
During the past 30 years, cancer research has been dominated by molecular biology, which has drastically overshadowed other credible approaches in the field. Metabolism appears to be the most etiolated field under the large umbrella of gene-centered cancer research. Unsurprisingly, the unique metabolic signatures of cancer cells, which were recognized nearly a century ago by Otto Warburg [1-3], have been frequently perceived as evolutionary conserved targets that are programmed by oncogenic gain-of-function events and the loss of tumor suppressors [4-6]. In the last few years, however, provocative evidence has accumulated to suggest that metabolism is not simply a consequence; rather, it might play a pivotal role in dictating the different phenotypic states exhibited by heterogeneous cancer cell populations, including those referred to as cancer stem (CS) cells [7-13]. CS cells have been causally implicated as drivers of primary tumor growth and are involved in seeding and establishing metastasis from most human epithelial carcinomas. On the one hand, cancer is now understood to be a disease of cellular reprogramming that involves progressive resetting of the metabolic infrastructure and metabolite levels concomitantly with changes in cellular differentiation [7, 14]. On the other hand, the modulation of metabolism and the associated signaling pathways has been increasingly implicated in cell identity determination during cellular reprogramming and oncogenesis [15-20]. Crucially, a transformation in cellular metabolism appears to precede changes in stemness; therefore, metabolic reprogramming may reflect the molecular dynamics fundamental to cell fate rearrangement and redirection.
Recent studies have revealed that a metabolic switch to glucose metabolism is a critical promotional event in the epithelial-to-mesenchymal (EMT)-driven CS-like phenotype. Epigenetic silencing of the gluconeogenic enzyme fructose-1,6-biphosphatase, which catalyzes the energy-consuming conversion of fructose 1,6-biphosphate to fructose-6-phosphate, is employed by CS cells as a mechanism of glucose flux maintenance via glycolysis and other associated biosynthetic pathways [21]. An increased reliance on glucose metabolism, in turn, reduces the level of reactive oxygen species (ROS) to promote EMT and the CS-like phenotype. Glucose also appears to act as an essential nutrient for CS cells, as its presence in the culture environment significantly increases the percentage of CS-like cells in the overall cancer cell population [22]; in contrast, glucose starvation is sufficient to cause a rapid depletion of the CS-like subpopulation in vitro. However, the ultimate metabolic features of CS cells and the effects of specific nutrients on CS cells remain largely unexplored. Moreover, although the important effects of the tumor tissue niche on CS cells have been recognized in recent years, the impacts of key tumor microenvironmental nutrients on CS cells have not yet been determined.
We recently suggested that CS cells might possess specific metabolic properties that distinguish them from the majority of tumor cells and that such metabotypes may constitute a basis for new therapeutic strategies that metabolically eliminate CS cells [7, 23-25]. Herein, we hypothesized that a systematic evaluation of the phenotypic variations of non-CS cells and CS isogenic cell types in response to multiple carbon and nitrogen substrates may unambiguously define a preliminary nutritional phenome of CS cells in terms of their energy-producing metabolic pathways. Stable sibling cell lines in which an EMT had been induced to stably propagate CS-like-enriched populations [26, 27] were subjected to Phenotype MicroArrays for Mammalian Cells (PMM) (Biolog, Hayward CA, USA), a standardized platform for high-throughput metabolic phenotyping [28-32]. EMT-induced CS-like cells and isogenic control cells were simultaneously screened for the ability to generate energy-rich NADH when cultured in wells that had been pre-loaded with >350 different carbon-energy and nitrogen-based substrates, including carbohydrates/starches, alcohols, fatty acids, ketones, carboxylic acids, amino acids, and bi-amino acids. We present herein the first-in-class nutritional-phenomic map confirming that the enhancement of certain catabolic energy-producing pathways in CS-like cells should be considered a specific nutritional property that phenotypically distinguishes CS cells from non-CS cells. Crucially, we present the first evidence of the existence of a cell-autonomous “reverse Warburg effect” in EMT-driven CS-like cells, where reprogramming to a CS-like cellular state appears sufficient to allow a cell-autonomous, vectorial transfer of energy-rich nutrients from the extracellular microenvironment to the CS-like cells’ energy-producing catabolic pathways.
Nutritional phenome of non-starved, EMT-induced CS-like cellular states
We used experimentally transformed HMLER breast cancer cells (human mammary epithelial cells [HMECs] overexpressing hTERT, SV40 T/t and H-RasV12) that had been modified to inhibit expression of the human CDH1 (E-cadherin) gene via short hairpin RNA (shRNA; HMLERshECad cells), which constitutes a valuable method for drastically enriching cells with CS-like properties [26, 27]. We simultaneously profiled these cells and the stable isogenic line HMLERshCntrol in four microplates (termed PM-Ms) in which the bottoms of the wells had been coated with substrate nutrients to create 367 unique culture conditions. PM-M1 contained primarily carbohydrate and carboxylate substrates, whereas PM-M2, M3, and M4 contained individual L-amino acids and most dipeptide combinations. The PM assay was conducted during a 2-day incubation period, and the HMLERshCntrol and HMLERshECad cells were incubated in Biolog IF-M1 medium (RPMI 1640 without glucose/glutamine; this medium provided all nutritional ingredients at sufficient levels other than major C- and N-sources, which were omitted) containing 5% serum.
Because the color formed from each substrate reflected the energy-producing activity of the associated catabolic pathway, it was clear that non-CS HMLERshCntrol and CS-like HMLERshEcad cells both exhibited strong reductive responses in wells containing D-glucose (Fig. 1 and Fig. 2; green boxes [positive controls], all panels) and little or no response in wells lacking any carbon source (Fig. 1 and Fig. 2; red boxes [negative controls], all panels). To quantitatively compare each state rapidly and systematically, we developed a scoring system based on the fold change in the optical density of each substrate at 590 nm (purple color) resulting from the accumulation of reduced dye over a 6-hour period after normalization of the values to those of the negative-control wells included in each of the PM-M plates. To quantify these comparisons, we also calculated a comparison score from the absolute ratio between the metabolic flows of the non-CS and CS-like cells upon comparison at the same time point (6 h).
