
INTRODUCTION
Acute lung injury (ALI) and its severe form acute respiratory distress syndrome (ARDS) are severe inflammatory reactions in the lung, which could cause alveolar damage and result in varied degrees of ventilation-perfusion mismatch, severe hypoxemia, decreased lung compliance and noncardiogenic pulmonary edema [1]. Despite various therapeutic strategies (e.g. lung-protective ventilation strategies [2], prone position [3] and fluid-conservative therapy [4]) have been proposed in the last decade, the mortality of ALI/ARDS remain as high as 40% [5]. Therefore, it is necessary to perform the mechanisms related studies that could be helpful in the therapeutic intervention of ALI.
It is generally accepted that the pathogenesis of ALI was caused by lung inflammation and cell apoptosis, including inflammatory cell accumulation, the aberrant level of proteases, reactive oxygen species (ROS), proinflammatory cytokines, dysfunction of alveolar capillary barrier, and death of pulmonary cells [6–8]. IL-17 is a proinflammatory cytokine produced by the memory CD4 + T cells after activation and has been shown to be invovled in the pulmonary cell emigration under the condition of both local gram-negative bacterial infection [9] and antigenic stimuli [10]. IL-17 overexpression or recombinant IL-17 administration into the lung could result in upregulation of chemokines, which recruit inflammatory cells to the airway [11]. Currently, IL-17 is considered to be an attractive target for inflammatory responses in the lung [12]. However, the exact role of IL-17 and Th17 related responses in ALI have not been defined yet.
Here, we assessed the clinical significance of IL-17 in patients with sepsis-induced ARDS and to investigate the effects and mechanism of IL-17 antibody administration in a mouse model of ALI. Our results showed that increased IL-17 was presented in patients with sepsis-induced ARDS and IL-17 may serve as a biomarker to indicate the severity of ARDS. Moreover, IL-17 blocking antibody administration could relieve the ALI symptom by affecting RAR-related orphan receptor gamma t (RORγt) level and phosphoinositide 3-kinase (PI3K) pathway.
RESULTS
Increased plasma IL-17 level was presented in patients with sepsis-related ARDS
A total of 35 sepsis-related-ARDS patients were included and the plasma level of IL-17 were determined by ELISA. According to the results, we found that significantly increased plasma IL-17 in these patients compared to healthy controls (ARDS: [26.5 ± 6.02] pg/mL vs. Healthy controls: [17.3 ± 3.07] pg/mL, p < 0.05) (Figure 1A). Furthermore, we also determined the plasma IL-17 level in survivors and non-survivors at the different time points and significantly increased IL-17 was found in non-survivors when compared to survivors at Day 7 ([24.9 ± 0.30] pg/mL vs. [22.2 ± 0.15] pg/mL, p < 0.05) (Figure 1B). In addition, we also found a correlation between plasma IL-17 level and the PaO2/FiO2 ratio of ARDS patients (r = −0.738, p < 0.01) (Figure 1C). The baseline characteristics and clinical data are shown in the supplement data (Supplementary Tables 1 and 2).
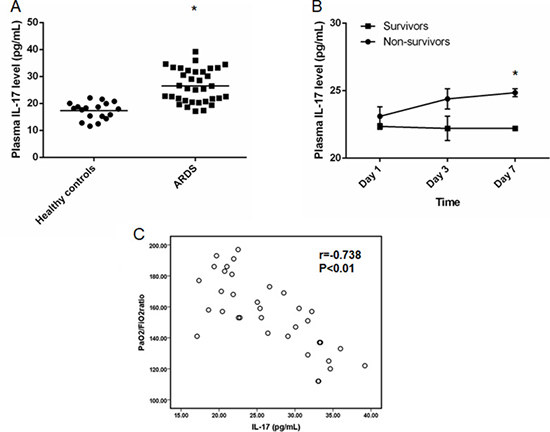
Figure 1: Elevated plasma IL-17 level in patients with sepsis-related ARDS. (A) Significantly increased IL-17 level in patients with sepsis-related ARDS (n = 35) compared to healthy controls (n = 18) (p < 0.05). (B) Plasma IL-17 level for survivors (n = 10) and non-survivors (n = 8) at different time points. Significantly increased plasma IL-17 level was found in non-survivors compared to that in survivors (p < 0.05). (C) Negative correlation was found between IL-17 level and PaO2/FiO2 ratio (r = -0.738, p < 0.01). *represents p < 0.05 after comparison between 2 groups.
