
INTRODUCTION
Venous thromboembolism (VTE), mainly including deep-vein thrombosis (DVT) and pulmonary embolism (PE), is a severe and complex disorder. With an estimated annual incidence rate of 0.13-0.57 per 1,000 individuals in Asians [1-3] and 1.04-1.83 per 1,000 individuals in Caucasians [4-7], VTE is the third leading cause of cardiovascular mortality after coronary artery disease and stroke, and a major global burden of disease [8]. Owing to the extension of life expectancy and population aging, the incidence of VTE in Asian countries has shown a steady increase [8]. Indeed, recent studies indicated that the rate of VTE after major surgery and in hospitalized medical patients is approaching that of Western populations [9]. Thus, persisting in studying the pathogenesis of VTE, which facilitates the identification and prophylaxis of patients at risk, will benefit human health.
VTE is a multifactorial disease. The interaction of genetic and environmental factors and life events plays a dominant role in the pathogenesis of this complex disease [10-12]. Using modern methodologies in genetic epidemiology, studies of twins and families have indicated that VTE is highly heritable, with an estimated heritability of approximately 0.50 and a familial standardized incidence ratio of around 2.50 [13-15]. Unfortunately, few genetic factors that significantly increase the risk of VTE have been identified [16]. Antithrombin deficiency was the first and is so far the strongest thrombophilic factor [16]. This is explained by the key inhibitory action that this anticoagulant serpin exerts on thrombin and other procoagulant proteases [17]. Patients with antithrombin deficiency have a 5-50-fold increased risk of thromboembolism, predominantly in the venous circulation [18, 19]. However, the incidence of antithrombin deficiency among patients with venous thrombosis is low, 1.9-3.5% in Caucasians and 4.1-8.1% in Chinese [20-25]. However, recent evidence suggests that antithrombin deficiency may be underestimated. Indeed, current functional methods to diagnose antithrombin deficiency fail to detect pathogenic mutations in SERPINC1, the gene encoding antithrombin [26], and certain mutations in SERPINC1 may only show pathogenicity under specific conditions [27, 28].
The aim of our study was to validate the potential thrombotic role of c.883G>A (p.Val295Met) mutation of SERPINC1 found in two patients upon sequencing this gene in 190 Chinese VTE patients with normal antithrombin levels [29]. Here, we report the identification of a recurrent hotspot in SERPINC1 that severely increases the risk of venous thrombosis in the Chinese population.
RESULTS
Baseline characteristics of the subjects
A total of 1304 VTE patients and 1344 healthy controls were enrolled in the current study. The demographic characteristics and genetic data of the study population were previously summarized in detail [30]. Briefly, the case and control groups had similar median ages (52 years), and included a slight predominance of women (51%). Malignancy and lupus anticoagulant were more prevalent in patients than controls (6.2% vs. 0.4% and 5% vs. 0.9% in cases and controls, respectively). Also, a higher incidence of cigarette smoking, immobility, oral contraceptives use, and hormone-replacement therapy was recorded in patients with VTE. In addition, three common polymorphisms, two in the protein C gene (PROC c.565C>T, PROC c.574_576del), and one in the thrombomodulin gene (THBD c.-151G>T), were more prevalent in patients than in controls.
SERPINC1 mutation identification
According to the results of restriction analysis done in the present case-control study, 19 patients with VTE and 2 healthy controls were found to carry an allele which could not be digested by BstUI, all in heterozygous state. Surprisingly, when these 21 positive samples were sequenced to validate the result, four different missense mutations were identified: c.883G>A (p.Val295Met) (rs201381904), detected in 11 VTE patients and 1 control; c.881G>T (p.Arg294Leu) (rs587776397), identified in 6 patients; c.880C>T (p.Arg294Cys) (rs747142328), found in 2 patients; and c.881G>A (p.Arg294His) (rs587776397), found in one healthy control (Figure 1). As expected, all these mutations disturbed the sequence recognized by BstUI, explaining why they all render the same restriction pattern. No additional prothrombotic polymorphisms (PROC c.565C>T, PROC c.574_576del or THBD c.-151G>T) were detected in any of the carriers of these SERPINC1 mutations.