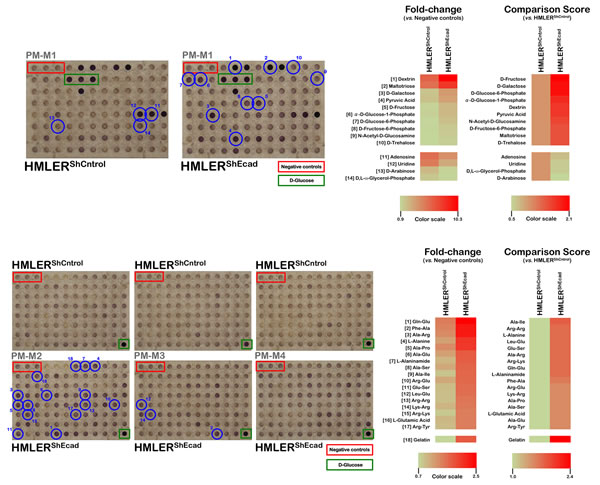
Figure 1: Metabolic fingerprint of non-starved, EMT-induced CS-like cellular states. Right panels. 50 µL per well of 400,000 cells/mL suspensions of non-CS HMLERshCntrol and CS-like HMLERshECad cells (20,000 cells per well) in Biolog IF-M1 medium, i.e., RPMI-1640 medium that lacked phenol red and depleted of carbon-energy sources (no glucose, low glutamine [0.3 mmol/L] and low FBS [5%]), were inoculated into Phenotype MicroArrays PM-M1 through PM-M4 (Biolog, Hayward, CA) which contained 367 biochemical substrates that could potentially be metabolized and provide energy for cells. After 48 h incubation in RPMI-1640 and glucose and was supplemented with penicillin/streptomycin and reduced levels of glutamine [0.3 mmol/L] and FBS, plates were incubated at 37 oC under air to assess dye reduction 6 h (Redox Dye Mix MA) and then photographed. This 2-days incubation should allow cells to use up residual carbon-energy sources in the 5% serum (e.g., 5% serum would contribute about 0.35 mmol/L glucose, plus lipids, and amino acids) and minimizes the background color in the negative control wells, which have no added biochemical substrate [30]. Furthermore, the 2-days incubation should allow cells to transition their metabolism to use the various substrates provided in the wells. The respective utilization of substrates to generate energy-rich NADH was measured as ODs at 590 nm. Negative controls (red boxes) have no substrate in the well. Wells containing D-glucose (green boxes) serve as positive controls. Thresholds were set to disregard small and insignificant changes, and all of the wells that exceed this threshold are marked with blue circles to denote differentially metabolized substrates for each cell line, which are described in the text. Parallel experimentation using conventional 96-well plates was performed using the MTT redox dye to ensure that the 2-days incubation with Biolog IF-M1 medium failed to significantly alter the baseline cell growth of non-CS HMLERshCntrol and CS-like HMLERshECad cells (data not shown). Importantly, whereas widely employed redox dyes such as MTT, MTS or XTT measure nonspecific cellular reductase activities, the redox chemistry employed in the PMM technology give very little non-specific dye reduction and are reduced in a manner that is strictly dependent on the presence of usable carbon-energy sources in the medium. Therefore, this redox dye approach accurately measures reductase activity due to energy- (i.e., NADH) producing catabolic pathways that use diverse biochemical substrates. Left panels. Phenetic maps of carbon and nitrogen utilization patterns of non-CS HMLERshCntrol and CS-like HMLERshECad cells under non-starved conditions. Phenotypes that are lost are colored green and phenotypes that are gained are colored red; the exact relative values are given by a corresponding color as indicated at the color scales.
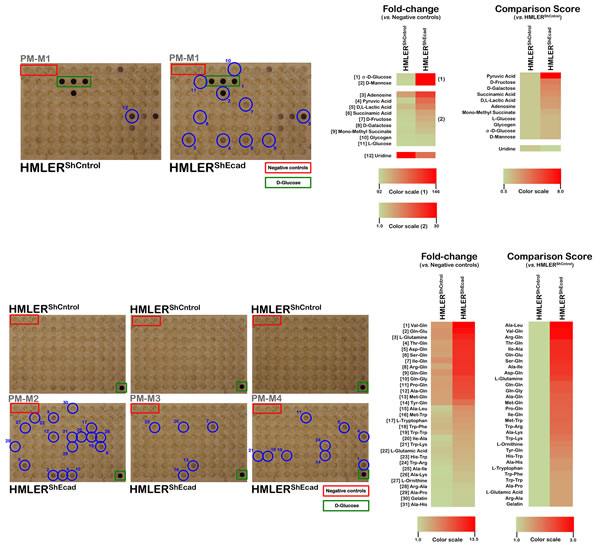
Figure 2: Metabolic fingerprint of the starved EMT-induced CS-like cellular state. Right panels. Experiments were conducted as in Figure 1 with the exception of the culture medium employed, which lacked serum, glucose, and amino acids. Left panels. Phenetic maps of carbon and nitrogen utilization patterns of non-CS HMLERshCntrol and CS-like HMLERshECad cells under starvation conditions. Phenotypes that are lost are colored green and phenotypes that are gained are colored red; the exact relative values are given by a corresponding color as indicated at the color scales.
Most changes that occurred following the acquisition of a CS-like cellular state (28 out of 31, 90%) were increases in the ability to generate energy from a higher extracellular nutrient level. Quantitatively, the greatest difference (2.1-fold) between the non-CS HMLERshCntrol and CS-like HMLERshEcad cells was observed for the ability of CS-like HMLERshEcad cell populations to metabolize monosaccharides such as D-fructose and D-galactose (Fig. 1, top panels). We observed higher sugar phosphate metabolism (D-glucose-6-phosphate, α-D-glucose 1-phosphate, and D-fructose-6-phosphate) by CS-like HMLERshEcad cells. CS-like HMLERshEcad cells also exhibited increased metabolism of the amide between glucosamine and acetic acid (N-acetyl-D-glucosamine), a monosaccharide glucose derivative. The metabolism of disaccharides (D-trehalose), trisaccharides (maltotriose), and mixtures of D-glucose (dextrin) polymers also increased in CS-like HMLERshEcad cells. Interestingly, the ketone pyruvic acid, the end product of glycolysis, was a substrate for which dye reduction significantly increased in CS-like HMLERshEcad cells relative to the basal metabolic flows observed in HMLERshCntrl cells in all cases.
