
INTRODUCTION
Colorectal cancer (CRC) is one of the most common solid tumor cancer types worldwide. The discovery of mutant KRAS as a predictor of resistance to EGFR monoclonal antibodies resulted in major progression in the treatment of metastatic CRC [1]. However, less than 20% of CRC patients with KRAS wild-type tumors may have durable responses to anti-EGFR treatment [2]. Therefore, all patients with metastatic CRC should be genotyped for KRAS, NRAS and BRAF mutations [3, 4]. Patients with mutations in these genes may not benefit from anti-EGFR targeted therapy.
Studies have identified several genetic alterations, including KRAS, NRAS and BRAF mutations, which are associated with resistance to anti-EGFR agents. A retrospective analysis confirmed the negative effects of KRAS, NRAS and BRAF mutations on the outcome after cetuximab treatment. The research also found that PIK3CA exon 20 mutations were significantly associated with a low response rate [4]. Furthermore, loss of PTEN expression was associated with a negative outcome of cetuximab-based treatment, although this result needs to be validated in more prospective studies [5–7]. In 174 advanced CRC patients with synchronous or metachronous distant metastasis, HER2 gene amplification was identified in 6.3% patients [8]. HER2 amplifications induced the activation of downstream signaling and led to cetuximab or panitumumab resistance [9, 10]. To examine the effect of somatic genetic changes in CRC that caused a response to anti-EGFR antibody therapy, a study was conducted to complete exome sequencing and copy number analyses of patient-derived tumor grafts and targeted genomic analyses of tumors, all of which were KRAS wild-type [11]. This study detected alterations in all genes previously thought to be involved in EGFR therapeutic resistance: NRAS codon 12 or 61 mutations, BRAF V600E mutation, MET amplification, HER2 amplification, PIK3CA exon 20 mutations and truncating or homozygous deletions of PTEN. Additionally, mutations in HER2, EGFR, FGFR1, PDGFRA, and MAP2K1 were also identified as potential mechanisms of primary resistance to anti-EGFR therapy.
As the number of validated anti-EGFR efficiency related genomic alterations increased, multigene panel molecular assays are needed to guide CRC treatment [12]. Hybrid capture-based next-generation sequencing (NGS) enables the simultaneous detection of multiple mutations in multiple genes. More importantly, NGS could discover novel targeted alterations and new available agents that could potentially be used in cancer treatment [13]. Using formalin-fixed paraffin embedded (FFPE) tumor tissues, NGS was well applied to guide precision cancer medicine in diverse cancer types [14, 15]. Several studies have already evaluated the clinical application of NGS in CRC, using panels covering 20-183 genes [16, 17].
In this study, a 508 gene panel-based NGS assay was used to identify anti-EGFR sensitivity-related mutations along with additional targetable genetic alterations from 57 CRC tissues. Comprehensive molecular profiling by NGS assays facilitates personalized therapy for CRC patients, which might be widely applied in the future.
RESULTS
NGS assay and genetic alterations
Fifty-seven CRC cases were selected for NGS assays, including 49 primary tumor tissue samples and 8 metastasis biopsies. The mean age was 60.4 years (range 35-83 years). Patient characteristics are listed in Supplementary Table 1. Approximately 14 days passed from DNA processing to the final report generation. The median average sequencing depth was 660.4±295.0 for tumor DNA from each sample. Genetic alterations were found in 204 genes in all 57 patients (mean 8.9±3.3, range 4-22). Seventeen genes, including those with targeted alterations and without available targeted agents, were observed in more than 5 samples (Figure 1). The most frequently mutated genes were APC (n=39, 68.4%), TP53 (n=32, 56.1%), KRAS (n=30, 52.6%), SMAD4 (n=14, 24.6%) and HER2 (n=10, 17.5%). Furthermore, 89.5% (51/57) of patients harbored at least one genetic alteration in APC, TP53 or KRAS. The detailed sequencing data are provided in Supplementary Table 2.