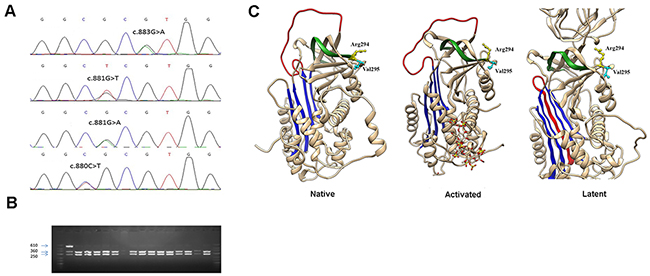
Figure 1: Identification of a mutational hotspot in SERPINC1. (A) Electropherogram of samples carrying all mutations affecting antithrombin Arg294 and Val295 residues. (B) Restriction fragments generated with BstUI on the PCR product of SERPINC1 exon 5 using the primers described in Materials and Methods. (C) Localization in the structure of antithrombin of affected residues in the native, activated, and latent configurations. The central A sheet is colored in blue, the reactive center loop in red, and s1B in green. Model building was performed by using SWISS-MODEL and the Swiss-PdbViewer programs [Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modelling. Electrophoresis 1997; 18: 2714–23] (http://www.expasy.ch/spdbv).
Globally, any mutation affecting this hotspot increased the risk of thrombosis 9.8-fold (95% CI: 2.3-42.4). The most recurrent mutation, c.883G>A, was strongly associated with increased risk of thrombosis (OR = 11.3; 95% CI: 1.5-88.0; P = 0.003). The statistical power for the study was approximately 0.855, with a significance level of 0.05.
Multivariate analysis, including age, gender, smoking, malignancy, lupus anticoagulant, immobility, and oral contraceptives/hormone-replacement therapy confirmed that all these SERPINC1 mutations significantly increased the risk of VTE (OR = 11.4; 95% CI: 2.6-49.4; P = 0.001), particularly the recurrent SERPINC1 c.883G>A mutation mentioned above (OR = 13.6; 95% CI: 1.7-107.1; P = 0.013).
The hotspot identified affects two residues strongly conserved among antithrombins from different species (Figure 2), but not among the serpin superfamily [31], located at the end of strand 1B (s1B) (Figure 1). None of these mutations had been previously described in patients with antithrombin deficiency (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SERPINC1).

Figure 2: Conservation of Arg294 and Val295 among antithrombins of 17 species. Alignments were performed using the Clustal Omega program. Data available at Uniprot (www.uniprot.org/uniprot/)
Plasma antithrombin levels and anticoagulant potential
Antithrombin antigen level, and two tests evaluating the anticoagulant activity of plasma antithrombin, i.e. anti-FXa and anti-FIIa assays, were done in 20 subjects carrying these four SERPINC1 mutations and in 30 healthy donors. Mutation carriers had normal antigen levels and anti-FXa activity, and although anti-FIIa activity was in most carriers within the normal range, 7 cases (4 with the c.883G>A mutation and 3 with the c.881G>T mutation) had values below the normal range (<80 U/dL). Consequently, anti-FIIa activity was overall slightly lower in carriers than in controls (Table 1).
Table 1: Functional studies in carriers and non-carriers of the SERPINC1 mutations
Control (n = 30) |
c.883G>A (n = 12) |
c.881G>T (n = 5)* |
c.880C>T (n = 2) |
c.881G>A (n = 1) |
|
|---|---|---|---|---|---|
AT antigen levels (%) |
102.3 ± 9.8 |
97.4 ± 11.7 |
93.8 ± 6.4 |
91.5 ± 13.4 |
81.2 |
(range) |
(82.6-121.5) |
(79.1-116.0) |
(87.3-101.6) |
(81.8-101.5) |
|
P = 0.1737 |
P = 0.0716 |
P = 0.1473 |
|||
Anti-IIa + UFH |
105.7 ± 11.3 |
89.2 ± 17.5 |
77.0 ± 10.8 |
90.5 ± 9.2 |
88 |
(range) |
(88-131) |
(65-124) |
(65-91) |
(84-97) |
|
P = 0.0008 |
P < 0.0001 |
P = 0.07 |
|||
Anti-Xa + LMWH |
113.5 ± 10.3 |
106.5 ± 16.1 |
124.3 ± 20.0 |
117.0 ± 21.2 |
ND |
(range) |
(86.4-128.3) |
(75.3-136.3) |
(103.7-146.2) |
(101.7-131.5) |
|
P =0.10 |
P = 0.36 |
P = 0.66 |
|||
Peak of thrombin |
258.0 ± 44.6 |
699.0 ± 509.4 |
513.6 ± 262.7 |
ND |
ND |
(range) |
(173.1-408.3) |
(224.0-1669.2) |
(222.5-733.1) |
||
P = 0.03 |
P = 0.23 |
||||
Time to peak |
4.48 ± 0.60 |
6.19 ± 1.64 |
6.30 ± 1.10 |
ND |
ND |
(range) |
(3.33-6.11) |
(4.17-9.33) |
(5.33-7.88) |
||
P = 0.01 |
P = 0.0005 |
||||
ETP |
1161.2 ± 188.0 |
3135.5 ± 2050.0 |
2324.2 ± 818.6 |
ND |
ND |
(range) |
(768.4-1640.9) |
(1141.1-7423.7) |
(1568.0-3193.5) |
||
P = 0.02 |
P = 0.12 |
*The plasma sample from a carrier of c.881G>T was not available. Values (mean ± SD) represent percentages of the result obtained in a pool of 100 healthy controls. AT, antithrombin; UFH, unfractionated heparin; LMWH, low-molecular-weight heparin. ETP: Endogenous thrombin potential
ND: Not Determined.
