumor immunity, 2) therapeutic vaccines that enhance antigen presentation and stimulate the generation of robust antitumor immune responses, 3) viruses engineered to selectively infect and destroy tumor cells, and 4) antibody inhibition of signaling through tumor-promoting pathways (VEGF, CTLA-4, PD-1 etc.) (Figure 1).
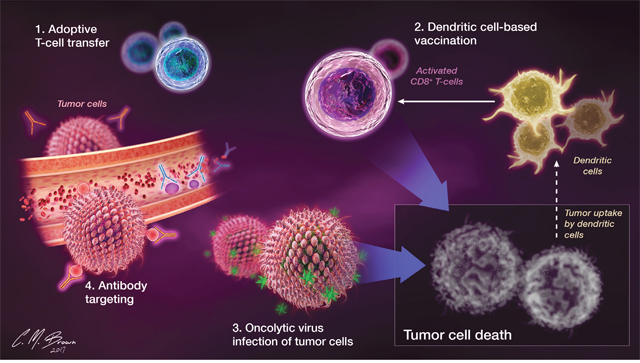
Figure 1: Commonly employed immunotherapeutic strategies enhance antitumor immunity by addressing different components of the immune response. 1) Antitumor immunity can be directly bolstered with the adoptive transfer of specialized and functional T cells. 2) Therapeutic vaccines that enhance dendritic cell function and presentation of tumor antigens promote more efficient activation of antigen specific CD8+ T cell responses. 3) Oncolytic viruses can selectively infect and lyse tumor cells, releasing tumor associated antigens into the glioma microenvironment which can be taken up by the resident DCs. 4) Monoclonal antibodies targeting immune cell (ex. CTLA-4, PD-1) and tumor-expressed (PD-L1, VEGF) molecules can be used to inhibit signaling through pathways that promote tumor cell growth or inhibit immune cell responses.
ENDOGENOUS PD PATHWAY
The PD pathway is an endogenous negative feedback mechanism for T-cell activity that functions in the healthy host to minimize tissue damage incurred with prolonged inflammatory responses and prevent the development of autoimmunity by inducing peripheral tolerance to self-antigens [7, 30]. An overview of the endogenous PD pathway is shown in Figure 2.
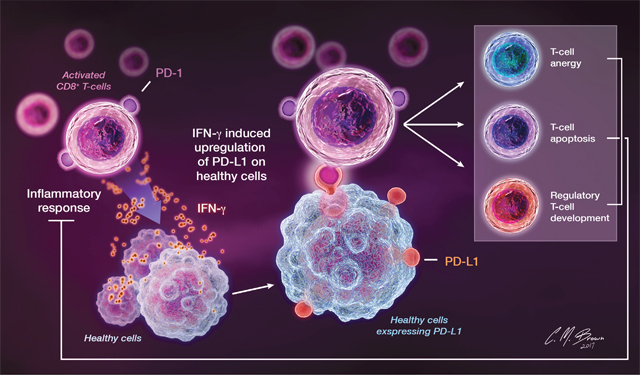
Figure 2: PD-1 is constitutively expressed on activated T cells, B cells, and other myeloid cells. Release of IFN-γ by activated T lymphocytes during inflammatory responses directly induces local PD-L1 expression on surrounding cells. Binding of PD-L1 to lymphocyte-expressed PD-1 transmits an inhibitory feedback signal that suppresses T cell proliferation and cytokine release and induces T cell anergy, apoptosis, and the development of regulatory T cells, thereby attenuating inflammatory responses.
PD PATHWAY IN CANCER
Seldom present on healthy cells in the steady state, PD-L1 is often expressed by both tumor cells and tumor-infiltrating lymphocytes (TILs) in a variety of human cancers, including GBM [31–34]. In a recent study of human GBM, 88% of tumors expressed PD-L1 [31], further supporting an exploration of the role of the PD pathway in malignant glioma. Induction of tumor PD-L1 expression likely occurs in response to inflammation induced by host antitumor immune responses [34] and through tumor-specific mutations, such as loss of the tumor-suppressor PTEN or enhanced ALK gene signaling [35, 36]. An overview of the PD-1 pathway in cancer is shown in Figure 3.
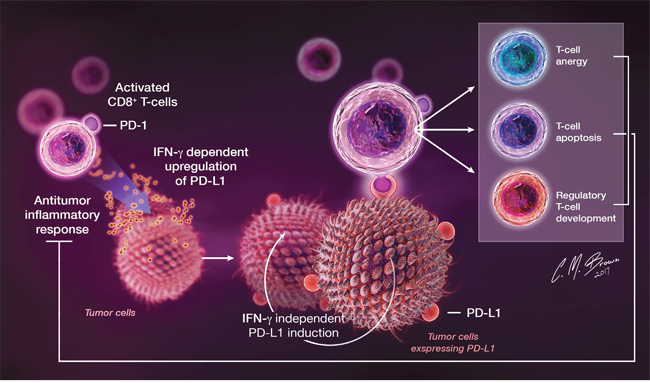
Figure 3: In the setting of cancer, PD-L1 is upregulated on tumor cells in response to IFN-γ released by infiltrating immune cells during antitumor immune responses, as well as through tumor-specific IFN-γ-independent mechanisms. PD-L1 serves as a receptor on cancer cells that, through interactions with PD-1expressing TIL, induces an intrinsic resistance to CTL killing and suppresses antitumor immune responses.