Increased IL-17 level was found in a mouse model of ALI
We then established a mouse of ALI and determined IL-17 level from different source of sample, including lung tissue, mBALF and plasma. The results showed that significantly increased IL-17 level was found in lung tissue lyse (Control: 6 h: 12 h: 24 h = (32.96 ± 12.26) : (121.3 ± 18.71) :(135.5 ± 27.1) : (124.5 ± 9.68) pg/mg protein; all the p < 0.05 vs. control) (Figure 2A), mBALF (Control: 6 h: 12 h: 24 h = (5.9 ± 1.32): (25.4 ± 4.67): (29.8 ± 4.07): (20.2 ± 4.31) pg/mL; all the p < 0.05 vs. control) (Figure 2B) and plasma (Control: 6h: 12h: 24 h = (10.6 ± 3.61) : (59.3 ± 8.19): (65.1 ± 8.43): (53.0 ± 6.75) pg/mL; all the p < 0.05 vs. control) (Figure 2C) at 6, 12 and 24 h after ALI when compared to the non-ALI controls.
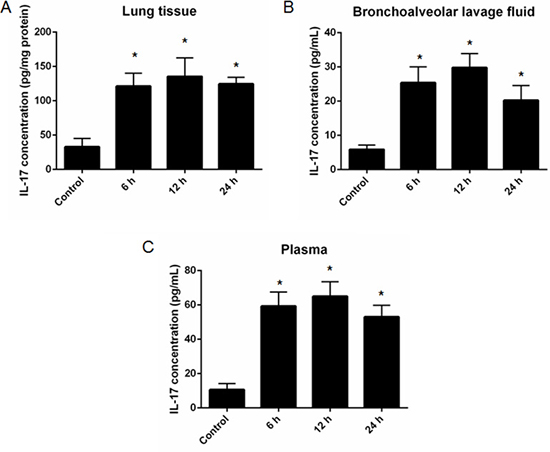
Figure 2: Measurement of IL-17 level in different samples collected from a mouse model of acute lung injury (ALI). Significantly increased IL-17 level was found in lung tissue lysates (A, n = 3), mouse bronchoalveolar lavage fluid (B, n = 3) and plasma (C, n = 3) at 6, 12 and 24 h after ALI . *represents p < 0.05 compared to the control mice which were challenged with physiological saline.
Effects of IL-17 blocking on LPS-induced ALI in vivo
Histological analysis (Figure 3A–3D) and lung injury score assessment (Figure 3E) of lung tissues showed that IL-17 antibody administration could significantly decrease injury such as pulmonary edema, hemorrhage and leukocytes infiltration into alveoli compared with mice in control group. IL-17 antibody administration could significantly decrease lung wet-to-dry ratio (LPS vs. LPS + IL-17 Ab = 6.04 ± 0.58 vs. 5.06 ± 0.11, p < 0.05 ) (Figure 3F), total cells (LPS vs. LPS + IL-17 Ab = [104 ± 7.2] × 104 vs. [75 ± 5.0] ×104 cells/mL , p < 0.05 ) (Figure 3G) and protein level (LPS vs. LPS + IL-17 Ab = [153 ± 23] vs. [96 ± 14] μg/mL, p < 0.05 ) (Figure 3H) in mBALF in mice from LPS + IL-17 Ab group compared to mice in LPS group.