Thrombin generation assays revealed really interesting information. VTE patients that carried the highly recurrent mutations (c.883G>A p.Val295Met and c.881G>T p.Arg294Leu) had a 2.0~2.7-fold higher thrombin generation in response to tissue factor than healthy controls (Table 1 and Figure 3). In agreement with these results, thrombin generation assays performed in relatives of a carrier of the c.880C>T (p.Arg294Cys) mutation confirmed that carriers (N = 5) in this family had significantly higher endogenous thrombin potential (ETP) than non-carriers (N = 3), while anti-FIIa activity was similar in all subjects (Figure 4).
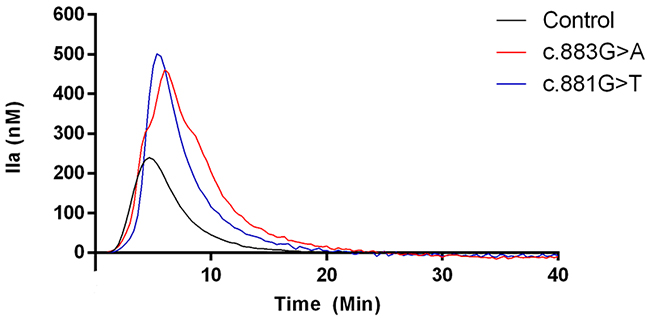
Figure 3: Representative thrombin generation of a control and two carriers of c.883G>A and c.881G>T mutations.
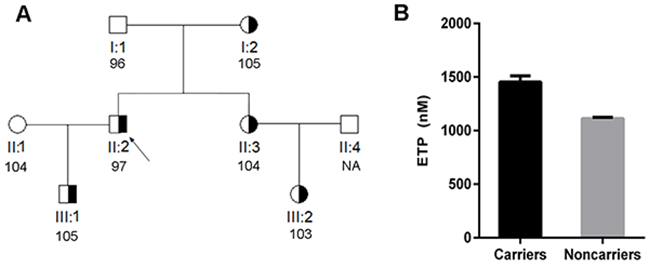
Figure 4: Anticoagulant capacity of carriers and non-carriers of the c. 880C>T mutation from a single family. (A) Pedigree of the family. The proband is identified with an arrow. The anti-FIIa activity of each subject is also shown. (B) Endogenous thrombin potential (ETP) of carriers and non-carriers of c.880C>T from this family. * P < 0.001.
Recombinant expression of SERPINC1 variants
To assess whether the p.Val295Met, p.Arg294Leu, p.Arg294His, and p.Arg294Cys SERPINC1 mutations identified in our study affected the expression of the anti-thrombin protein transcript, recombinant expression of these four variants was evaluated in HEK cells expressing the Epstein Barr Nuclear Antigen 1 (HEK-EBNA). Results showed that the expression of the four recombinant proteins was comparable to that of wild type antithrombin. Moreover, all these recombinant molecules were able to form thrombin-antithrombin complexes in the presence of UFH (Figure 5).
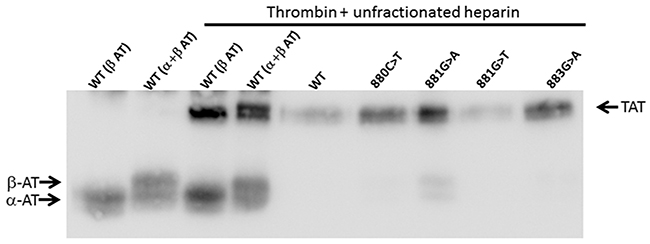
Figure 5: Formation of thrombin-antithrombin complexes by recombinant wild type and variant antithrombins. Conditioned medium of HEK-EBNA cells transfected with plasmids carrying different SERPINC1 mutations was incubated for 5 min with human thrombin and UFH. Antithrombin (AT) glycoforms (α and β) and thrombin-antithrombin complex (TAT) pointed by arrows were detected after SDS-PAGE by Western blot using a polyclonal antibody against human antithrombin.
DISCUSSION
Despite modern methods of massive genetic screening, new prothrombotic mutations have been rarely identified in the last decade [32, 33]. As most of these studies have been restricted to the Caucasian population, screening of other populations may help to identify not only new genetic risk factors potentially affecting millions of people in societies where the rate of venous thrombosis is increasing, but also to describe new mechanisms underlying VTE.