There were only three carbon sources to which CS-like HMLERshEcad cells exhibited a decreased catabolic response relative to the HMLERshCntrl cells. CS-like HMLERshEcad cells were slightly less efficient than HMLERshCntrl cells in the catabolism of D, L-α-glycerol-3-phosphate, an intermediate common to both the lipid and carbohydrate metabolic pathways. CS-like HMLERshEcad cells also exhibited reduced uridine metabolism and notably reduced by half the ability to catabolize the monosaccharide D-arabinose relative to HMLERshCntrl cells (Fig. 1, top panels).
Despite a low absolute rate of dye reduction, CS-like HMLERshEcad cells demonstrated enhanced catabolic responses to the amino acids alanine, glutamic acid, and arginine in addition to the dipeptides containing these amino acids (Fig. 1, bottom panels). Notably, whereas HMLERshCntrl cells completely failed to produce energy from gelatin, a mixture of peptides and proteins produced by partial collagen hydrolysis, CS-like HMLERshEcad cells efficiently used gelatin as a catabolic substrate to generate energy-rich NADH.
Nutritional phenome of the starved EMT-induced CS-like cellular state
Cell metabolism under standard culture conditions may differ significantly from in vivo conditions because the cancer cell microenvironment is generally characterized by poor blood perfusion, hypoxia, and nutrient limitations. To evaluate how the cell-autonomous changes in nutrient utilization observed in EMT-induced CS-like cells would be affected by the stressful conditions of the tumor microenvironment, we partially recapitulated this specific metabolic milieu by re-testing the abilities of CS-like HMLERshEcad cells and non-CS HMLERshCntrol parental cells to produce energy-rich NADH from the same panel of 367 substrate nutrients in serum-, glucose-, and amino acid-free Biolog IF-M2 medium (RPMI 1640 without glucose/glutamine/amino acids) (Fig. 2). As the cell response to the nutrients over a 2-day incubation period mostly comprised rapid cell death, similar to the responses observed in wells with no substrates, this assay format can be considered to evaluate cell survival responses under different nutrient supply conditions.
Similarly to non-starved conditions, most of the changes that occurred following the acquisition of a CS-like cellular state upon starvation comprised increases in the ability to generate energy from a broader range of extracellular nutrients (42 out of 43, 98%). Remarkably, the greatest quantitative difference was in pyruvic acid metabolism, wherein starved CS-like HMLERshEcad cells exhibited a metabolic level 8.0-fold higher than that observed in starved non-CS HMLERshCntrol cells (Fig. 2, top panels). Moreover, pyruvic acid was not the sole glycolysis end product efficiently metabolized by the starved CS-like HMLERshEcad cells; D, L-lactic acid, the most common accumulated acidic waste in the extracellular tumor tissue microenvironment, was efficiently utilized as an energetic fuel 2.6-fold faster in starved CS-like HMLERshEcad cells than in starved non-CS HMLERshCntrol cells. In response to starvation, the CS-like HMLERshEcad cell populations exhibited a further increase in the ability to metabolize the monosaccharides D-fructose and D-galactose in addition to D-glucose. The metabolism of succinamic acid and its monomethyl ester was notably increased in starved CS-like HMLERshEcad cells relative to starved HMLERshCntrl cells. Intriguingly, the energy-generating capabilities of starved CS-like HMLERshEcad cells appeared to not be restricted to D-sugars, as these cells were able to generate energy-rich NADH in a minimal medium that contained L-glucose as the sole carbon source. Starved CS-like HMLERshEcad cells also significantly gained the ability to use the purine nucleoside adenosine as an energy-generating metabolic substrate (2.5-fold increase relative to starved HMLERshCntrl cells).
Interestingly, as observed in the non-starved CS-like HMLERshEcad cells, starved CS-like HMLERshEcad cells reduced by half their ability to catabolize the nucleoside base uridine relative to starved HMLERshCntrl cells (Fig. 2, top panels).
Starved CS-like HMLERshEcad cells notably acquired the ability to catalyze glutamine and multiple glutamine-containing dipeptides (e.g., 13) to generate energy-rich NADH (Fig. 2, bottom panels). Starved CS-like HMLERshEcad cells additionally gained the ability to catabolize the amino acid tryptophan and tryptophan-containing dipeptides.
EMT-induced CS-like cells strongly catabolize exogenous mitochondrial fuel
To further corroborate the above-mentioned findings suggesting that EMT-induced CS-like cellular states could cell-autonomously enhance the vectorial transfer of energy-rich nutrients from the extracellular microenvironment to the energy-producing catabolic pathways in CS-like cells, we used the PM-M TOX1 MicroPlate to re-assess the differential utilization of eight carbon substrates by cellular mitochondria that are fed into the electron transport chain at different points (Fig. 3). The hexose D-galactose is metabolized to NADH via mitochondrial activity, whereas α-D-glucose can bypass these mitochondrial functions. Glucose-1-phosphate is metabolized differently from glucose and galactose, whereas ribose-containing inosine and xylitol are both metabolized (albeit differently) via the pentose phosphate pathway. α-ketoglutarate directly enters the tricarboxylic acid cycle (TCA) cycle, whereas the ketones β-hydroxybutyrate and pyruvic acid enter the TCA cycle upon metabolism and linkage to coenzyme A.
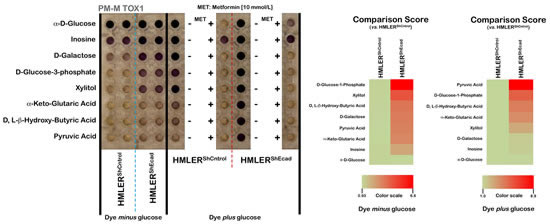
Figure 3: Alternative mitochondrial fuel of EMT-induced CS-like cellular states. Right panels. Suspensions of non-CS HMLERshCntrol and CS-like HMLERshECad cells in RPMI-1640 that lacked phenol red and glucose were dispensed into Biolog PM-M TOX1 MicroPlate (Biolog, Hayward, CA) wells whose rows contain different carbon/energy sources as indicated. After two days of incubation, Biolog Redox Dye Mix MB without or containing glucose was added, plates were incubated at 37 oC under air to assess dye reduction 6 h (Redox Dye Mix MA) and then photographed. The respective utilization of substrates to generate energy-rich NADH was measured as ODs at 590 nm. Left panels. Phenetic maps of the utilization patterns of mitochondrial substrates in non-CS HMLERshCntrol and CS-like HMLERshECad cells. Phenotypes that are lost are colored green and phenotypes that are gained are colored red; the exact relative values are given by a corresponding color as indicated at the color scales.