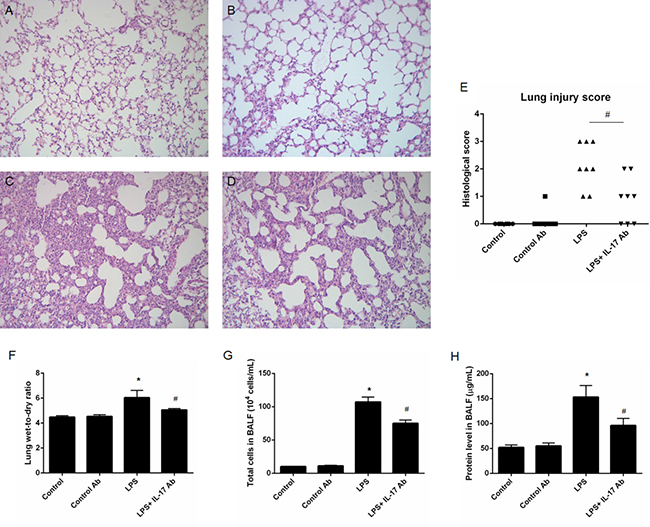
Figure 3: Effects of IL-17 blocking antibody administration on LPS-induced acute lung injury (ALI) in vivo. Histological analysis of lung tissue collected from the ALI mice at 24 post-injury and control mice. (A) Control mice; (B) Mice treated with control antibody; (C) LPS-induced ALI mice; (D) LPS-induced ALI mice treated with IL-17 mAb. (E) Lung injury score; (F) Lung wet-to-dry ratio; (G) Total cell in mouse bronchoalveolar lavage fluid (mBALF) collected from different groups of mice; (H) Protein level in mBALF collected from different groups of mice. All the pathological images were 40× magnification. * represents p < 0.05 when compared to the mice treated with control antibody while # represents p < 0.05 when compared to LPS-induced ALI mice.
IL-17 blocking decrease the level of inflammatory cytokines in ALI
We then measured the inflammatory cytokines in mBALF and plasma in different group of mice. The results showed that IL-17 antibody administration could significantly decrease the TNF-α level in mBALF (LPS vs. LPS + IL-17 Ab = [200.5 ± 15.50] vs. [148.8 ± 10.63] pg/mL, p < 0.05) (Figure 4A) and plasma (LPS vs. LPS + IL-17 Ab = [150.4 ± 22.67] vs. [105.6 ± 10.61] pg/mL, p < 0.05) (Figure 4B) in mice from LPS + IL-17 Ab group compared to mice in LPS group. Moreover, IL-17 blocking antibody administration could also significantly increase IL-10 level in mBALF (LPS vs. LPS + IL-17 Ab = [70.0 ± 7.80] vs. [94.1 ± 7.38] pg/mL, p < 0.05) (Figure 4C) and plasma (LPS vs. LPS + IL-17 Ab = [54.9 ± 4.63] vs. [70.6 ± 5.61] pg/mL, p < 0.05) (Figure 4D) in mice from LPS + IL-17 Ab group compared to mice in LPS group.
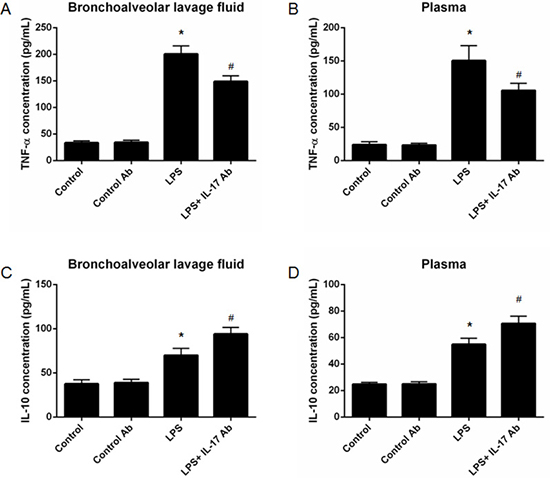
Figure 4: Measurement of inflammatory cytokines in mouse bronchoalveolar lavage fluid (mBALF) and plasma. IL-17 blocking antibody administration could significantly decrease the tumor necrosis factor-α (TNF-α) level in mBALF (A) and plasma (B). IL-17 blocking antibody administration could significantly increase interleukin-10 (IL-10) level in mBALF (C) and plasma (D). *represents p < 0.05 when compared to the mice treated with control antibody while # represents p < 0.05 when compared to LPS-induced ALI mice.
RORγt and activity of PI3K-Akt pathway were involved in the effects of IL-17 antibody administration
To further explore the mechanism, we performed immunohistological analysis of Th-17 related key transcription factor RORγt and employed Western-blot to analyze the RORγt and activity of PI3K-Akt pathway. The results showed that IL-17 blocking antibody administration could decrease the expression of RORγt (Figure 5A–5D by immunohistological assay and p < 0.05 according to Figure 5E and 5F by western-blotting) and Akt phosphorylation (p < 0.05; Figure 5E and 5G by western blotting) were found in in mice from LPS + IL-17 Ab group compared to mice in LPS group.