There are evident ethnic differences between Asians and Caucasians in the genetic features of VTE. Three prothrombotic SERPINC1 polymorphisms found in Caucasians, namely factor V Leiden, prothrombin G20210A, and antithrombin Cambridge II (p.Ala416Ser), are not found or rarely reported among Asians [20, 34, 35]. In contrast, two prothrombotic polymorphisms in PROC seem to be restricted to the Asian population [36, 37]. There are also significant differences concerning severe thrombophilia. The prevalence of antithrombin, protein C, and protein S deficiencies has been reported to be higher in the general population or in patients with VTE from China, Japan, and Thailand than in Caucasians [9]. In this study, we have identified a hotspot in SERPINC1 that significantly and severely increases the risk of VTE in the Chinese population. According to our large case-control study, three close mutations, c.883G>A (p.Val295Met), c.881G>T (p.Arg294Leu), and c.880C>T (p.Arg294Cys) increase almost 20-fold the risk of VTE. Among them, c.880C>T (p.Arg294Cys) was once reported in a Japanese patient with VTE [23]. However, they are nearly absent in the Caucasian cohort including 160 antithrombin deficiency patients, 100 VTE patients and 100 healthy subjects.
More interesting than this association is, in our opinion, the fact that these mutations are not detected by current methods to diagnose antithrombin deficiency: antigenic assays and functional methods evaluating anti-FXa and anti-FIIa activities. In agreement with our results, the anticoagulant activity of the Japanese carrier of c.880C>T (p.Arg294Cys) mutation was also within the normal range.[23] In addition, the recombinant model supports that these mutations have negligible consequences on SERPINC1 expression and anti-thrombin activity, at least under our experimental conditions. These data are consistent with recent studies indicating that other pathological mutations affecting SERPINC1 are not detected by functional methods [26]. Interestingly, the thrombin generation assay revealed that these mutations significantly impair the anticoagulant capacity, leading to increased generation of thrombin, which explains the associated risk of thrombosis. It is now apparent that current functional methods, based on the inhibition of a single protease under specific conditions (with predetermined concentrations and incubation conditions for specific cofactors) fail in unmasking the deleterious consequences of some SERPINC1 mutations. Indeed, the p.Val295Met mutation was determined to be ‘damaging’ and ‘probably damaging’ according to, respectively, SIFT and PolyPhen-2 predictions. Modification of these residues, located at the end of s1B, might impair the interaction of antithrombin with target proteases, particularly with thrombin; the resulting reduction in its anticoagulant capacity is not revealed by current functional methods but can be instead detected by thrombin generation assays. Actually, the closely located residues Tyr285 and Glu287 belong to an exosite that augments protease interaction mediated by allosteric activation of antithrombin by heparin [38]. It is hence possible that mutations in Arg294 or Val295 impair the interaction of native antithrombin with certain proteases, particularly thrombin. Further studies are required to verify this hypothesis.
All these data, together with recent evidence that highlights the limitations of current functional methods to detect pathological mutations in SERPINC1 [23, 26], as well as the presence of SERPINC1 mutations that only achieve pathogenic consequences under specific conditions [39], strongly suggest that defects on antithrombin as the underlying cause of VTE are actually underestimated. Antithrombin is critical in the physiopathology of the hemostatic system because of its highly efficient anticoagulant mechanism and the wide variety of procoagulant proteases that is able to inhibit. The risk of VTE is increased not only by severe, but also by mild antithrombin deficiency [40] or even by low borderline plasma levels of antithrombin [41, 42], which have been associated with a small but statistically significant increased risk of VTE. Our study supplies further evidence for the potential contribution of antithrombin polymorphisms to VTE, by stressing that mutations that apparently have minor or no functional effect may significantly increase its risk; moreover, current laboratory diagnostic tests are often insufficient to predict whether these mutations carry deleterious consequences. Therefore, development of new functional methods to diagnose antithrombin deficiency is warranted, and the thrombin generation assay may be an excellent platform in this regard. Additionally, the use of molecular diagnostic methods in cases where current functional methods yielded negative results should help clarify the real prevalence of antithrombin defects in VTE.
MATERIALS AND METHODS
Subjects
All the participants gave written informed consent to enter the study, which was approved by the institutional review boards of the Ethics Committee of the Union Hospital affiliated with Huazhong University of Science and Technology, or the Hospital Reina Sofia, University of Murcia, Spain. The demographic features, sample collection, and criteria for diagnosis of VTE have been previously described in detail [29, 30]. Briefly, blood samples were taken at least one month after the acute VTE event, and none of the subjects received anticoagulant therapy for at least two weeks prior to the time of admission. Objective diagnosis of VTE was mandatorily based on clinical manifestations, D-dimer levels, and the results of compression ultrasonography, ventilation/perfusion lung scan, or spiral computed tomography (CT). Patients with heparin treatment, chronic liver diseases, or nephrotic syndrome were excluded. Age- and sex-matched healthy volunteers, who had no documented personal or family history of thrombosis in first-degree relatives, were enrolled as controls.