PD signaling in the setting of cancer represents an adaptation through which tumors can exploit endogenous cellular feedback mechanisms to suppress antitumor immune responses in a mechanism termed “adaptive resistance” [7]. Tumor PD-L1 binds to PD-1 receptors on infiltrating effector T-cells, inhibiting their cytotoxic activity and thereby rendering malignant cells resistant to CTL-mediated destruction [37]. Elevated tumor PD-L1 expression has been associated with aggressive disease and poor prognoses in several cancers including renal [38], pancreatic [34], breast [32], ovarian [39], esophageal [40], and gastric cancers [41]. Patients with PD-L1-positive tumors displayed local evidence of impaired cellular immune responses, including a paucity of TILs or an abundance of heavily immunosuppressed and dysfunctional effector cells [34, 39]. However, in other studies of NSCLC [42], colorectal cancer [33], and melanoma [43], tumor PD-L1 expression was associated with evidence of strong ongoing antitumor immune responses and correlated with a positive prognosis. These patients seem to be benefitting from robust inflammatory antitumor responses that, in turn, induce PD-L1 expression. Furthermore, in some of these cases, infiltrating effector cells lacked PD-1 expression entirely, rendering them functionally insensitive to PD-L1-mediated inhibition [33].
With respect to GBM, although the immunosuppressive effects of the PD pathway have been well documented in preclinical studies, the overall relationship between baseline tumor PD-L1 expression and patient prognosis remains unclear. Several studies have correlated elevated pretreatment levels of PD-L1 with a worse prognosis [44, 45], whereas others have found no prognostic predictive value [31].
CLINICALLY TARGETING PD PATHWAY IN CANCER
Given the observed role of the PD-1/PD-L1 immune checkpoint in the pathophysiology of tumor-induced immunosuppression, blockade of these interactions represents a promising anticancer treatment strategy. Human ex vivo studies have demonstrated reversal of PD pathway-mediated T-cell exhaustion and enhancement of lymphocyte proliferation and cytokine production after administration of monoclonal antibodies targeting either PD-1 or PD-L1 [46–51]. Preclinical studies in mouse tumor models have established the in vivo safety and efficacy of these agents, yielding significant tumor regression and prolonged animal survival in the setting of many cancers, including GBM [14, 52, 53]. In phase III clinical trials, anti-PD pathway therapies have produced substantial clinical responses in a subset of patients with variety of cancers [9–12, 54–56], culminating in FDA approval of two immune checkpoint inhibitors, pembrolizumab and nivolumab, both anti-PD-1 monoclonal antibodies, in the treatment of unresectable or metastatic melanoma (pembrolizumab and nivolumab) and NSCLC (nivolumab) [57, 58]. A list of all currently active clinical trials of PD-1/PD-L1 inhibitors in patients with malignant glioma is shown in Table 1 [59–68].
Table 1: Clinical trials with PD-1/PD-L1 blockade in malignant glioma
Malignancy |
Phase |
N |
Name of trial |
Therapeutic compounds |
Clinical trial identifier |
Status |
References |
|---|---|---|---|---|---|---|---|
Recurrent High Grade Glioma |
I |
26* |
Hypofractionated Stereotactic Irradiation With Nivolumab in Patients With Recurrent High Grade Gliomas |
Nivolumab, hfSRT |
NCT02829931 |
Recruiting |
[59] |
Recurrent Malignant Glioma |
I |
46* |
Hypofractionated Stereotactic Irradiation (HFSRT) With Pembrolizumab and Bevacizumab for Recurrent High Grade Gliomas |
Pembrolizumab, bevacizumab, hfSRT |
NCT02313272 |
Recruiting |
|
Malignant Glioma |
I |
66* |
Nivolumab With DC Vaccines for Recurrent Brain Tumors (AVERT) |
Nivolumab, DC vaccine |
NCT02529072 |
Recruiting |
|
Glioblastoma, Gliosarcoma |
II |
48* |
Combination Adenovirus + Pembrolizumab to Trigger Immune Virus Effects (CAPTIVE) |
DNX-2401, pembrolizumab |
NCT02798406 |
Recruiting |
|
Glioblastoma |
I/II |
60* |
A Phase 1/2 Safety Study of Intratumorally Dosed INT230-6 (IT-01) |
INT230-6, anti-PD-1 antibody |
NCT03058289 |
Recruiting |
|
Glioblastoma |
II |
205* |
A Dose Escalation and Cohort Expansion Study of Anti-CD27 (Varlilumab) and Anti-PD-1 (Nivolumab) in Advanced Refractory Solid Tumors |
Varlilumab, nivolumab |
NCT02335918 |
Recruiting |
[62] |
Recurrent/Progressive Glioblastoma |
Pilot |
30* |
A Pilot Surgical Trial To Evaluate Early Immunologic Pharmacodynamic Parameters For The PD-1 Checkpoint Inhibitor, Pembrolizumab (MK-3475), In Patients With Surgically Accessible Recurrent/Progressive Glioblastoma |
Pembrolizumab |
NCT02852655 |
Recruiting |
|
Glioblastoma, Gliosarcoma, Recurrent Brain Neoplasm |
I |
68* |
Anti-LAG-3 or Urelumab Alone and in Combination With Nivolumab in Treating Patients With Recurrent Glioblastoma |
Anti-LAG-3, urelumab, nivolumab |
NCT02658981 |
Recruiting |
|
Glioblastoma, other select advance solid tumors |
I |
280* |
Study of FPA008 in Combination With Nivolumab in Patients With Selected Advanced Cancers (FPA008-003) |
FPA008, nivolumab |
NCT02526017 |
Recruiting |
[63] |
Glioblastoma |
I |
6* |
Intra-tumoral Ipilimumab Plus Intravenous Nivolumab Following the Resection of Recurrent Glioblastoma (GlitIpNi) |
Ipilimumab, nivolumab |
NCT03233152 |
Recruiting |
|
Glioblastoma |
II |
43* |
Avelumab With Hypofractionated Radiation Therapy in Adults With Isocitrate Dehydrogenase (IDH) Mutant Glioblastoma |
Avelumab |
NCT02968940 |
Recruiting |
|
Glioblastoma, other select advance solid tumors |
I/II |
291* |
A Study of the Safety, Tolerability, and Efficacy of Epacadostat Administered in Combination With Nivolumab in Select Advanced Cancers (ECHO-204) |