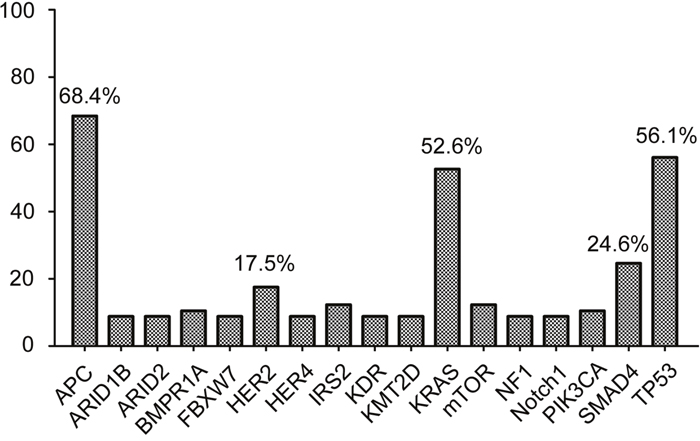
Figure 1: Cancer-related genes altered in more than 5 samples. Seventeen mutant genes, including genetic alterations with or without available targeted agents, were observed in more than 5 samples. The top 5 most frequently mutant genes were APC, TP53, KRAS, SMAD4 and HER2.
Molecular classification of CRC identified by NGS
In total, 47 of 57 (82.5%) patients had at least one potentially actionable alteration. The mean number of potentially actionable alterations per patient was 1.1 (1.1±0.8, range 0-4). Molecular classification of the 57 patients identified by the NGS assay is shown in Figure 2. The specific alterations that were observed in the most common actionable targets included the following: KRAS (n=29, 50.9%), NRAS (n=2, 3.5%), BRAF (n=2, 3.5%), PIK3CA (n=1, 1.8%), PTEN (n=2, 3.5%), HER2 (n=7, 12.3%), NF1 (n=1, 1.8%), BRCA1 (n=1, 1.8%) and CCND2 (n=2, 3.5%) (Figure 2). Finally, 17.5% (10/57) of patients had no actionable somatic alterations.
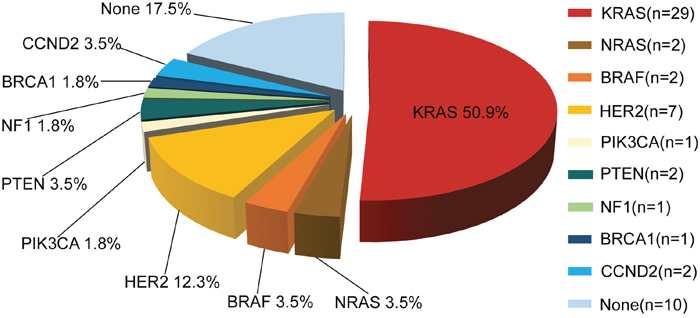
Figure 2: Molecular profiling of actionable genetic alterations in 57 CRC patients.
The specific mutation sites of KRAS observed in 29 patients are shown in Figure 3. Of the KRAS mutations, 89.7% (26/29) were located in codon 12, 6.9% (2/29) were in codon 13 and 3.4% (1/29) were in codon 61.
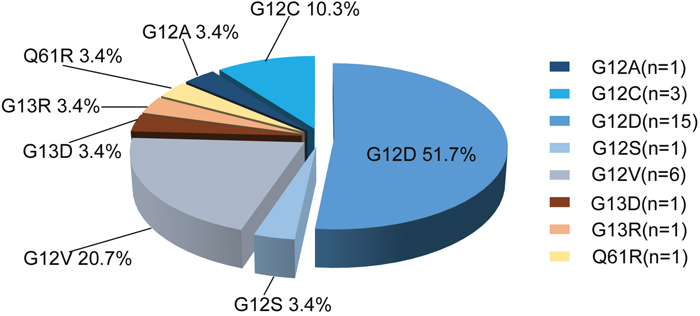
Figure 3: Distribution of KRAS mutation sites in 29 CRC specimens.
Clinical implications directed by the NGS assay
Considering the anti-EGFR therapy response and targeted drug options for the actionable alterations, clinical implications of the 57 CRC patients are shown in Table 1. Of the tumors harboring KRAS, NRAS or BRAF mutations, 57.9% (33/57) did not benefit from anti-EGFR agents, such as cetuximab or panitumumab. Furthermore, 17.5% (10/57) patients harboring somatic mutations or copy number variations in PIK3CA, PTEN (mutation or loss) and HER2 (mutation or amplification) might be associated with a low response to anti-EGFR therapy. Finally, for the remaining 24.6% (14/57) of the patients, cetuximab or panitumumab could be used according to the NCCN guidelines.