When these 8 substrates were tested to probe the differential utilization of bioenergetic pathways to generate NADH using tetrazolium dye with omission of glucose (the so-called “substrate metabolism assay”), we first confirmed that CS-like HMLERshEcad cells metabolized the sugar phosphate D-glucose-1-phosphate and the monosaccharide D-galactose faster (approximate 7-fold and 3.5-fold increases, respectively) than HMLERshCntrl parental cells. Whereas non-CS HMLERshCntrl parental cells were fully unable to generate energy-rich NADH from the non-fermentable sugar xylitol, xylitol was found to be a very efficient energy source for the CS-like HMLERshEcad cells (approximate 4.5-fold increase). CS-like HMLERshEcad cells notably acquired the ability to efficiently generate energy-rich NADH (approximate 3.5-fold increase versus HMLERshCntrl parental cells) from D, L-β-hydroxy-butyric acid, a ketone body that is produced during ketosis and used as an energy source by the brain under conditions of low blood glucose. α-keto-glutaric acid, one of the two ketone derivatives of glutaric acid, was also catabolized by CS-like HMLERshEcad cells but not by HMLERshCntrl parental cells.
When Biolog Redox Dye MB, which contains glucose, was added to detect the metabolic activity of the cells that remained viable after a 48-h incubation with the 8 different carbon substrates (the so-called “cell viability assay”), a highly divergent pattern was again observed between non-CS HMLERshCntrl parental cells and CS-like HMLERshEcad cells. Whereas CS-like HMLERshEcad cells already catabolized pyruvate, the end product of glycolysis, approximately 3-fold more rapidly than HMLERshCntrl parental cells in the absence of exogenous glucose, this capability was enhanced further in the presence of exogenous glucose (approximate 9-fold increase versus HMLERshCntrl parental cells). Intriguingly, re-addition of exogenous glucose was not sufficient to rescue the very low levels of catabolism observed in non-CS HMLERshCntrl parental cells after a 48-h exposure to the ketones pyruvic acid, D, L-β-hydroxy-butyric acid, and α-keto-glutaric acid. Conversely, exogenous glucose supplementation notably augmented the catabolic generation of energy-rich NADH in CS-like HMLERshEcad cells, regardless of the nature of the carbon substrates employed.
To further study the mechanistic aspects of these dramatic changes observed in the dye-reduction assay, we tested non-CS HMLERshCntrl parental cells and CS-like HMLERshEcad cells in the PM-M TOX1 MicroPlate in the presence of high concentrations of the mitochondrial poison metformin. The re-addition of glucose was apparently insufficient to rescue the cells from the cytotoxic effects of metformin, which completely blocked dye reduction by substrates that would presumably be oxidized mitochondrially but partially rescued the production of NADH from inosine.
The nutritional phenotype for the EMT-induced CS-like cellular states
A new perspective in cancer research. Universal phenotyping techniques that could discriminate among the various dynamically co-existing cellular states in heterogeneous cancer tissues may radically amend the current perception of cancer disease treatment [33-35]. Such techniques are a crucial research theme for CS cell-cellular state identification and monitoring because carcinoma aggressiveness derives not from the pre-existing CS cell content but rather from the proclivity of tumor tissues to generate new CS cells from non-CS cell populations [36-38]. A crucial challenge for the application of functional genomics to CS cells is the identification of genotypes associated with a specific phenotype. However, in contrast to well-established genotyping platforms, standard quantitative phenotyping methods are currently not available. Because normal and neoplastic epithelial cells have been shown to possess sufficient plasticity to re-enter the stem cell state via the interpretation of multiple microenvironmental signals [39-45], we recently reasoned that certain extracellular nutrients may be major determinants of the maintenance and/or expansion of the metabolic states characteristic of a CS-like cellular state. A comprehensive evaluation of the phenotypic variations among non-CS and CS-like isogenic cells based on a single-assay metabolic profile of several hundred nutrient sources may thus constitute a novel approach to the precise identification of CS-like cellular states and a starting point for uncovering the microecological nutritional niches of CS cells. In this scenario, we envisioned that PMM (Phenotype MicroArrays for Mammalian Cells) technology, which has been frequently used to analyze bacterial, yeast, and fungal mutant strains [28-32], would be well suited to a nutrient-response profile assessment of human CS-like cells. Indeed, the use of this standardized high-throughput metabolic phenotyping platform in the present study provided us with the first nutritional phenome for the EMT-driven CS-like cellular state with regard to energy-producing pathways.
Even at a gross level, PM-based global nutritional profiling immediately suggested important physiological consequences of acquiring a CS-like cellular state, as evidenced by the sole and specific loss of the epithelial marker E-cadherin. Both the qualitative and quantitative metabolic differences for the 367 substrates provided a simple but highly informative metabolic characterization that reflected the singular bioenergetics of acquired stemness in a cancer cell population. First, the CS-like HMLERshEcad cells were much more metabolically active under both non-starved (28 additional energy-generating substrates) and starved (42 additional energy-generating substrates) conditions than their non-CS HMLERshCntrol isogenic counterparts. Second, the types of bioenergetic fuels that could be employed by EMT-driven CS-like cells strictly corresponded to the availability of nutrients and energy sources in the extracellular milieu. For example, whereas an enhanced ability to generate energy from alanine and alanine-containing peptides appeared to be a metabolic property acquired by CS-like cells regardless of the microenvironmental condition (starved or non-starved), starved CS-like HMLERshEcad cells exhibited a notable reduction in their ability to catabolize glutamate and glutamate-containing peptides, an acquired ability that was initially discovered in non-starved CS-like HMLERshEcad cells. Alternatively, starved CS-like HMLERshEcad cells rapidly acquired the ability to generate energy-rich NADH from glutamine and glutamine-containing peptides. Because tetrazolium dye reduction through nitrogen source utilization is measured indirectly via a linkage with carbon energy metabolism, these findings indirectly revealed that the coordination of NADH-producing metabolic pathways was drastically altered in EMT-driven CS-like cellular states relative to non-CS cellular states in an otherwise isogenic background.