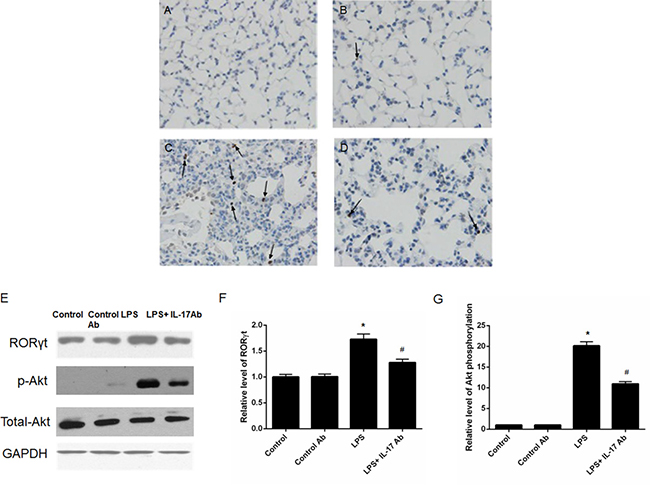
Figure 5: IL-17 blocking attenuated the expression of RORγt and activity of PI3K-Akt pathway. Immunohistological analysis of Th-17 related key transcription factor RORγt. (A) Control mice; (B) Mice treated with control antibody; (C) LPS-induced ALI mice; (D) LPS-induced ALI mice treated with IL-17 blocking antibody. (E) Western-blot analysis of RORγt and activity of PI3K-Akt pathway. IL-17 blocking antibody administration could decrease the expression of RORγt and Akt phosphorylation. (F) Semi-quantitation of the RORγt level. (G) Semi-quantitation of the Akt phoshorylation level. The arrows showed the positive staining of RORγt.* represents p < 0.05 when compared to the mice treated with control antibody while # represents p < 0.05 when compared to LPS-induced ALI mice.
DISCUSSION
As a form of inflammatory disease, ALI could cause alveolar damage and result in varied degrees of ventilation-perfusion mismatch, severe hypoxemia, decreased lung compliance and noncardiogenic pulmonary edema, and finally even death. Recent studies have suggested that the immune regulation disorder may be an important factor in initiating inflammation. Treg and Th17 cells, which belong to CD4 + T cells, have gained much attention. Treg, characterized as a CD4 + CD25 + FOXP3 + T cell, is the master of immune system through a) suppressing a wide array of effector immune cells, including Th cells, B cells dendritic cells and macrophages, and b) secreting of immunosuppressive cytokines, such as TGF-β and IL-10 [13, 14]. Th17 cells, a recently detected effective subset of CD4 + T cells and the major source of IL-17, play an important role in defending a host against microorganisms, such as staphylococcus [15, 16]. A previous study showed that patients with infection-induced ALI/ARDS appeared to have obvious activation and proliferation of T-cells, particularly the presence of Th17 cells [17]. However, the exact role of Th17 has not been determined yet.
In present study, our results demonstrated that significantly increased IL-17 level in patients with sepsis-related ARDS compared to healthy controls while significantly increased plasma IL-17 level was found in non-survivors compared to that in survivors (In the survivor and non-survivors setting, we only detected 18 patients due to the first 17 patients was lost to follow when we performed the Day 3 and Day 7 plasma collection). Significantly increased IL-17 level was found in lung tissue lysate, mBALF and plasma at 6, 12 and 24 h after ALI. Histological analyses revealed that reduced sign of pathological changes in the lungs after IL-17 blocking antibody administration. Reduced level of proinflammatory TNF-α and increased level of anti-inflammatory factor IL-10 were found in both mBALF and plasma. Moreover, IL-17 blocking antibody administration attenuated the expression of RORγt and activity of PI3K-Akt pathway. To the best of our knowledge, this is the first in vivo study to elucidate the role of IL-17 in ALI.