Our study included 1,304 VTE individuals and 1,334 controls from China. The acquired risk factors for VTE, including smoking, malignancy, immobility, pregnancy or puerperium, oral contraceptives, hormone-replacement therapy, and lupus anticoagulant, were also documented in cases and controls.
Additionally, a cohort of Caucasian subjects was also enrolled in this study: 160 patients with antithrombin deficiency, 100 patients with VTE but no antithrombin deficiency, and 100 healthy controls.
Genotyping and sequencing
Our first aim was to genotype in the whole case control study the SERPINC1 c.883G>A (p. Val295Met) mutation identified in a previous study [29] in two out of 190 VTE patients with normal antithrombin activity (normal range: 80–120 U/dL). We therefore amplified exon 5 of SERPINC1 using the following oligonucleotide primers: forward primer, 5’-TCATTCTGACACAGCCATT-3’; and reverse primer, 5’-CCTGACTTGTTGCTCCTTT-3’. Identification of the c.883G>A mutation was done by restriction analysis of the 649-bp PCR product with BstUI (New England Biolabs, Ipswich, MA, USA). Briefly, 10 μL PCR product was completely digested with 2 U of BstUI during 2 hours at 60°C. The digestion products were separated by electrophoresis in 2% agarose gels. Two fragments of 371 and 278-bp were generated by the wild type G allele, while the A allele, which disturbs the sequence recognized by BstUI, generated a single band of 649-bp (Figure 1). All cases with a potentially mutated allele were verified by direct sequencing of the PCR product.
Three common prothrombotic polymorphisms in the Chinese population, two affecting the gene encoding protein C (PROC c.565C>T, and c.574_576del) and one affecting the gene encoding thrombomodulin (THBD c.-151G>T) were also genotyped in patients and controls, as reported previously [30, 36, 37].
Plasma antithrombin measurements
Plasma anti-FIIa activity was measured with the commercial reagent STA-STACHROM® AT III on the STA-R evolution automatic coagulation analyzer (Diagnostica Stago, Asnières, France), in the presence of unfractionated heparin (UFH) following the manufacturer’s instructions. Anti-FXa activity with low molecular weight heparin (LMWH) was measured using a chromogenic substrate method (Hyphen Biomed, Neuville-sur-Oise, France). Plasma from 100 healthy donors was pooled to generate the reference sample used to build the standard curve for the assays.
Immunochemical determination of antithrombin antigen levels was performed using a Human Antithrombin III ELISA Kit (AssayPro, St. Charles, MO, USA).
Functional and antigenic assays were done in plasma from 20 carriers of SERPINC1 mutations affecting the hotspot identified in this study, as well as in 30 healthy controls.
Thrombin generation assay
The thrombin generation assay was performed to evaluate the influence of the SERPINC1 mutations c.883G>A and c.881G>T on blood coagulation with a calibrated automated thrombography system (Thrombinoscope BV, Maastricht, Netherlands), as previously described [29]. Briefly, in each well of a 96-well round-bottomed plate, 80 μL of plasma was mixed with 20 μL of PPP reagent (tissue factor 5 pM; phospholipids 4 μM). Coagulation was initiated by adding 100 mM calcium chloride (20 μL) in a custom BSA buffer containing 2.5 mM fluorogenic substrate. Thrombin generation was followed for 40 min at 37 °C.
Recombinant expression of antithrombin variants
Recombinant expression of the four SERPINC1 mutations identified in the present study was done in Human Embryonic Kidney cells expressing the Epstein Barr Nuclear Antigen 1 (HEK-EBNA). The SERPINC1 mutations were generated into the SERPINC1 cDNA (SERPINC1 open reading frame expression-ready clone; GeneCopoeia, Rockville, MD, USA) by site-directed mutagenesis using appropriate primers (Supplementary Table 1). The resulting plasmids were sequenced to confirm correct mutagenesis. HEK-EBNA cells were grown in DMEM with GlutaMAX-I medium (Invitrogen, Prat de Llobregat, Barcelona, Spain) supplemented with 5% fetal bovine serum (Sigma-Aldrich) to 60% confluence at 37 ºC and 5% CO2 in a humidified incubator. Then, 200 μg/ml of wild-type or mutant plasmids were transfected for 30 min in OptiMEM with Lipofectamine LTX (Invitrogen), following the manufacturer’s recommendations. After 24 hours, cells were washed with PBS and exchanged into CD-CHO medium (Invitrogen) supplemented with 4 mM L-glutamine and 0.25 mg/ml Geneticin (Invitrogen). Cells were grown for 2 days and culture medium was collected. Cells were extensively washed with sterile PBS and then lysed with 50 μl of lysis buffer (10 mM Tris-HCl, 0.5 mM DTT, 0.035% SDS, 1 mM EGTA, 50 mM sodium fluoride, 50 μM sodium orthovanadate, 5 mM benzamidine, and 20 mM phenylmethylsulfonyl fluoride).