Nivolumab, epacadostat |
NCT02327078 |
Recruiting |
[64] |
Malignant Glioma, Recurrent Glioblastoma |
II |
36* |
Tremelimumab and Durvalumab in Combination or Alone in Treating Patients With Recurrent Malignant Glioma |
Durvalumab, tremelimumab, surgical procedure |
NCT02794883 |
Recruiting |
|
Recurrent Malignant Glioma |
I/II |
52* |
MK-3475 in Combination With MRI-guided Laser Ablation in Recurrent Malignant Gliomas |
MK-3475, MRI-guided laser ablation |
NCT02311582 |
Recruiting |
|
Glioblastoma Multiforme |
I |
20* |
Pilot Study of Autologous Chimeric Switch Receptor Modified T Cells in Recurrent Glioblastoma Multiforme |
Anti-PD-L1 CSR T cells, cyclophosphamide, fludarabine |
NCT02937844 |
Recruiting |
|
Glioblastoma |
I/II |
62* |
A Study Evaluating the Association of Hypofractionated Stereotactic Radiation Therapy and Durvalumab for Patients With Recurrent Glioblastoma (STERIMGLI) |
Durvalumab, hfSRT |
NCT02866747 |
Recruiting |
|
Glioblastoma |
II |
159 |
Phase 2 Study of MEDI4736 in Patients With Glioblastoma |
MEDI4736, radiotherapy, bevacizumab |
NCT02336165 |
Active, Not Recruiting |
|
Recurrent Glioblastoma |
II |
82 |
Pembrolizumab +/- Bevacizumab for Recurrent GBM |
Pembrolizumab, bevacizumab |
NCT02337491 |
Active, Not Recruiting |
[67] |
Recurrent Glioblastoma |
II |
30* |
Autologous Dendritic Cells Pulsed With Tumor Lysate Antigen Vaccine and Nivolumab in Treating Patients With Recurrent Glioblastoma |
Autologous DCs pulsed with tumor lysate antigen vaccine, nivolumab |
NCT03014804 |
Not Yet Recruiting |
|
Glioblastoma Multiforme |
II |
29 |
Neoadjuvant Nivolumab in Glioblastoma (Neo-nivo) |
Nivolumab |
NCT02550249 |
Completed |
|
Recurrent High-Grade Gliomas |
|
20 |
OS09.5 Synergistic effect of reirradiation and PD-1 inhibitors in recurrent high-grade gliomas |
PD-1 Inhibitors, reirradiation |
|
|
[68] |
hfSRT – hypofractionated stereotactic irradiation.
* Estimated/Anticipated sample size.
Given that the checkpoint inhibitors exert their antitumor effects by preventing PD-1/PD-L1 interactions thereby allowing for uninhibited effector T-cell activity, positive clinical responses to treatment would be expected to correlate with evidence of active antitumor immune responses. Supporting this assertion, numerous studies involving various cancers have associated patient response to treatment with the selective expansion and activation of antigen-specific CD8+ CTLs, reduced Treg cell activity, IFN-γ secretion and expression of IFN-γ-inducible genes, and subsequent IFN-γ-induced upregulation of PD-L1 expression on both tumor cells and TILs [9, 69–75]. Biopsies of regressing lesions demonstrate dense intratumoral CD8+ infiltrates [69, 73], and elevations in peripheral blood lymphocyte counts are observed as well [71, 75, 76]. Patients not responding to treatment characteristically have lacked this evidence of immune activation and possess reduced numbers of peripheral blood antigen-specific T-cells, instead accumulating immunosuppressive regulatory T-cells [77].
Immune checkpoint inhibition has been shown to be particularly effective in patients with elevated pretreatment levels of tumor PD-L1 expression [9, 11, 55, 70, 71, 74, 77–83]. In one such study of NSCLC, treatment with the anti-PD-1 antibody nivolumab was shown to be more effective than docetaxel chemotherapy in patients with PD-L1 positive, but not PD-L1 negative, tumors at all classified levels of PD-L1 expression (≥1%, ≥5%, and ≥10% of tumor cells) [11]. Similarly, in a phase I trial of nivolumab in 90 patients with unresectable melanoma, overall response rates were observed in 67% of patients with PD-L1-positive tumors (≥5%) as compared to 19% of patients with PD-L1-negative tumors [9]. However, even patients with PD-L1-negative tumors have been shown to receive some survival benefit from anti-PD-1 therapy [12, 54, 56, 70]. For example, in another phase III clinical trial of nivolumab vs docetaxel in NSCLC patients, prolonged survival was seen in patients treated with nivolumab, regardless of tumor cell PD-L1 expression [12]. The possibility exists that in this and other similar cases, PD blockade exerts therapeutic effects, at least partially, by preventing TILs from interacting with PD-L1 expressed by other infiltrating immune cells, such as dendritic cells (DCs), rather than tumor cells. This assertion is supported by the results of a study of the anti-PD-L1 antibody atezolizumab (MPDL3280A) in multiple solid tumors, in which positive treatment responses were significantly associated with higher baseline levels of TIL-expressed PD-L1; in this study, there was no correlation between treatment response and PD-L1 expression on tumor cells. [70] Other studies have similarly documented associations between PD-L1 expression by peripheral blood lymphocytes, like CD4+ and CD8+ T-cells, and treatment response to immune checkpoint blockade [71]. However so far, there are no published studies evaluating T-cell response following treatment with checkpoint inhibitors in GBM.
Atypical responses have also been observed in the use of immune checkpoint inhibitors and must also be considered as well. In some cases, anti-PD therapy has been associated with accelerated disease progression and reduced OS, in a phenomenon known as hyperprogressive disease (HPD) [84–87]. Observed in a small subset of patients, HPD occurs in many different tumor types and has been shown to be independent of initial tumor burden. Although the mechanism of HPD remains unknown, it has been linked with several genetic changes and is associated with advanced patient age, raising concerns for PD blockade in the elderly population [84, 85].