Table 1: NGS results and clinical implication in 57 CRC patients
Actionable alterations in 57 patients |
Response to anti-EGFR therapy |
|---|---|
BRAF G469A + PI3KCA E545K |
Resistant to anti-EGFR therapy: 33/57 (57.9%) |
BRAF V600E |
|
KRAS G12A + PI3KCA E542K; KRAS G12C + PI3KCA E542K; KRAS G12V + PI3KCA E546K |
|
KRAS G12D + FBXW7 W237* |
|
KRAS G12V + mTOR S2215Y |
|
KRAS Q61R + PTEN loss+ HER2 T862A |
|
KRAS G12C/G12D/G12S/G12V/G13D/G13R |
|
NRAS G12D + NF1 F843Sfs*35 + ATM c.[3577-1G>T] |
|
NRAS Q61R |
|
PIK3CA H1047R |
Associated with low-response to Anti-EGFR therapy: 10/57 (17.5%) |
PTEN L100Tfs* |
|
PTEN loss + NF1 S1864Rfs*3 + NF1 D1866Rfs*17 |
|
HER2 G776V + FBXW7 R658* |
|
HER2 V842I + PIK3CA E545K |
|
HER2 amplification |
|
HER2 amplification + ERG-TMPRSS2 fusion |
|
BRCA1 Q148* |
Approved anti-EGFR therapy: 14/57 (24.6%) |
CCND2 amplification |
|
NF1 R1306* + EGFR amplification |
|
None actionable alterations detected |
Association between actionable alterations
Notably, more than one actionable alteration was identified in 22.8% (13/57) of the patients (Figure 4). Consequently, several targeted agents, including both approved drugs and experimental agents, might be simultaneously available for these patients. In this case, only the actionable alteration with the best clinical option, such as on-label agents will be exhibited in molecular classification (Figure 2). The additional overlapping actionable alterations included the following: PIK3CA (n=5), FBXW7 (n=2), NF1 (n=2), PTEN (n=1), HER2 (n=1), mTOR (n=1), ATM (n=1), EGFR (n=1) and ERG-TMPRSS2 (n=1) (Figure 4).
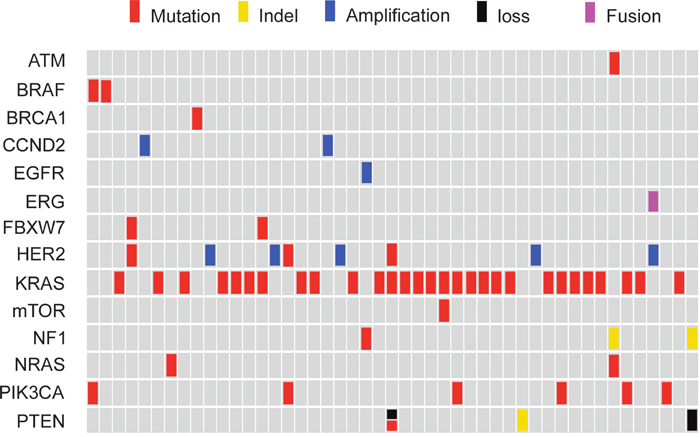
Figure 4: Comprehensive annotation of actionable genetic alterations identified by NGS assay in 57 CRC.
KRAS and NRAS mutations were mutually exclusive, as were KRAS and BRAF mutations, and NRAS and BRAF mutations (Figure 4). Of the KRAS mutant tumors, 20.7% (6/29) harbored mTOR pathway-related genetic alterations (FBXW7, PTEN, PIK3CA, and mTOR), of which PIK3CA exon 9 mutations accounted for half (n=3). One HER2 T862A mutation concurred with KRAS Q61R, while all cases of HER2 amplification found in this research (8.8%, 5/57) were exclusive with mutations of KRAS, NRAS and BRAF.
DISCUSSION
As more anti-EGFR resistance related and candidate therapeutically targeted genetic alterations are identified, comprehensive molecular diagnostic approaches are needed to match CRC patients with the appropriate therapies [13]. Here, we described a targeted sequencing test using broad, hybrid capture-based NGS technology across 508 cancer-related genes in Chinese CRC patients. The most frequently mutated genes identified in this study were APC, TP53 and KRAS. In total, 89.5% of the patients harbored at least one genetic alteration in the above genes. Our results discovered that 82.5% of CRC patients harbored at least one actionable genetic alteration, which suggests that NGS could provide comprehensive and robust genetic alteration data to guide personalized CRC care. Several studies have already evaluated the clinical application of NGS in CRC [18–24]. Compared to these studies, the molecular profiling of representative actionable genetic mutations found in Chinese CRC patients was similar.