Antithrombin in lysates and conditioned media was evaluated by SDS-PAGE and Western blot. Briefly, after separation, proteins were transblotted onto a polyvinylidene difluoride membrane. Antithrombin was immunostained with a rabbit anti-human antithrombin polyclonal antibody (Sigma-Aldrich) followed by donkey anti-rabbit IgG–horseradish peroxidase conjugate (GE Healthcare, Madrid, Spain), and signal revealed with an ECL kit (Amersham Biosciences, Piscataway, NJ, USA). The anticoagulant activity of the variant secreted to the medium was assayed by evaluating the formation of thrombin-antithrombin complexes by SDS-PAGE and Western blot after incubation of medium with human thrombin and UFH for 5 minutes.
Statistical analysis
Statistical analyses were performed with SPSS 13.0 for Windows (SPSS Inc, Chicago, IL, USA). Continuous variables were expressed as means ± standard deviation (SD), and discrete variables were expressed as counts (percentages). The unpaired Student’s t test or Mann-Whitney U test were performed to compare continuous variables, and the Pearson’s chi-square test or the Fisher exact test were used to compare allele and genotype distributions in patients and controls. Multivariate analysis was performed using multiple logistic regression and including all the significant covariates. The coefficients obtained from the logistic regression were expressed in terms of odds ratios (ORs) with 95% confidence intervals (CIs). All the results are expressed as 2-tailed values. Statistical significance level was set at P < 0.05.
Abbreviations
VTE, venous thromboembolism; DVT, deep vein thrombosis; PE, pulmonary embolism; UFH, unfractionated heparin; LMWH, low molecular weight heparin; ETP, endogenous thrombin potential
Author contributions
Y.H. was the principal investigator of the study. Y.H. and J.C. coordinated and oversaw the study. W. Z, B.H. and L.T. collected samples, performed experimentation, and analyzed data; Y.-Y.Y. collected samples, participated in sample processing, and performed bioinformatic and data analyses. T.M and M.-B.M. performed and analyzed recombinant experiments and collected and analyzed samples from Spain. Z.W., B.H., L.T., J.C., and Y.H. wrote the manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
This study was funded by the National Natural Sciences Foundation of China (No. 81370622, No. 81400099 and No. 81600108).
REFERENCES
1. Cheuk BL, Cheung GC, Cheng SW. Epidemiology of venous thromboembolism in a Chinese population. Br J Surg. 2004; 91: 424-8. https://doi.org/10.1002/bjs.4454.
2. Molina JA, Jiang ZG, Heng BH, Ong BK. Venous thromboembolism at the National Healthcare Group, Singapore. Ann Acad Med Singapore. 2009; 38: 470-8.
3. Jang MJ, Bang SM, Oh D. Incidence of venous thromboembolism in Korea: from the Health Insurance Review and Assessment Service database. J Thromb Haemost. 2011; 9: 85-91. https://doi.org/10.1111/j.1538-7836.2010.04108.x.
4. Silverstein MD, Heit JA, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med. 1998; 158: 585-93.
5. Oger E. Incidence of venous thromboembolism: a community-based study in Western France. EPI-GETBP Study Group. Groupe d’Etude de la Thrombose de Bretagne Occidentale. Thromb Haemost. 2000; 83: 657-60.
6. White RH, Zhou H, Murin S, Harvey D. Effect of ethnicity and gender on the incidence of venous thromboembolism in a diverse population in California in 1996. Thromb Haemost. 2005; 93: 298-305. https://doi.org/10.1160/TH04-08-0506.
7. Tagalakis V, Patenaude V, Kahn SR, Suissa S. Incidence of and mortality from venous thromboembolism in a real-world population: the Q-VTE Study Cohort. Am J Med. 2013; 126: 832 e13-21. https://doi.org/10.1016/j.amjmed.2013.02.024.
8. Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar A, Konstantinides SV, McCumber M, Ozaki Y, Wendelboe A, Weitz JI, et al. Thrombosis: a major contributor to global disease burden. Arterioscler Thromb Vasc Biol. 2014; 34: 2363-71. https://doi.org/10.1161/ATVBAHA.114.304488.
9. Angchaisuksiri P. Venous thromboembolism in Asia--an unrecognised and under-treated problem? Thromb Haemost. 2011; 106: 585-90. https://doi.org/10.1160/TH11-03-0184.