CHECKMATE 143 TRIAL
CheckMate 143 (NCT 02017717), sponsored by Bristol-Myers Squibb (BMS), was the first large-scale randomized clinical trial of PD pathway inhibition in the setting of malignant glioma. It was designed to evaluate the safety and efficacy of nivolumab in the treatment of patients with GBM and included a study of nivolumab monotherapy as compared to bevacizumab in the setting of recurrent disease. Patients received either nivolumab 3mg/kg or bevacizumab 10mg/kg IV every 2 weeks, until disease progression or unacceptable toxicity. At the time of final analysis, this phase III clinical trial enrolled 369 patients with first recurrence of GBM, previously treated with combination radiation and TMZ. At baseline, a significant proportion of patients in both treatment arms, 40% (nivolumab) and 43% (bevacizumab), required glucocorticoid therapy, with 14% (nivolumab) and 15% (bevacizumab) receiving ≥ 4 mg/day.
Results reported in April of 2017 at the WFNOS conference revealed a failure of nivolumab to extend OS in patients with recurrent GBM as compared to bevacizumab, and this arm of the trial was prematurely terminated. In both treatment groups, the 12-month OS was 42%. The median OS for patients treated with nivolumab was 9.8 months as compared to 10 months for those receiving bevacizumab monotherapy. Median PFS was 1.5 months (nivolumab) and 3.5 months (bevacizumab). Overall treatment response rates were 8% (nivolumab) and 23% (bevacizumab) and median durations of response were 11.1 months (nivolumab) and 5.3 months (bevacizumab). Treatment-related adverse events (TRAEs) occurred in 57% (nivolumab) and 58% (bevacizumab) of patients, with the most common being fatigue (21% vs 14%) and hypertension (1% vs 22%). Grade 3–4 TRAEs occurred in 18% (nivolumab) and 15% (bevacizumab) of patients and TRAEs leading to discontinuation of therapy were reported in 10% (nivolumab) and 15% (bevacizumab) of patients [15].
BMS is currently conducting two additional trials of combination nivolumab and RT with or without TMZ in patients with newly diagnosed MGMT-unmethylated, CheckMate-498 (NCT02617589), and MGMT-methylated, CheckMate-548 (NCT02667587), GBM, both of which are recruiting participants.
INSIGHT INTO TRIAL FAILURE
Dependent upon interactions with PD-1-expressing T-cells, one possible etiology of treatment failure is an impairment of the interaction between nivolumab and PD-1 receptors on patient lymphocytes. This could be related to a systemic lymphopenia, reduced T-cell expression of PD-1, or the presence of structural barriers preventing T-cell-antibody interactions (Figure 4). Although baseline information regarding the immune status of patients enrolled in this trial is not available, it is widely known that patients suffering from GBM experience global immune dysfunction and possess reduced levels of circulating CD4+ and CD8+ lymphocytes, an effect compounded by lymphocyte-depleting treatments such as chemotherapy [88, 89]. The treatment of CNS pathology, including malignant glioma, has been historically limited by poor drug penetration of the BBB [4], which is significantly prohibitive for compounds of size greater than 400-600 Da [90]. The calculated molecular mass of nivolumab is 146 kDa [57], supporting the assertion that antibody-mediated inhibition of the PD-1/PD-L1 axis occurs external to tumor sites and that effector T-cells, activated against tumor-associated antigens in peripheral lymphoid tissues, enter the TME precoated with anti-PD-1 antibodies [52]. With tumor progression and particularly in the setting of recurrent disease, any PD-1-expressing lymphocytes activated against tumor antigens would be expected to have already migrated to tumor sites, where they are inaccessible to monoclonal antibodies.
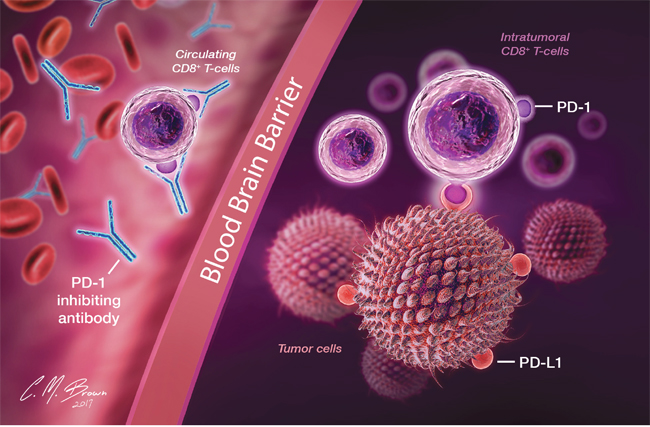
Figure 4: In recurrent disease, efficacy of nivolumab is limited by its inability to cross the blood brain barrier and a paucity of functional circulating T-cells with which to interact and form a protective barrier against subsequent possible PD-1/PD-L1 interactions. Exposed to numerous immunosuppressive influences within the glioma microenvironment, including uninhibited PD-1/PD-L1 interactions, T-cells already sequestered within the TME are expected to be heavily dysfunctional and unable to be rescued solely with immune checkpoint inhibition.
Upon introduction to tumor sites, effector T-cells are exposed to a multitude of concentrated immunosuppressive factors within the surrounding milieu, including PD-L1. As a result, antigen-specific T-cells in patients with GBM are heavily dysfunctional and often are rendered permanently anergic, or tolerogenic towards tumor-associated antigens. Studies of T-cell function in the setting of chronic antigen exposure have suggested that these “exhausted” T-cells expressing high levels of PD-1 have poor effector functions that may not be fully restored with PD-1 blockade [91–93]. PD-1 blockade can “remove the brakes” placed on host immune responses by PD pathway signaling at tumor sites, however the ultimate antitumor effect is constrained by the host’s ability to generate adaptive immune responses against tumor-associated antigens, an ability that is often significantly impaired in GBM. “Removing the brakes” from a dysfunctional, inadequate immune response leaves an uninhibited, though still dysfunctional and inadequate, immune response; thus, given the highly complex network of immunosuppression commonly present in GBM, single-agent therapy with PD-1 inhibition is unlikely to address and alleviate all factors contributing to T-cell dysfunction, and therefore would not be expected to result in durable, sustained tumor regression.