The nutritional phenome of CSCs: A new perspective on the “reverse Warburg effect”
A landmark study reported by Sonveaux et al. in 2008 [46, 47] suggested a “symbiotic relationship” among cancer cells in the tumor microenvironment where cancer cells distal to a blood vessel would be deprived of oxygen by cancer cells located proximal to the blood vessel. These distal hypoxic cells would then exhibit a different metabolic profile than the proximal cells and, indeed, hypoxic cells distal to the blood vessel have been shown to convert glucose to lactate, which could then be imported into aerobic cells and converted to pyruvate for mitochondrial oxidation. This model has been recently extended by the Lisanti group to suggest that the Warburg effect occurs in stromal cells rather than in cancer cells that feed off of stromal cell-generated lactate; this is in contrast to normal differentiated cells, which rely primarily on mitochondrial oxidative phosphorylation (OXPHOS) to generate the energy needed for cellular processes, as most cancer tissues instead rely on glycolysis even in the presence of sufficient oxygen. The provocative view submitted by the Lisanti group established that in cancer cells, mitochondrial OXPHOS can induce oxidative stress in adjacent fibroblasts (cancer-associated fibroblasts [CAFs]) via H2O2 and reactive oxygen species (ROS), thus resulting in the onset of an autophagic phenotype in CAFs. This autophagic phenotype subsequently leads to a loss of mitochondria via mitophagy and forces CAFs to undergo aerobic glycolysis (“reverse Warburg effect”) [48-65]. The increased aerobic glycolysis resulting from enhanced mitochondrial turnover in stromal cells generates excessive lactate, pyruvate, and other ketones, which are secreted into the intracellular space. The products of aerobic glycolysis are then re-used by cancer cells for OXPHOS, resulting in an increased mitochondrial mass. Importantly, the utilization by cancer cells of the high-energy autophagic stromal metabolites pyruvate, lactate, and ketones may increase the transcriptional expression of gene profiles normally associated with stemness, including genes commonly upregulated in embryonic stem cells. In addition to the resulting efficient net energy transfer from the tumor stroma to epithelial cancer cells in a unilateral and vectorial manner, this host-parasite intercellular cooperation between the tumor stroma and the epithelial cancer cells, which has been designated “the autophagic tumor stromal model of cancer cell metabolism”, “battery-operated tumor growth”, “stromal-epithelial metabolic coupling”, and the “reverse Warburg effect”, may therefore promote the dynamic appearance of a CS-like cellular state, thus resulting in a significant decrease in patient survival. Overall, the literature strongly suggests that cancer cells cannot effectively use ketones for fuel. Indeed, mostly in animal models of malignant glioma, ketogenic diets have been suggested as potential non-toxic treatments or adjuvant therapies to standard care for patients with systemic metastatic disease [66-70]. As there is apparently no known metabolic pathway by which fibroblasts can produce ketone bodies from glucose and as it is widely accepted that ketone bodies are produced nearly exclusively via the β-oxidation of fatty acids in hepatocytes, the “reverse Warburg effect” has been viewed as an artifactual phenomenon that occurs solely in the genetically engineered co-culture system used by the Lisanti group because it does not account for many observations that support cell-autonomous changes in cancer cell metabolism and excludes the possibility that cancer cells could utilize ketone bodies as efficient energy substrates.
We now challenge the above-mentioned views by revealing that the acquisition of stemness traits by cancer cells undergoing EMT is sufficient to cell-autonomously enable the vectorial transfer of energy-rich nutrients from the extracellular microenvironment to energy-producing catabolic pathways in the CS-like cells. We present the first evidence for the existence of a cell-autonomous “reverse Warburg effect” where CS cell reprogramming pre-activates energy-producing pathways that can efficiently metabolize bona fide ketone bodies such as β-hydroxy-butyric acid in vitro (Fig. 4). Furthermore, the drastic increase in the ability of cells with EMT-induced CS-like cellular states to take up not only ketone bodies but other high-energy metabolites such as lactate [71] and, more importantly, pyruvate from the extracellular milieu and use these metabolites to feed mitochondrial energy production, especially upon starvation, is notable. In addition, cells with EMT-driven CS-like cellular states appear to acquire the ability to supra-additively generate energy-rich NADH upon re-exposure to an adequate source of exogenous glucose, regardless of the specific disposable nutrient source in the extracellular milieu. Cancer tissues are known to adapt to tumor microenvironment components such as hypoxia, nutrient deficiency, acidosis, and reactive oxygen species via altered cancer metabolism and CSC enrichment. Thus, long-term hypoxia and nutrient starvation contribute to tumor aggressiveness and recurrence because hypoxic and nutrient-starved tumor microenvironments suppress cell cycle progression but enrich non-proliferating CS cells [72-78]. In this scenario, the response to oxygen or nutrient deficiency and acidosis can be attained via metabolic alterations to glycolysis, glutamine metabolism, and other metabolic pathways. We now provide evidence that the EMT-driven switch from a non-CS- to a CS-like cellular state results in the pre-activation of a metabolic infrastructure that permits vectorial energy transfer from a broader range of extracellular nutrients, including high-energy metabolites such as pyruvate, lactate, and ketones, under stressful microenvironmental conditions. Metabolic reprogramming may thus constitute an efficient adaptive strategy through which these pre-programmed CS-like cellular states would rapidly obtain an advantage in hostile milieus such as those that follow the inhibition of tumor angiogenesis, a state wherein heterogeneous cancer cell populations are transiently exposed to both hypoxia and nutrient starvation; this state is known to stimulate tumor aggressiveness. The metabolic reprogramming of EMT-driven CS-like cellular states can elicit rapid adaptations to the metabolic tumor microenvironmental milieu that are likely responsible for the failures of antiangiogenic therapy with respect to metastatic tumors. Moreover, because alterations to the appropriate balance of fuels and/or signal transduction pathways that handle nutrient utilization have been suggested to underlie the cancer predisposition associated with metabolic diseases such as diabetes and obesity, our current findings offer a new perspective regarding how whole-body metabolism may directly interact with CS cells metabolism and therefore may better define these risks; for example, lactic acidosis and ketoacidosis could be viewed as systemic pre-neoplastic/pre-metastatic and possibly EMT-CS cell-enriching conditions [79-81].