10. Bovill EG, Hasstedt SJ, Leppert MF, Long GL. Hereditary thrombophilia as a model for multigenic disease. Thromb Haemost. 1999; 82: 662-6.
11. Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999; 353: 1167-73.
12. Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005; 293: 715-22. https://doi.org/10.1001/jama.293.6.715.
13. Souto JC, Almasy L, Borrell M, Gari M, Martinez E, Mateo J, Stone WH, Blangero J, Fontcuberta J. Genetic determinants of hemostasis phenotypes in Spanish families. Circulation. 2000; 101: 1546-51.
14. Heit JA, Phelps MA, Ward SA, Slusser JP, Petterson TM, De Andrade M. Familial segregation of venous thromboembolism. J Thromb Haemost. 2004; 2: 731-6. https://doi.org/10.1111/j.1538-7933.2004.00660.x.
15. Zoller B, Li X, Sundquist J, Sundquist K. Age- and gender-specific familial risks for venous thromboembolism: a nationwide epidemiological study based on hospitalizations in Sweden. Circulation. 2011; 124: 1012-20. https://doi.org/10.1161/CIRCULATIONAHA.110.965020.
16. Martinelli I, De Stefano V, Mannucci PM. Inherited risk factors for venous thromboembolism. Nat Rev Cardiol. 2014; 11: 140-56. https://doi.org/10.1038/nrcardio.2013.211.
17. Bjork I, Olson ST. Antithrombin. A bloody important serpin. Adv Exp Med Biol. 1997; 425: 17-33.
18. Mateo J, Oliver A, Borrell M, Sala N, Fontcuberta J. Increased risk of venous thrombosis in carriers of natural anticoagulant deficiencies. Results of the family studies of the Spanish Multicenter Study on Thrombophilia (EMET study). Blood Coagul Fibrinolysis. 1998; 9: 71-8.
19. Rosendaal FR. Risk factors for venous thrombotic disease. Thromb Haemost. 1999; 82: 610-9.
20. Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med. 2001; 344: 1222-31. https://doi.org/10.1056/NEJM200104193441607.
21. Chen TY, Su WC, Tsao CJ. Incidence of thrombophilia detected in southern Taiwanese patients with venous thrombosis. Ann Hematol. 2003; 82: 114-7. https://doi.org/10.1007/s00277-002-0603-z.
22. Angchaisuksiri P, Atichartakarn V, Aryurachai K, Archararit N, Rachakom B, Atamasirikul K, Tiraganjana A. Risk factors of venous thromboembolism in thai patients. Int J Hematol. 2007; 86: 397-402. https://doi.org/10.1532/IJH97.A20715.
23. Miyata T, Sato Y, Ishikawa J, Okada H, Takeshita S, Sakata T, Kokame K, Kimura R, Honda S, Kawasaki T, Suehisa E, Tsuji H, Madoiwa S, et al. Prevalence of genetic mutations in protein S, protein C and antithrombin genes in Japanese patients with deep vein thrombosis. Thromb Res. 2009; 124: 14-8. https://doi.org/10.1016/j.thromres.2008.08.020.
24. Fischer R, Sachs UJ, Heidinger KS, Eisenburger D, Kemkes-Matthes B. Prevalence of hereditary antithrombin mutations is higher than estimated in patients with thrombotic events. Blood Coagul Fibrinolysis. 2013; 24: 444-8. https://doi.org/10.1097/MBC.0b013e32835cc143.
25. Ding Q, Wang M, Xu G, Ye X, Xi X, Yu T, Wang X, Wang H. Molecular basis and thrombotic manifestations of antithrombin deficiency in 15 unrelated Chinese patients. Thromb Res. 2013; 132: 367-73. https://doi.org/10.1016/j.thromres.2013.07.013.
26. Corral J, Vicente V. Puzzling questions on antithrombin: Diagnostic limitations and real incidence in venous and arterial thrombosis. Thromb Res. 2015; 135: 1047-8. https://doi.org/10.1016/j.thromres.2015.04.012.
27. Bruce D, Perry DJ, Borg JY, Carrell RW, Wardell MR. Thromboembolic disease due to thermolabile conformational changes of antithrombin Rouen-VI (187 Asn-->Asp). J Clin Invest. 1994; 94: 2265-74. https://doi.org/10.1172/JCI117589.
28. Beauchamp NJ, Pike RN, Daly M, Butler L, Makris M, Dafforn TR, Zhou A, Fitton HL, Preston FE, Peake IR, Carrell RW. Antithrombins Wibble and Wobble (T85M/K): archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood. 1998; 92: 2696-706.