FUTURE DIRECTION
Despite failing to prolong OS in patients with recurrent GBM, nivolumab may still have a place in the successful treatment of this disease. A small subset (8%) of patients in the CheckMate 143 trial did respond to nivolumab, and with a much longer duration of response (11.1 months) than seen with bevacizumab therapy (5.3 months). Further analysis of this subgroup in terms of immune status and tumor biomarkers will provide key insight regarding treatment failure and pave a pathway for future success.
Bevacizumab, the alternative treatment to nivolumab in the Check Mate 143 trial, is used regularly in the treatment for patients with recurrent GBM and will likely remain a component of standard therapy. In both primary and recurrent GBM, bevacizumab has been shown to increase PFS and improve peritumoral edema, reducing the need for immunosuppressive glucocorticoids known to interfere with the efficacy of immunotherapy [94]. Although not shown to extend OS, bevacizumab may confer additional benefits in the setting of immunotherapy. High levels of intratumoral VEGF are strongly immunosuppressive, promoting the activity of Treg cells, shifting DC populations towards an immature phenotype, and inducing apoptosis in CD8+ T-cells, effects abrogated with the use of anti-VEGF therapy [95]. The vasculature-normalizing effects of anti-VEGF therapy have also been shown to improve delivery of chemotherapeutic agents to tumor sites and enhance intratumoral immune cell infiltration, improving the efficacy of therapies like adoptive T-cell transfer [95, 96]. Furthermore, in a recent phase I trial of patients with metastatic melanoma, bevacizumab enhanced intratumoral lymphocyte infiltration and humoral immune responses in combination with CTLA-4 blockade [97]. Although bevacizumab independently has immune-modulating functions and can provide some synergistic effects in combination with nivolumab, this synergy may be further enhanced with the addition of other immune-modulating strategies.
The addition of therapies addressing other immunosuppressive pathways, like CTLA-4 blockade, is one promising treatment strategy for recurrent GBM. In contrast to PD-L1, which suppresses existing immune responses, CTLA-4 signaling inhibits initial immune cell activation. Combination anti-PD-1 and anti-CTLA-4 treatment in several studies has resulted in better clinical outcomes than seen with either agent alone [79, 98]. Furthermore, CTLA-4 and PD-1 expression in peripheral blood and tumor-infiltrating lymphocytes increases with immune checkpoint inhibition, reflecting the expansion of effector T-cell populations that would otherwise have been apoptotic or dysfunctional. Additionally, this increased expression of immune checkpoint molecules renders patients more susceptible to immune checkpoint blockade and represents another indication for combination treatment [99]. Combining immune checkpoint blockade with other chemotherapeutic agents has also been shown to relieve tumor-induced immunosuppression and improve immune and clinical outcomes. Combination treatment with CT-011, an anti-PD-1 antibody, and cyclophosphamide resulted in a significant decrease in intratumoral Treg cell infiltration as well as an increase in the presence of antigen-specific CD8+ T-cells [100]. Mkrtichyan and colleagues also reported that treating mice with combination cyclophosphamide and a PD-1 inhibitor reduced the presence of exhausted PD-1-expressing immune cells within the TME, allowing infiltration and proliferation of non-exhausted, functional, PD-1-deficient T-cells [101].
Another strategy to augment the efficacy of nivolumab in patients with GBM is combination treatment with immunotherapies that actively stimulate immune responses. Radiotherapy, a component of the standard treatment for GBM, induces phenotypic changes in tumor cells that enhance their susceptibility to immune-mediated destruction, such as increased expression of death receptors, costimulatory molecules, stress ligands, adhesion molecules, and MHC class I molecules [102]. RT also promotes intracellular protein degradation and broadens the peptide repertoire of available tumor antigens by inducing the production of novel proteins [103]. Although GBM is known to have fewer mutations and tumor associated antigens (TAA) compared to various other cancers [104], future immunostimulatory strategies will involve targeting predominantly tumor-generated neo-antigens instead of TAA. Treatment with TMZ and fractionated RT have specifically been shown to increase IFN-γ release, leading to upregulation of PD-L1 in in vitro GBM cell lines [105]. Tumor cell death induced by RT and chemotherapy releases inflammatory tumor cell debris and tumor-associated antigens into the TME, leading to increased antigen presentation and activation of adaptive immune responses [102, 106]. Other therapies to consider that promote the activation and recruitment of inflammatory cells to the TME include DC-based vaccination, oncolytic virotherapy (OVT), and adoptive T-cell transfer [107–109]. Tumor cell PD-L1 expression has been shown to preclude the effectiveness of adoptive T-cell therapy by promoting apoptosis of transferred cells, an effect that can be abrogated with the addition of PD-1 blocking antibodies [110]. In a preclinical study of mice bearing B7-H1/SCCVII tumors treated with adoptive T-cell transfer, anti-PD-1 therapy, or both, combination treatment was required to achieve ultimate tumor regression and prolonged animal survival [108]. Given the mechanisms underlying PD-L1 upregulation, patients with stronger IFN-γ-releasing adaptive immune responses and more intense intra- and peritumoral inflammation would be expected to exhibit higher levels of PD-L1 expression, and therefore increased susceptibility to anti-PD therapy. This represents another mechanism of synergy whereby immunotherapies that enhance IFN-γ secretion, such as OVT, will locally sensitize tumors to PD blockade [109]. In a recent study of combination OVT and PD blockade, an oncolytic measles virus was shown to upregulate expression of PD-L1 in human GBM cells, and combination therapy led to prolonged survival of C57BL/6 mice bearing syngeneic orthotopic GL261 gliomas. Tumor analysis in treated mice revealed an elevated influx of inflammatory immune cells, particularly antigen-specific CD8+ CTLs [111]. Treatment with nivolumab has also been associated with activation of a variety of genes associated with innate immunity and IFN-γ-releasing natural killer (NK) cell function, introducing the possibility of combination treatment with NK cell-directed therapies as well [73, 98, 100].