The major advantage of NGS is that multiple genetic alterations can be profiled within a single assay. Our research found that 57.9% of CRC harbor mutations in KRAS, NRAS or BRAF. Additionally, another 17.5% of patients with wild type versions of the above genes were identified with genetic mutations in PTEN, PIK3CA and HER2. Consequently, up to 75.4% of patients might not benefit from anti-EGFR therapy. Meta-analysis has suggested that mutations in KRAS, NRAS, BRAF, PIK3CA and non-functional PTEN predict resistance to anti-EGFR therapy [25]. Therefore, it is necessary to screen mutations in genes other than RAS and BRAF to optimize the identification of patients who will benefit from anti-EGFR treatment. Multiple gene panel analysis by NGS could be widely applied in the future to improve cancer therapy.
HER2 amplifications or mutations are commonly found in a wide range of solid cancers. In this study, HER2 amplifications and mutations were detected in 14.0% of the CRC patients. All of the HER2 amplifications identified were exclusive with KRAS, NRAS and BRAF. In CRC, HER2 amplifications induced the activation of downstream signaling even when cetuximab was bound to EGFR, thus leading to drug resistance [26]. HER2 amplification was present in 6.0% to 13.8% of the tumors and was correlated with negative responses to anti-EGFR targeted therapy in KRAS wild-type CRC [10]. Therefore, HER2 amplification contributes to the resistance of anti-EGFR therapy in partial RAS and RAF wild-type CRC patients and should be routinely tested. Pre-clinical and clinical data provide a rationale for the inclusion of trastuzumab, pertuzumab and pan-ERBB inhibitors in the treatment strategy for HER2 amplification or mutation cancer patients. Response of CRC patients with HER2 amplification or HER2 overexpression to treatment with the combination of trastuzumab plus lapatinib was tested in a phase 2 clinical trial, the HERACLES trial [27]. Researchers screened 914 patients who were refractory to chemotherapy and had KRAS wild-type CRC, identifying 46 patients as having HER2-positive disease and enrolling 27 eligible patients into the study. Twenty patients (74.1%) achieved either a complete response, partial response, or stable disease. The median response duration was 9.5 months, median progression-free survival was 5.2 months, and median overall survival was 11.5 months. The phase II MyPathway trial evaluated agents targeting the HER2, BRAF, Hedgehog, or EGFR pathways in patients with advanced cancer. The most promising efficacy was seen among patients with HER2-amplified bladder, biliary, and CRC [28]. The HERACLES trial, as well as the MyPathway basket trials, showed the potential of HER2 as a promising target in the treatment of CRC. HER2 amplifications should be defined as a novel molecular subtype of advanced CRC.
KRAS and NRAS mutations are most commonly located in codons 12, 13 and 61, and lead to the constitutive activation of RAS [29]. Our analysis revealed that 57.9% of samples had mutations in the oncogenes KRAS, NRAS and BRAF, and the majority of KRAS mutations were found in codon 12. A previous study used the Ion PGM and AmpliSeq cancer panel to identify genetic mutations in 91 rectal cancer patients. Of the samples, 69.2% had mutations in the RAS signaling pathway [20]. Consistent with this study, concurrence of KRAS, NRAS, and BRAF mutations with PTEN/PI3K/AKT/mTOR pathway genetic alterations, including PTEN, NF1, FBXW7, PIK3CA and mTOR, were frequently detected in our research. No targeted therapies are available for KRAS-mutant CRC that progresses after all approved standard therapies have been given. Clinical trials showed that inhibitors of MEK1/2, downstream of the RAS pathway, had promising efficiency in KRAS mutant NSCLC and NRAS mutant melanoma [30, 31]. However, clinical activity with single MEK inhibitors had limited efficiency and acquired resistance was easily developed. PI3K signaling was important for cell survival in NRAS mutant melanoma when MEK was inhibited [32]. Combined targeting of the MEK/ERK and PI3K/mTOR pathways had antitumor activity and might serve as a therapeutic option in the treatment of NRAS mutant melanoma [32, 33]. Preclinical data supported dual targeted inhibition of MEK and one or more of the PI3K/AKT pathway effectors in metastatic CRC, which was superior to a single agent alone [34]. Future clinical trials with molecular stratification by NGS have the potential to improve outcomes for CRC patients, especially for patients with additional actionable alterations other than RAS and RAF.