29. Zeng W, Tang L, Jian XR, Li YQ, Guo T, Wang QY, Liu H, Wu YY, Cheng ZP, Hu B, Lu X, Yu JM, Deng J, et al. Genetic analysis should be included in clinical practice when screening for antithrombin deficiency. Thromb Haemost. 2015; 113: 262-71. https://doi.org/10.1160/TH14-05-0446.
30. Tang L, Wang HF, Lu X, Jian XR, Jin B, Zheng H, Li YQ, Wang QY, Wu TC, Guo H, Liu H, Guo T, Yu JM, et al. Common genetic risk factors for venous thrombosis in the Chinese population. Am J Hum Genet. 2013; 92: 177-87. https://doi.org/10.1016/j.ajhg.2012.12.013.
31. Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 2000; 10: 1845-64.
32. Germain M, Chasman DI, de Haan H, Tang W, Lindstrom S, Weng LC, de Andrade M, de Visser MC, Wiggins KL, Suchon P, Saut N, Smadja DM, Le Gal G, et al. Meta-analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet. 2015; 96: 532-42. https://doi.org/10.1016/j.ajhg.2015.01.019.
33. Su J, Shu L, Zhang Z, Cai L, Zhang X, Zhai Y, Liu J. A small deletion in SERPINC1 causes type I antithrombin deficiency by promoting endoplasmic reticulum stress. Oncotarget. 2016; 7: 76882-90. https://doi.org/10.18632/oncotarget.12349.
34. Corral J, Hernandez-Espinosa D, Soria JM, Gonzalez-Conejero R, Ordonez A, Gonzalez-Porras JR, Perez-Ceballos E, Lecumberri R, Sanchez I, Roldan V, Mateo J, Minano A, Gonzalez M, et al. Antithrombin Cambridge II: an underestimated genetic risk factor for venous thrombosis. Blood. 2007; 109: 4258-63. https://doi.org/10.1182/blood-2006-08-040774.
35. Zhang GS, Tang YM, Tang MQ, Qing ZJ, Shu C, Tang XQ, Deng MY, Tan LM. Antithrombin Cambridge II(A384S) mutation frequency and antithrombin activity levels in 120 of deep venous thrombosis and 150 of cerebral infarction patients in a single center in Southern China. Blood Coagul Fibrinolysis. 2010; 21: 588-91. https://doi.org/10.1097/MBC.0b013e32833dbe68.
36. Tang L, Guo T, Yang R, Mei H, Wang H, Lu X, Yu J, Wang Q, Hu Y. Genetic background analysis of protein C deficiency demonstrates a recurrent mutation associated with venous thrombosis in Chinese population. PLoS One. 2012; 7: e35773. https://doi.org/10.1371/journal.pone.0035773.
37. Tang L, Lu X, Yu JM, Wang QY, Yang R, Guo T, Mei H, Hu Y. PROC c.574_576del polymorphism: a common genetic risk factor for venous thrombosis in the Chinese population. J Thromb Haemost. 2012; 10: 2019-26. https://doi.org/10.1111/j.1538-7836.2012.04862.x.
38. Izaguirre G, Aguila S, Qi L, Swanson R, Roth R, Rezaie AR, Gettins PG, Olson ST. Conformational activation of antithrombin by heparin involves an altered exosite interaction with protease. J Biol Chem. 2014; 289: 34049-64. https://doi.org/10.1074/jbc.M114.611707.
39. Navarro-Fernandez J, de la Morena-Barrio ME, Padilla J, Minano A, Bohdan N, Aguila S, Martinez-Martinez I, Sevivas TS, de Cos C, Fernandez-Mosteirin N, Llamas P, Asenjo S, Medina P, et al. Antithrombin Dublin (p.Val30Glu): a relatively common variant with moderate thrombosis risk of causing transient antithrombin deficiency. Thromb Haemost. 2016; 116. https://doi.org/10.1160/TH15-11-0871.
40. Di Minno MN, Dentali F, Lupoli R, Ageno W. Mild antithrombin deficiency and risk of recurrent venous thromboembolism: a prospective cohort study. Circulation. 2014; 129: 497-503. https://doi.org/10.1161/CIRCULATIONAHA.113.003756.
41. Bucciarelli P, Passamonti SM, Biguzzi E, Gianniello F, Franchi F, Mannucci PM, Martinelli I. Low borderline plasma levels of antithrombin, protein C and protein S are risk factors for venous thromboembolism. J Thromb Haemost. 2012; 10: 1783-91. https://doi.org/10.1111/j.1538-7836.2012.04858.x.
42. Di Minno MN, Dentali F, Veglia F, Russolillo A, Tremoli E, Ageno W. Antithrombin levels and the risk of a first episode of venous thromboembolism: a case-control study. Thromb Haemost. 2013; 109: 167-9. https://doi.org/10.1160/TH12-09-0663.