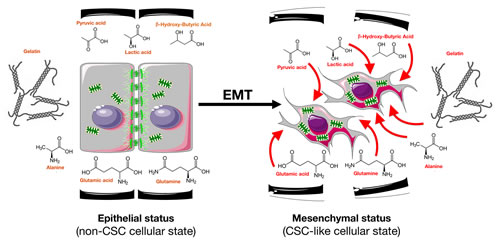
Figure 4: The nutritional phenotype of EMT-induced CS-like cellular states. EMT-induced cancer stemness activates a metabolic infrastructure enabling the usage of extracellular, high-energy nutrients such as glycolysis end products (pyruvate, lactate) and bona fide ketone bodies (β-hydroxybutyrate) to support mitochondrial energy production. Metabolic reprogramming might constitute an efficient adaptive strategy through which pre-programmed EMT-CS-like phenotypes would rapidly obtain an advantage in hostile conditions such as nutrient starvation following the inhibition of tumor angiogenesis.
From nutritional phenotypes of EMT-CS-like cells to systemic “metabolic nichotherapies”
Our current results provide additional insights into the commonly oversimplified view of cancer metabolism. Global nutrient utilization analyses can offer a previously unrecognized complement to the phenotypic characterization of highly plastic CS-like cellular states, and it may provide a better understanding of the “genotype-phenotype” maps with respect to cancer pathogenesis and metastasis. A definitive elucidation of the metabolo-phenomic maps of CS cells may reveal an unforeseen route to the development of therapies against not only the intrinsic metabolic energy-generating machinery of CS cells but also their nutritional niches. The emerging discipline of nutritional genomics or nutrigenomics includes both the study of the effects of diet on an individual’s gene activity and health and the study of how genetic composition affects nutrient metabolism [82-85]. Through an understanding of the unique roles of specific nutrients and their possible roles in boosting EMT-CS-like cell phenotypes, it might be possible to customize “smart foods” or systemic “metabolic nichotherapies” tailored to the specific nutritional phenomes possessed by cells with CS-like cellular states. Our findings also present a new CS cells-centered mechanistic perspective on the current investigations of dietary supplements and strict glucose control as adjuncts for managing the cachexia associated with excess tumor nutrient consumption. Moreover, simultaneous evaluations of the human plasma levels of energy-rich nutrients such as lactic acid, pyruvic acid, and 3-hydroxybutyric acid to facilitate the clinical monitoring of lactic acidosis and ketone body formation may provide a new strategy for predicting the whole-body metabolic regulation with respect to cancer risk and EMT-CS-like cellular phenotype-based metastatic progression. Nevertheless, because new techniques such as multi-isotope imaging mass spectrometry permit the high-resolution tracking of heavy isotope-labeled molecules upon utilization by specific types of cells [86, 87], the unique metabolic fluxes generated by the catabolic energy-producing machinery of CS cells could be implemented in a novel manner to monitor the spatiotemporal distributions and functionality of CS cellular states in real time.
Metabolic phenomics of CS Cells
A practical corollary. As a practical corollary of this perspective it is necessary to bear in mind that we have taken advantage of the landmark Weinberg observation that breast cancer cell populations experimentally induced into an EMT drastically increase the proportion of CS-like cells [26, 27]. V12H-Ras-transformed derivatives of immortalized mammary epithelial cells (HMLER) driven to undergo the EMT by E-cadherin knockdown (HMLERshECad) exhibit characteristics of proliferative CS-like cells or cancer stemloids [26, 27, 88], which has been successfully exploited as a valuable screening method for preliminarily identifying agents with specific toxicity towards proliferating CS-like cells with stemness properties [27]. We confirmed that HMLERshECad cells acquire a mesenchymal breast cancer phenotype and display an increase in the proportion of cells with a CD44highCD24low marker profile [89], which was originally associated with human mammary CS cells (approx. a 10-fold increase) when compared with HMLERshCntrl cells. HMLERshECad cells also showed a dramatic increase in tumorsphere-forming ability relative to HMLERshCntrl cells, which mostly failed to form bona fide mammospheres, an in vitro assay that gauges stemness (data not shown). Using a colorimetric assay extremely simple to perform, which does not require any prior knowledge of the energy pathways present and active in a given cell line, our current execution of the PM device with the Weinberg’s conceptual modeling of EMT-induced CS-like cells clearly exemplifies the usefulness of the PMM technology for researchers wanting to study metabolic pathways activities preferentially owned by CS cells.
While metabolomic assay of pool levels might be better for detecting the presence of pathways and detecting the site of metabolic pathway blockages and the use of quantitative isotope tracking might be better to measuring pathway fluxes in CS-like cellular states, the PMM technology might provide researchers with a simple tool for a global perspective of the range with advantages for measuring in vivo metabolic fluxes and regulation of multiple energy-producing pathways, thus enabling a wide range of rapid studies relating tumor genotype to CS cells phenotypes. However, although EMT-induced CS-like cells have been evidenced in many cancer types, it is certainly a simplistic view to consider equivalent the bona fide CS cells, which is able to support the growth and heterogeneity of an entire tumor, with the ability of differentiated tumor cells to generate, through EMT, proliferating CS-like cell populations. Cancer stemness must be viewed as a multifaceted evolutionary hallmark of selected cancer cells endowed with the most competitive properties (e.g., immortality, dormancy, chemo- or radioresistance, and EMT) resulting from several epigenetic mechanisms and signaling programs. In this complex scenario in which cancer stemness is an acquired and reversible trait that is quantitative rather than qualitative and that results from a stochastic rather than deterministic process, we acknowledge that the incorporation of robust metabolic phenomics, i.e. the systematic acquisition and objective documentation of cancer metabolic data at the level of CSC cellular states, might revolutionize the advancement of CSC-related cancer precision medicine. Because CS cellular states should involve changes in the capacity (enzyme abundance) and kinetics (enzyme activity) of certain metabolic nodes that might generate CS cell-associated onco-metabolites or metabolomic flux imprints, such a unambiguous, qualitative and quantitative phenomic representation of the CSC metabo-phenome will require a systematic combination of biology, biochemistry, genetics, metabolomics, fluxomics, and mathematical approaches. Nevertheless, the preliminary generation of carbon and nitrogen utilization “phenetic maps” upon the acquisition of CS-like cellular states, such as those presented here, might certainly accelerate our current speed of knowledge in the unique metabolic infrastructure of CS cells.