Finally, if the mechanism of the CheckMate trial failure involves an inability of nivolumab to reach TILs already sequestered in the recurrent tumor microenvironment, it may be expected to function better in patients with newly diagnosed GBM, where newly activated circulating T-cells would be available for interaction with nivolumab prior to their migration to tumor sites. Additionally, surgical resection and radiation therapy employed in the treatment of primary disease provide tumor debulking leading to GBM cell death, elaboration of tumor-associated antigens, and the release of TILs into the periphery, increasing their availability for interaction with circulating nivolumab [112].
CONCLUSION
The use of immune checkpoint inhibitors, like nivolumab, has resulted in improved clinical outcomes for patients with a variety of cancers. In order to successfully extend the application of immune checkpoint blockade therapy to patients with GBM, future studies must consider the unique aspects of these tumors and the immune status observed in patients with primary and recurrent disease. An improved understanding of how the quantity and quality of TILs influence antitumor responses and PD blockade in the setting of primary vs. recurrent GBM, and a better analysis of the relative contributions of other determinants of patient responses to therapy will be imperative in designing an optimal treatment regimen. The ideal pairing of anti-PD pathway treatment with immunotherapies that counteract tumor-induced immunosuppression and/or enhance the generation of antitumor responses will likely confer the strongest benefit in immune and clinical outcomes for patients with both primary and recurrent GBM. The unique combination of these therapies will both “remove the brakes” and “step on the gas” of the vehicle that is the host antitumor immune response, working together to advance the field of glioma immunotherapy.
Author contributions
M.D: Contributed to article conception, writing and over all supervision.
A.C.F and M.H: Contributed to literature review and writing.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the NIH NRCDP K-12 and K08NS092895 grant (MD). Authors would like to thank Christopher Brown MS for his help with the figure illustrations.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352: 987-96.
2. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009; 10: 459-66.
3. Chen Y, Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev. 2012; 64: 640-65.
4. Miura Y, Takenaka T, Toh K, Wu S, Nishihara H, Kano MR, Ino Y, Nomoto T, Matsumoto Y, Koyama H, Cabral H, Nishiyama N, Kataoka K. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano. 2013; 7: 8583-92.
5. Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. 1999; 100: 216-32.
6. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005; 5: 263-74.
7. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015; 125: 3384-91.
8. Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, Weller M, Wiendl H. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003; 63: 7462-7.
9. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, Linette GP, Thomas L, Lorigan P, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015; 16: 375-84.
10. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015; 373: 23-34.
11. Borghaei H, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015; 373: 1627-39.
12. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015; 373: 123-35.
13. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, Han Y, Lesniak MS. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014; 20: 5290-301.
14. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, Pradilla G, Ford E, Wong J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013; 86: 343-9.
15. Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, Phuphanich S, de Souza P, Ahluwalia MS, Lim M, Vlahovic G, Sampson J. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncol. 2017; 19: iii21.
16. Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, Taylor LP, Lieberman F, Silvani A, Fink KL, Barnett GH, Zhu JJ, Henson JW, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA. 2015; 314: 2535-43.
17. Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro Oncol. 2013; 15: 4-27.
18. Perry JR, Bélanger K, Mason WP, Fulton D, Kavan P, Easaw J, Shields C, Kirby S, Macdonald DR, Eisenstat DD, Thiessen B, Forsyth P, Pouliot JF. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol. 2010; 28: 2051-7.
19. Lamborn KR, Yung WK, Chang SM, Wen PY, Cloughesy TF, DeAngelis LM, Robins HI, Lieberman FS, Fine HA, Fink KL, Junck L, Abrey L, Gilbert MR, et al. Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol. 2008; 10: 162-70.
20. Ballman KV, Buckner JC, Brown PD, Giannini C, Flynn PJ, LaPlant BR, Jaeckle KA. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol. 2007; 9: 29-38.
21. Yung WK, Albright RE, Olson J, Fredericks R, Fink K, Prados MD, Brada M, Spence A, Hohl RJ, Shapiro W, Glantz M, Greenberg H, Selker RG, et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000; 83: 588-93.
22. Chamberlain MC. Bevacizumab for the treatment of recurrent glioblastoma. Clin Med Insights Oncol. 2011; 5: 117-29.
23. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014; 370: 699-708.
24. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, Cloughesy T. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014; 370: 709-22.
25. Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Dowell JM, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Wagner M, Bigner DD, Friedman AH, Friedman HS. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007; 13: 1253-9.
26. Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, Wagner M, Bailey L, Bigner DD, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007; 25: 4722-9.
27. Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, Park J, Albert PS, Fine HA. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009; 27: 740-5.
28. Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, Cloughesy T. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009; 27: 4733-40.
29. Mrugala MM, Ruzevick J, Zlomanczuk P, Lukas RV. Tumor treating fields in neuro-oncological practice. Curr Oncol Rep. 2017; 19: 53.
30. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH, Freeman GJ. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003; 33: 3117-26.
31. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, Kurscheid S, Hegi ME, Zielinski CC, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015; 17: 1064-75.
32. Ghebeh H, Mohammed S, Al-Omair A, Qattan A, Lehe C, Al-Qudaihi G, Elkum N, Alshabanah M, Bin Amer S, Tulbah A, Ajarim D, Al-Tweigeri T, Dermime S, et al. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in breast cancer patients with infiltrating ductal carcinoma: correlation with important high-risk prognostic factors. Neoplasia. 2006; 8: 190-8.
33. Droeser RA, Hirt C, Viehl CT, Frey DM, Nebiker C, Huber X, Zlobec I, Eppenberger-Castori S, Tzankov A, Rosso R, Zuber M, Muraro MG, Amicarella F, et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur J Cancer. 2013; 49: 2233-42.
34. Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, Nakamura S, Enomoto K, Yagita H, Azuma M, Nakajima Y. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007; 13: 2151-7.
35. Song M. Chen D, Lu B, Wang C, Zhang J, Huang L, Wang X, Timmons CL, Hu J, Liu B, Wu X, Wang L, Wang J, Liu H. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS One. 2013; 8: e65821.
36. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007; 13: 84-8.
37. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009; 114: 1537-44.
38. Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, Krejci KG, Lobo JR, Sengupta S, Chen L, Zincke H, Blute ML, Strome SE, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004; 101: 17174-9.
39. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, Higuchi T, Yagi H, Takakura K, Minato N, Honjo T, Fujii S. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007; 104: 3360-5.
40. Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N, Mizuno T, Yoriki R, Kashizuka H, Yane K, Tsushima F, Otsuki N, Yagita H, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005; 11: 2947-53.
41. Wu C, Zhu Y, Jiang J, Zhao J, Zhang XG, Xu N. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem. 2006; 108: 19-24.
42. Velcheti V, Schalper KA, Carvajal DE, Anagnostou VK, Syrigos KN, Sznol M, Herbst RS, Gettinger SN, Chen L, Rimm DL. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest. 2014; 94: 107-16.
43. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012; 4: 127ra37.
44. Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, Ling X, Zhou S, Ivan C, Chen JQ, Burks JK, Fuller GN, Calin GA. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016; 18: 195-205.
45. Liu Y, Carlsson R, Ambjørn M, Hasan M, Badn W, Darabi A, Siesjö P, Issazadeh-Navikas S. PD-L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. J Neurosci. 2013; 33: 14231-45.
46. Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003; 170: 1257-66.
47. Blank C, Kuball J, Voelkl S, Wiendl H, Becker B, Walter B, Majdic O, Gajewski TF, Theobald M, Andreesen R, Mackensen A. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer. 2006; 119: 317-27.
48. Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol. 2009; 21: 1065-77.
49. Wong RM, Scotland RR, Lau RL, Wang C, Korman AJ, Kast WM, Weber JS. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int Immunol. 2007; 19: 1223-34.
50. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, David O, Burow M, Gordon A, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003; 9: 562-7.
51. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005; 65: 1089-96.
52. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, Wen PY, Van Den Abbeele AD, Hodi FS, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016; 4: 124-35.
53. Huang BY, Zhan YP, Zong WJ, Yu CJ, Li JF, Qu YM, Han S. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS One. 2015; 10: e0134715.
54. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbé C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015; 372: 320-30.
55. Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S, Logan TF, Margolin KA, Plimack ER, Lambert AM, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol. 2015; 33: 1430-7.
56. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015; 372: 2521-32.
57. Bristol-Myers Squibb Company. Opdivo (nivolumab) [prescribing information]. 2014.
58. Merck & Co., Inc. Keytruda (pembrolizumab) [prescribing information]. 2015.
59. Sahebjam S, Forsyth PA, Arrington J, Tran ND, Jaglal MV, Mokhtari S, Long W, Macaulay RJ, Wicklund M, Drury-Sibiga A, Gatewood T, Robinson TJ, Raval RR, et al. Nivolumab combined with hypofractionated stereotactic irradiation (HFSRT) for patients with recurrent high grade gliomas: a phase I trial (NCT02829931). J Clin Oncol. 2017; 35: TPS2084.
60. Sahebjam S, Johnstone PA, Forsyth P, Arrington J, Jaglal M, Tran ND, Vrionis FD, Etame AB, Wicklund M, Elie AL, Gatewood T, Macaulay R, Chinnaiyan P, Yu M. ATIM-15. A phase I trial of hypofractionated stereotactic irradiation (hfsrt) with pembrolizumab and bevacizumab in patients with recurrent high grade gliomas. Neuro Oncology. 2016; 18: vi21.
61. Sahebjam S, Johnstone PA, Forsyth PA, Arrington J, Vrionis FD, Etame AB, Tran N, Dalvi P, Kim S, Macaulay R, Chinnaiyan P, Yu M. Safety and antitumor activity of hypofractionated stereotactic irradiation (HFSRT) with pembrolizumab (Pembro) and bevacizumab (Bev) in patients (pts) with recurrent high grade gliomas: Preliminary results from phase I study. J Clin Oncol. 2016; 34: 2041.
62. Sanborn RE, Pishvaian MJ, Kluger HM, Callahan MK, Weise AM, Lutzky J, Yellin MJ, Rawls T, Vitale L, Halim A, Keler T, Davis T, Rizvi NA. Clinical results with combination of anti-CD27 agonist antibody, varlilumab, with anti-PD1 antibody, nivolumab, in advanced cancer patients. J Clin Oncol. 2017; 35: 3007.
63. Brahmer J, Rasco D, Chen M, Masteller E, Qazi I, Rogers S, Sankar N, Sikorski R, Hambleton J, Hodi FS. Abstract B143: A phase 1a/1b study of FPA008 in combination with nivolumab in patients with selected advanced cancers. Cancer Immunol Res. 2016; 4: B143.
64. Perez RP, Riese MJ, Lewis KD, Saleh MN, Daud A, Berlin J, Lee JJ, Mukhopadhyay S, Zhou L, Serbest G, Hamid O. Epacadostat plus nivolumab in patients with advanced solid tumors: preliminary phase I/II results of ECHO-204. J Clin Oncol. 2017; 35: 3003.
65. Reardon DA, Dietrich J, Kaley TJ, Gan HK, Dunn GP, Cloughesy TF, Lim M, Clarke JL, Park AJ, Pan LS, Lai DW, Karakunnel JJ, Robbins PB, et al. Phase II study to evaluate the clinical efficacy and safety of MEDI4736 in patients with glioblastoma (GBM). J Clin Oncol. 2015; 33.
66. Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn GP, Lim M, Cloughesy TF, Gan HK, Park AJ, Schwarzenberger P, Ricciardi T, Macri MJ, Ryan A, et al. Phase 2 study to evaluate safety and efficacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: An update. J Clin Oncol. 2017; 35: 2042.
67. Reardon DA, De Groot J, Colman H, Jordan J, Daras M, Clarke J, Nghiemphu P, Gaffey S, Peters K. Safety of pembrolizumab in combination with bevacizumab in recurrent glioblastoma (rGBM). J Clin Oncol. 2016; 34: 2010.
68. Iwamoto FM, Donovan L. OS09.5 Synergistic effect of reirradiation and PD-1 inhibitors in recurrent high-grade gliomas. Neuro Oncology. 2017; 19: iii19.
69. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013; 369: 134-44.
70. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515: 563-7.
71. Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, Romaguera J, Hagemeister F, Fanale M, Samaniego F, Feng L, Baladandayuthapani V, Wang Z, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014; 15: 69-77.
72. Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, Snyder ES, Ricart AD, Balakumaran A, Rose S, Moskowitz CH. Programmed death-1 blockade with pembrolizumab in patients with Classical Hodgkin Lymphoma after brentuximab vedotin failure. J Clin Oncol. 2016.
73. Choueiri TK, Fishman MN, Escudier B, McDermott DF, Drake CG, Kluger H, Stadler WM, Perez-Gracia JL, McNeel DG, Curti B, Harrison MR, Plimack ER, Appleman L, et al. Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma. Clin Cancer Res. 2016; 22: 5461-71.
74. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014; 515: 568-71.
75. Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, Kaminski MS, Holland HK, Winter JN, Mason JR, Fay JW, Rizzieri DA, Hosing CM, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013; 31: 4199-206.
76. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, Koren-Michowitz M, Shimoni A, Nagler A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008; 14: 3044-51.
77. Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, Zhao X, Martinez AJ, Wang W, Gibney G, Kroeger J, Eysmans C, Sarnaik AA, Chen YA. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013; 31: 4311-8.
78. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366: 2443-54.
79. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013; 369: 122-33.
80. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, Shen X, Boyd Z, Hegde PS, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014; 515: 558-62.
81. Rizvi NA, Mazières J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, Lena H, Minenza E, Mennecier B, Otterson GA, Campos LT, Gandara DR, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015; 16: 257-65.
82. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372: 2018-28.
83. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014; 20: 5064-74.
84. Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017; 23: 4242-50.
85. Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, Chaput N, Eggermont A, Marabelle A, Soria JC, Ferté C. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2017; 23: 1920-8.
86. Saada-Bouzid E, Defaucheux C, Karabajakian A, Coloma VP, Servois V, Paoletti X, Even C, Fayette J, Guigay J, Loirat D, Peyrade F, Alt M, Gal J, Le Tourneau C. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2017; 28: 1605-11.
87. Hodi FS, Hwu WJ, Kefford R, Weber JS, Daud A, Hamid O, Patnaik A, Ribas A, Robert C, Gangadhar TC, Joshua A, Hersey P, Dronca R, et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol. 2016; 34: 1510-7.
88. Mirzaei R, Sarkar S, Yong VW. T cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol. 2017; 38: 104-15.
89. Gustafson MP, Lin Y, New KC, Bulur PA, O'Neill BP, Gastineau DA, Dietz AB. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010; 12: 631-44.
90. Banks WA. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009; 9: S3.
91. Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res. 2013; 19: 1021-34.
92. Zhang Y, Huang S, Gong D, Qin Y, Shen Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell Mol Immunol. 2010; 7: 389-95.
93. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008; 105: 15016-21.
94. Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer. 2015; 113: 232-41.
95. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010; 70: 6171-80.
96. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, Leblanc P, Munn LL, Huang P. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012; 109: 17561-6.
97. Wu X, Giobbie-Hurder A, Liao X, Lawrence D, McDermott D, Zhou J, Rodig S, Hodi FS. VEGF Neutralization plus CTLA-4 blockade alters soluble and cellular factors associated with enhancing lymphocyte infiltration and humoral recognition in melanoma. Cancer Immunol Res. 2016; 4: 858-68.
98. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R, Dhodapkar MV, Dhodapkar KM. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015; 194: 950-9.
99. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010; 107: 4275-80.
100. Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, Cook L, Khleif SN. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol. 2011; 41: 2977-86.
101. Mkrtichyan M, Najjar YG, Raulfs EC, Liu L, Langerman S, Guittard G, Ozbun L, Khleif SN. B7-DC-Ig enhances vaccine effect by a novel mechanism dependent on PD-1 expression level on T cell subsets. J Immunol. 2012; 189: 2338-47.
102. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013; 105: 256-65.
103. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, Griekspoor A, Mesman E, Verreck FA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006; 203: 1259-71.
104. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502: 333-9.
105. Derer A, Spiljar M, Bäumler M, Hecht M, Fietkau M, Frey B, Gaipl US. Chemoradiation increases PD-L1 expression in certain melanoma and glioblastoma cells. Front Immunol. 2016; 7: 610.
106. Workenhe ST, Mossman KL. Mossman, oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther. 2014; 22: 251-6.
107. Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, Yagita H, Overwijk WW, Lizée G, Radvanyi L, Hwu P. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 2012; 72: 5209-18.
108. Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, Salomao D, Cheville J, Hirano F, Lin W, Kasperbauer JL, Ballman KV, Chen L. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003; 63: 6501-5.
109. Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001; 19: 65-91.
110. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002; 8: 793-800.
111. Hardcastle J, Mills L, Malo CS, Jin F, Kurokawa C, Geekiyanage H, Schroeder M, Sarkaria J, Johnson AJ, Galanis E. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2017; 19: 493-502.
112. Heisel SM, Ketter R, Keller A, Klein V, Pallasch CP, Lenhof HP, Meese E. Increased seroreactivity to glioma-expressed antigen 2 in brain tumor patients under radiation. PLoS One. 2008; 3: e2164