In human ARDS patients, very little has been reported related to IL-17. Recently, Li et al showed that found significant elevation of IL-17A in BALF from patients with ARDS and recombinant IL-17A directly increased permeability across cultured human alveolar epithelial monolayers [18]. Furthermore, Yan et al. also detected increased plasma IL-17 level in ARDS patients [19]. Here, we obtained the similar results as these study, we found increased IL-17 level in ARDS patients and correlation between IL-17 level and ARDS survival. In addition, we detected a significantly increased IL-17 level in mBALF from ALI mice.
The differentiation of Th17 cells is moderated by many pathways. RORγt is one of the most important nuclear transcription factors in this regulation process [20, 21]. Here, we examined the expression of RORγt in lung tissue by immunohistological analysis and western-blot. Both results suggested that ALI could increase the expression of RORγt. Moreover, our results also indicated that IL-17 mAb treatment could attenuate the severity of ALI by affecting RORγt. In addition, Studies have shown that PI3K-Akt signaling positively regulates Th17 differentiation via multiple mechanisms including the regulation of hypoxia-inducible factor 1α (HIF-1 α) expression, Signal transducer and activator of transcription 3 (STAT3) phosphorylation, Gfi1 downregulation, and the nuclear translocation of RORγt [22–24]. Therefore, the Akt phosphorylation could be considered as the initiator during the Th17 differentiation. Here, our results showed that decreased Akt phosphorylation in IL-17 mAb treated ALI mice, supporting the involvement of PI3K-Akt signaling during Th17 cell differentiation.
MATERIALS AND METHODS
Ethical approval of the study protocol
All research involving human participants was approved by the Institutional Review Board of the First Affiliated Hospital of Soochow University School of Medicine, Suzhou, China and written informed consent was obtained from each participating individual. The study protocol was also approved by the Institutional Animal Care and Use Committee of Soochow University School of Medicine as adherent to generally accepted international guidelines for animal experimentation.
Patients
The cohort consisted of 35 sepsis-related-ARDS patients admitted to the respiratory intensive care unit (RICU) between June 1, 2012 and May 31, 2015 at the First Affiliated Hospital of Soochow University. The inclusion criteria, primarily as previously described [25], were performed according to the Berlin definition [26]. The exclusion criteria were as follows: 1) 18 years of age and younger, 2) previously underwent immunosuppressant therapy, 3) received long term glucocorticoid therapy (0.5 mg/Kg/D, ≥ 1 months), 4) confirmed or suspected malignant tumor history, and 5) previously included in other studies. Blood samples were collected within 24 hours (n = 35) and at another 2 time points (n = 18) after the patient met the inclusion criteria. Briefly, peripheral venous blood samples were drawn on an agreed upon time point and centrifuged at 3000 rpm for 15 minutes. The plasma was stored at −80°C for ELISA testing. The plasma levels of IL-17 (Shanghai Yanhui Biological Technology Co., Ltd) was measured using commercially available sandwich ELISA kits.
Animals
Male C57BL/6 mice (6–8 weeks old) were purchased from the Experimental Animal Center of the Soochow University and were maintained in cages at temperature of 24–26°C under a 12 hr light/dark cycle with free access to food and water.
Acute lung injury (ALI) model and mice grouping
Mice were randomly divided into 4 groups as follows: Control group (PBS, n = 8), Control antibody group (treated with 1μg control antibody, n = 8), LPS group (treated with 3 mg/kg LPS, n = 8) and LPS + IL-17 antibody (treated with 3 mg/kg LPS and 1μg IL-17 antibody, n = 8). After grouping, these mice were anesthetized through intraperitoneal administration of sodium pentbarbital (40 mg/kg) and intratracheally instilled with 3 mg/kg LPS (Sigma, prepared from Escherichia coli 0111:B4) in 50 μ L PBS or sterile PBS alone (as the control group), and control antibody and IL-17 antibody were infused through tail vein injection after LPS instillation for 6 h. The mice in these 4 groups were sacrificed by cervical dislocation at the time point of 24 h post-exposure to evaluate the severity of the ALI. Since all the mice were sacrificed at 24 h post the LPS installation. No mice mortalities was found.