ACKNOWLEDGMENTS
We are greatly indebted to Prof. Robert A. Weinberg (Whitehead Institute for Biomedical Research, Cambridge, MA, USA) for providing the HMLERshCntrol and HMLERshEcad cells used in this work. This work was financially supported by the Ministerio de Ciencia e Innovación (SAF2012-38914), Plan Nacional de I+D+I, MICINN, Spain.
CONFLICT OF INTEREST STATEMENT
The authors of this manuscript have no conflicts of interest to declare.
REFERENCES
1. WARBURG O. On the origin of cancer cells. Science. 1956; 123:309-314.
2. Warburg, O. On respiratory impairment in cancer cells. Science. 1956; 124: 269-270.
3. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011; 11:325-337.
4. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472-482.
5. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009; 23: 537-548.
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
7. Menendez JA, Joven J, Cufí S, Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, Martin-Castillo B, López-Bonet E, Alarcón T, Vazquez-Martin A. The Warburg effect version 2.0: metabolic reprogramming of cancer stem cells. Cell Cycle. 2013; 12:1166-1179.
8. Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, Zhang J. Autophagy in stem cells. Autophagy. 2013;9:830-849.
9. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, Reue K, Christofk H, Mischel PS, Pajonk F. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108:16062-16067.
10. Goidts V, Bageritz J, Puccio L, Nakata S, Zapatka M, Barbus S, Toedt G, Campos B, Korshunov A, Momma S, Van Schaftingen E, Reifenberger G, Herold-Mende C, Lichter P, Radlwimmer B. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene. 2012; 31:3235-3243.
11. Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, Cai S, Scheeren FA, Kuo AH, Diehn M. Targeting Unique Metabolic Properties of Breast Tumor Initiating Cells. Stem Cells. 2014 Feb 4. doi: 10.1002/stem.1662.
12. Palorini R, Votta G, Balestrieri C, Monestiroli A, Olivieri S, Vento R, Chiaradonna F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J Cell Biochem. 2014;115:368-379.
13. Mihaylova MM, Sabatini DM, Yilmaz ÖH. Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell. 2014;14:292-305.
14. Menendez JA, Alarcón T, Corominas-Faja B, Cuyàs E, López-Bonet E, Martin AG, Vellon L. Xenopatients 2.0: reprogramming the epigenetic landscapes of patient-derived cancer genomes. Cell Cycle. 2014;13:358-370.
15. Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11:589-595.
16. Rafalski VA, Mancini E, Brunet A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J Cell Sci. 2012; 125:5597-5608.
17. Riggs JW, Barrilleaux BL, Varlakhanova N, Bush KM, Chan V, Knoepfler PS. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev. 2013;22:37-50.
18. Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013; 140:2535-2547.
19. Ochocki JD, Simon MC. Nutrient-sensing pathways and metabolic regulation in stem cells. J Cell Biol. 2013; 203:23-33.
20. Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15:243-256.
21. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, Lorkiewicz P, St Clair D, Hung MC, Evers BM, Zhou BP. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316-331.
22. Liu PP, Liao J, Tang ZJ, Wu WJ, Yang J, Zeng ZL, Hu Y, Wang P, Ju HQ, Xu RH, Huang P. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014;21:124-135.
23. Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B, Menendez JA. Metformin: multi-faceted protection against cancer. Oncotarget. 2011; 2:896-917.
24. Vazquez-Martin A, López-Bonetc E, Cufí S, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. Repositioning chloroquine and metformin to eliminate cancer stem cell traits in pre-malignant lesions. Drug Resist Updat. 2011; 14:212-223.
25. Vazquez-Martin A, Cufi S, Lopez-Bonet E, Corominas-Faja B, Oliveras-Ferraros C, Martin-Castillo B, Menendez JA. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci Rep. 2012;2:964.
26. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704-715.
27. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009; 138:645-659.
28. Bochner BR, Gadzinski P, Panomitros E. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 2001; 11:1246-1255.
29. Bochner BR. Global phenotypic characterization of bacteria. FEMS Microbiol Rev. 2009; 33:191-205.
30. Bochner BR, Siri M, Huang RH, Noble S, Lei XH, Clemons PA, Wagner BK. Assay of the multiple energy-producing pathways of mammalian cells. PLoS One. 2011; 6:e18147.
31. Shea A, Wolcott M, Daefler S, Rozak DA. Biolog phenotype microarrays. Methods Mol Biol. 2012; 881:331-373.
32. Mackie AM, Hassan KA, Paulsen IT, Tetu SG. Biolog Phenotype Microarrays for phenotypic characterization of microbial cells. Methods Mol Biol. 2014; 1096:123-130.
33. Houle D, Govindaraju DR, Omholt S. Phenomics: the next challenge. Nat Rev Genet. 2010;11:855-866.
34. Lanktree MB, Hassell RG, Lahiry P, Hegele RA. Phenomics: expanding the role of clinical evaluation in genomic studies. J Investig Med. 2010; 58:700-706.
35. Zbuk KM, Eng C. Cancer phenomics: RET and PTEN as illustrative models. Nat Rev Cancer. 2007;7:35-45.
36. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA, Weinberg RA. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013; 154:61-74.
37. Marjanovic ND, Weinberg RA, Chaffer CL. Poised with purpose: cell plasticity enhances tumorigenicity. Cell Cycle. 2013;12:2713-2714.
38. Ansieau S. EMT in breast cancer stem cell generation. Cancer Lett. 2013; 338:63-68.
39. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013; 501:328-337.
40. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013; 59:168-179.
41. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009; 139:693-706.
42. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011; 146:633-644.
43. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, Weinberg RA. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011; 108:7950-7955.
44. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011; 108:1397-1402.
45. Polytarchou C, Iliopoulos D, Struhl K. An integrated transcriptional regulatory circuit that reinforces the breast cancer stem cell state. Proc Natl Acad Sci U S A. 2012; 109:14470-14475.
46. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008; 118:3930-3942.
47. Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest. 2008; 118:3835-3837.
48. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009; 8:3984-4001.
49. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn UE, Howell A, Sotgia F, Lisanti MP. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY). 2010; 2:185-199.
50. Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, Witkiewicz AK, Vander Heiden MG, Migneco G, Chiavarina B, Frank PG, Capozza F, Flomenberg N, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle. 2010; 9:1960-1971.
51. Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, Flomenberg N, Tsirigos A, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle. 2010; 9:2412-2422.
52. Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, Flomenberg N, Howell A, Pestell RG, Knudsen ES, Sotgia F, Lisanti MP. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010; 9:3256-3276.
53. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010; 9:3506-3514.
54. Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, Birbe RC, Howell A, Pavlides S, Gandara R, Pestell RG, Sotgia F, Philp NJ, Lisanti MP. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011; 10:1772-1783.
55. Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, Chiarugi P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130-5140.
56. Salem AF, Whitaker-Menezes D, Lin Z, Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, Howell A, Pestell RG, Sotgia F, Lisanti MP. Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle. 2012; 11:2545-2556.
57. Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Andò S, Martinez-Outschoorn U, Sotgia F, Lisanti MP. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle. 2012; 11:3019-3035.
58. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle. 2012;11:3956-3963.
59. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone body utilization drives tumor growth and metastasis. Cell Cycle. 2012; 11:3964-3971.
60. Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, Goldberg AF, Pestell RG, Howell A, Sneddon S, Birbe R, Tsirigos A, Martinez-Outschoorn U, Sotgia F, Lisanti MP. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012; 11:2285-2302.
61. Lisanti MP, Martinez-Outschoorn UE, Sotgia F. Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle. 2013; 12:2723-2732.
62. Sanchez-Alvarez R, Martinez-Outschoorn UE, Lamb R, Hulit J, Howell A, Gandara R, Sartini M, Rubin E, Lisanti MP, Sotgia F. Mitochondrial dysfunction in breast cancer cells prevents tumor growth: understanding chemoprevention with metformin. Cell Cycle. 2013; 12:172-182.
63. Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Cancer metabolism: new validated targets for drug discovery. Oncotarget. 2013; 4:1309-1316.
64. Martinez-Outschoorn U, Sotgia F, Lisanti MP. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Semin Oncol. 2014;41:195-216.
65. Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 2014; 25:47-60.
66. Zhou W, Mukherjee P, Kiebish MA, Markis WT, Mantis JG, Seyfried TN. The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr Metab (Lond). 2007; 4:5.
67. Stafford P, Abdelwahab MG, Kim do Y, Preul MC, Rho JM, Scheck AC. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr Metab (Lond). 2010;7:74.
68. Maurer GD, Brucker DP, Bähr O, Harter PN, Hattingen E, Walenta S, Mueller-Klieser W, Steinbach JP, Rieger J. Differential utilization of ketone bodies by neurons and glioma cell lines: a rationale for ketogenic diet as experimental glioma therapy. BMC Cancer. 2011;11:315.
69. Abdelwahab MG, Fenton KE, Preul MC, Rho JM, Lynch A, Stafford P, Scheck AC. The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLoS One. 2012; 7:e36197.
70. Champ CE, Palmer JD, Volek JS, Werner-Wasik M, Andrews DW, Evans JJ, Glass J, Kim L, Shi W. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J Neurooncol. 2014;117:125-131.
71. Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, Landon CD, Chi JT, Pizzo S, Schroeder T, Dewhirst MW. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS One. 2013; 8:e75154.
72. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005; 8:299-309.
73. Shen R, Ye Y, Chen L, Yan Q, Barsky SH, Gao JX. Precancerous stem cells can serve as tumor vasculogenic progenitors. PLoS One. 2008; 3:e1652.
74. Cabarcas SM, Mathews LA, Farrar WL. The cancer stem cell niche--there goes the neighborhood? Int J Cancer. 2011; 129:2315-2327.
75. Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB, Oborn CJ, Park YY, Erez B, Jacoby JJ, Lee JS, Lin HY, Ciardiello F, Herbst RS, Langley RR, Heymach JV. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest. 2011; 121:1313-28.
76. Osawa T, Shibuya M. Targeting cancer cells resistant to hypoxia and nutrient starvation to improve anti-angiogeneic therapy. Cell Cycle. 2013; 12:2519-2520.
77. Philip B, Ito K, Moreno-Sánchez R, Ralph SJ. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis. 2013; 34:1699-1707.
78. Mathonnet M, Perraud A, Christou N, Akil H, Melin C, Battu S, Jauberteau MO, Denizot Y. Hallmarks in colorectal cancer: Angiogenesis and cancer stem-like cells. World J Gastroenterol. 2014; 20:4189-4196.
79. Ming Valerie Lin, Gene Bishop, Maria Benito-Herrero. Diabetic Ketoacidosis in Type 2 Diabetics: A Novel Presentation of Pancreatic Adenocarcinoma. J Gen Intern Med. 2010; 25: 369–373.
80. Martinez-Outschoorn UE, Whitaker-Menezes D, Valsecchi M, Martinez-Cantarin MP, Dulau-Florea A, Gong J, Howell A, Flomenberg N, Pestell RG, Wagner J, Arana-Yi C, Sharma M, Sotgia F, Lisanti MP. Reverse Warburg effect in a patient with aggressive B-cell lymphoma: is lactic acidosis a paraneoplastic syndrome? Semin Oncol. 2013; 40:403-418.
81. Bijland LR, Bomers MK, Smulders YM. Smelling the diagnosis: a review on the use of scent in diagnosing disease. Neth J Med. 2013; 71:300-307.
82. Riscuta G, Dumitrescu RG. Nutrigenomics: implications for breast and colon cancer prevention. Methods Mol Biol. 2012; 863:343-358.
83. Ferguson LR, Schlothauer RC. The potential role of nutritional genomics tools in validating high health foods for cancer control: broccoli as example. Mol Nutr Food Res. 2012;56:126-146.
84. Lundstrom K. Past, present and future of nutrigenomics and its influence on drug development. Curr Drug Discov Technol. 2013; 10:35-46.
85. Levesque JP, Winkler IG, Rasko JE. Nichotherapy for stem cells: there goes the neighborhood. Bioessays. 2013; 35:183-290.
86. Steinhauser ML, Bailey AP, Senyo SE, Guillermier C, Perlstein TS, Gould AP, Lee RT, Lechene CP. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature. 2012; 481:516-519.
87. Steinhauser ML, Lechene CP. Quantitative imaging of subcellular metabolism with stable isotopes and multi-isotope imaging mass spectrometry. Semin Cell Dev Biol. 2013; 24:661-667.
88. Blagosklonny MV. Cancer stem cell and cancer stemloids: from biology to therapy. Cancer Biol Ther. 2007; 6:1684-1690.
89. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003; 100:3983-3988.