Mouse bronchoalveolar lavage (mBAL)
mBAL was performed with the whole lung. Three hundred microliter aliquots of 37°C, sterile, pyrogen-free, 0.9% saline were flushed through the tracheotomy tube and this process was repeated for 5 times. The five fractions were recovered and pooled. The total number of cells was counted using a standard hemocytometer. The mBAL fluid (mBALF) was then centrifuged at 600× g for 5 min. The supernatant was collected and stored at −80°C. Protein quantification was determined by using BCA Protein Assay Kit (Pierce, Rockford, IL, USA).
Lung wet-to-dry weight ratio
To measure the total amount of lung water, the animals were dissected under deep sevoflurane anesthesia, and the lung weight was measured immediately after its excision (wet weight). The lung tissue was then dried in an oven at 60°C for 5 days and re-weighed as dry weight. The wet-to-dry lung weight ratio (W: D ratio) was calculated by dividing the wet weight by the dry weight.
Histology
Lung from different treatment group were collected 6, 24, 48 h after LPS induced lung injury and fixed with 10% formalin. After fixation, the lungs were embedded in paraffin, cut into 5 μm section and stained with hematoxylin-eosin staining (H&E staining) and observed under by microscopy (Zeiss, Gottingen, Germany). Slides were blinded and scored by an experienced pathologist in a semiquantitative manner according to the relative degree of inflammatory infiltration. Inflammation was scored as follows: 0, no inflammation; 1, perivascular cuff of inflammatory cells; 2, mild inflammation, extending throughout 25% of the lung; 3, moderate inflammation covering 25–50% of the lung; 4, severe inflammation involving over one-half of the lung.
Immunohistochemistry
Lung tissue were processed and immunostained as previously described [27]. Negative controls, prepared by omission of primary antibody or substitution with a non-immune, isotype control primary antibody were examined to confirm the specificity of primary antibodies. Th17 cells were detected with rabbit anti-RORγt IgG (Cell Signaling Technology, Cambridge, MA, USA) and HRP-conjugated goat anti-rabbit IgG. Finally, the sections were stained with oxidase diaminobenzidine (DAB) (ZSGB-BIO, China) for 5 min, followed by hematoxylin staining for 2 min.
Measurement of IL-10 and TNF-α
The concentrations of interleukin 10 (IL-10) and Tumor necrosis factor-α (TNF-α) in mBALF and plasma were quantified by enzyme-linked immunosorbent assay (ELISA), using standard commercially available ELISA kits (R&D Systems, Minneapolis, MN, USA).
Western blotting
Frozen lung tissues obtained from model mice were lysed in RIPA buffer, followed by high-speed centrifugation and quantification using bicinchoninic acid. Cellular proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidenedifluoride membranes. After blocking, membranes were incubated with total or phospho-Akt and RORγt monoclonal primary antibodies (Cell Signaling Technology, Cambridge, MA, USA). GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as the loading control. Appropriate horseradish peroxidase-conjugated secondary antibodies were applied. The protein bands were detected with SuperSignal Ultra Chemiluminescent Substrate (Pierce, Rockford, IL, USA) on X-ray films (Kodak, Tokyo, Japan). The quantitation of western-blot was fulfilled by using Image J software (National Institutes of Health).
Statistical analysis
All statistical analyses were performed using the SPSS13.0 software (SPSS Inc., Chicago, IL, USA). The results were presented as means ± standard deviation (SD). One way ANOVA or student t test was used to examine the differences among multiple groups or between 2 groups. Correlation analysis was performed by spearman test. P < 0.05 was considered as statistically significance.
CONCLUSIONS
In conclusion, we found here that increased IL-17 was presented in patients with sepsis-induced ARDS and IL-17 may serve as a biomarker to indicate the severity of ARDS. Moreover, IL-17 blocking antibody administration could relieve the ALI symptom by affecting RORγt level and PI3K pathway.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
This work was supported by grants from the National Natural Science Foundation of China (31270940), Clinical Medical Center of Suzhou (SZZX201502) and Clinical Key Speciality Project of China. Suzhou Municipal Natural Science Foundation (SYSD2015107).
REFERENCES
1. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005; 353:1685–1693.
2. Brower RG, Matthay MA, Morris A, Schoenfeld D, Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000; 342:1301–1308.
3. Guérin C, Reignier J, Richard JC, Beuret P, Gacouin A, Boulain T, Mercier E, Badet M, Mercat A, Baudin O, Clavel M, Chatellier D, Jaber S, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med. 2013; 368:2159–2168.
4. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012; 122:2731–2740.
5. Zambon M, Vincent JL. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest. 2008; 133:1120–1127.
6. Chopra M, Reuben JS, Sharma AC. Acute lung injury:apoptosis and signaling mechanisms. Exp Biol Med (Maywood). 2009; 234:361–371.
7. Mendelson CR. Role of transcription factors in fetal lung development and surfactant protein gene expression. Annu Rev Physiol. 2000; 62:875–915.
8. Rooney SA. Regulation of surfactant secretion. Comp Biochem Physiol A Mol Integr Physiol. 2000; 129:233–243.
9. Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, Kolls JK. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001; 25:335–340.
10. Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, Mathieu C, Ceuppens JL. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003; 28:42–50.
11. Laan M, Cui ZH, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC, Skoogh BE, Lindén A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999; 162:2347–2352.
12. Jones CE, Chan K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-alpha, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am J Respir Cell Mol Biol. 2002; 26:748–753.
13. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Pillars article: immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 2011; 186:3808–3821.
14. Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology. 2006; 117:433–442.
15. Kim MR, Hong SW, Choi EB, Lee WH, Kim YS, Jeon SG, Jang MH, Gho YS, Kim YK. Staphylococcus aureus-derived extracellular vesicles induce neutrophilic pulmonary inflammation via both Th1 and Th17 cell responses. Allergy. 2012; 67:1271–1281.
16. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011; 34:149–162.
17. Risso K, Kumar G, Ticchioni M, Sanfiorenzo C, Dellamonica J, Guillouet-de Salvador F, Bernardin G, Marquette CH, Roger PM. Early infectious acute respiratory distress syndrome is characterized by activation and proliferation of alveolar T-cells. Eur J Clin Microbiol Infect Dis. 2015; 34:1111–1118.
18. Li JT, Melton AC, Su G, Hamm DE, LaFemina M, Howard J, Fang X, Bhat S, Huynh KM, O'Kane CM, Ingram RJ, Muir RR, McAuley DF, et al. Unexpected Role for Adaptive alphabetaTh17 Cells in Acute Respiratory Distress Syndrome. J Immunol. 2015; 195:87–95.
19. Yan Z, Xiaoyu Z, Zhixin S, Di Q, Xinyu D, Jing X, Jing H, Wang D, Xi Z, Chunrong Z, Daoxin W. Rapamycin attenuates acute lung injury induced by LPS through inhibition of Th17 cell proliferation in mice. Sci Rep. 2016; 6:20156.
20. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directsthe differentiation program of proinflammatory IL-17 + T helper cells. Cell. 2006; 126:1121–1133.
21. Eberl G, Littman DR. The role of the nuclear hormone receptor RORgammat in the development of lymph nodes and Peyer’s patches. Immunol Rev. 2003; 195:81–90.
22. Nagai S, Kurebayashi Y, Koyasu S. Role of PI3K/Akt and mTOR complexes in Th17 cell differentiation. Ann N Y Acad Sci. 2013; 1280:30–34.
23. Okkenhaug K, Patton DT, Bilancio A, Garçon F, Rowan WC, Vanhaesebroeck B. The p110delta isoform of phosphoinositidekinase controls clonal expansion and differentiation of Th cells. J Immunol. 2006; 177:5122–5128.
24. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008; 28:454–467.
25. Tang L, Zhao Y, Wang D, Deng W, Li C, Li Q, Huang S, Shu C. Endocan levels in peripheral blood predict outcomes of acute respiratory distress syndrome. Mediators Inflamm. 2014;2014:625180.
26. Ferguson ND, Fan E, Camporota L, Antonelli M, Anzueto A, Beale R, Brochard L, Brower R, Esteban A, Gattinoni L, Rhodes A, Slutsky AS, Vincent JL, et al. The Berlin definition of ARDS: an expanded rationale, justification, and supplementary material. Intensive Care Med. 2012; 38:1573–1582.
27. Ji Y, Strawn TL, Grunz EA, Stevenson MJ, Lohman AW, Lawrence DA, Fay WP. Multifaceted role of plasminogen activator inhibitor-1 in regulating early remodeling of vein bypass grafts. Arterioscler Thromb Vasc Biol. 2011; 31:1781–1787.