In conclusion, our results showed that 82.5% of CRC tissues harbored at least one actionable genetic alteration identified by NGS testing. In addition to the 57.9% of CRC harboring KRAS, NRAS and BRAF mutations, another 17.5% patients with genetic mutations in genes including PTEN, PIK3CA and HER2 might not benefit from anti-EGFR therapy. HER2 amplification or mutation, which was identified in 12.3% of the tissues, defined a unique molecular subtype of CRC. This study highlights the potential of NGS to further help identify specific CRC patients who could benefit from anti-EGFR treatment. NGS provides a high-throughput and systematic method to identify all genetic alterations and to define novel molecular targets, which might be routinely used to guide precision cancer medicine in the future.
MATERIALS AND METHODS
Patients and Samples
From June 2014 to June 2016, 57 CRC tissues were collected. The diagnosis of CRC was confirmed by hematoxylin and eosin staining. Patient characteristics are shown in Supplementary Table 1. This study was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University, and the investigations were carried out following the rules of the Declaration of Helsinki of 1975 (https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/), revised in 2013. Signed informed consent was obtained from all patients included in the study, and all the experiments were carried out in accordance with the National Health and Family Planning Commission of the PRC’s guidelines.
NGS-based assay
Tumor DNA was extracted from FFPE samples using a QIAamp DNA FFPE Tissue Kit and a Qiagen’s DNEasy Blood and Tissue Extraction Kit (Qiagen), according to the manufacturer's instructions. Genomic DNA from 2ml peripheral blood was purified using a QIAamp DNA Blood Mini Kit (Qiagen). All FFPE tissue samples were reviewed by a qualified pathologist to ensure >70% tumor content. DNA purity and concentration were determined by the NanoDrop2000 spectrophotometer and Qubit 2.0 Fluorometer with Quant-IT dsDNA HS Assay Kit (Thermo Fisher Scientific), respectively. The quality of genomic DNA from tumor tissue and peripheral blood was assessed by agarose gel electrophoresis, and the size distribution of circulating DNA was evaluated on a 2100 Bioanalyzer using the DNA 1000 Kit (Agilent).
Library construction with tumor tissue DNA and paired peripheral blood DNA was performed using 1μg of DNA sheared by an ultrasonoscope to generate fragments with a peak of 250 bps, followed by end repair, A-tailing and ligation to the Illumina-indexed adapters according to the standard library construction protocol. Target enrichment was performed on a custom sequence capture-probe (Nimblegen, USA) that targeted 7,708 exons of 508 cancer-related genes and 78 introns from 19 genes recurrently rearranged in solid tumor, representing ~1.7 Mb of the human genome in total (Supplementary Table 3). Sequencing was performed with 2x101 bp paired-end reads and 8-bp index reads on an Illumina Hiseq 2500 platform (Illumina, San Diego, USA) using the manufacturer’s protocols.
Primary sequence data were first processed by filtering adaptor sequences and removing low-quality reads using the SOAPnuke software (http://soap.genomics.org.cn/) developed by BGI, and aligned to build hg19 of the NCBI reference genome assembly using BWA aligner v0.6.2-r126. PCR duplicate reads were removed by PICARD v1.98. Local realignment and base quality score recalibration were performed using GATK v2.3-9, and poorly mapped reads were removed based on the recalibration result. SNVs were detected by Mutect and SOMATK-SNV (developed by BGI, manuscript in preparation), and Indel (small insertions and deletions) were detected by GATK and SOMATK-INDEL (developed by BGI, manuscript in preparation). A minimal amplicon coverage of 300 was defined, and a variant allele frequency of 1% as theminimal threshold was used to provide reliable diagnostic analysis. CNV calling was done by CONTRA v2.0.4 [35]. We defined a gene as showing copy number gain when its coverage fold ratio was ≥2.0 and loss when ≤0.5.
Statistical analysis
The experimental data are presented as the mean±SEM and were analyzed by a two-tailed Student’s t test. The threshold of P < 0.05 was considered statistically significant.
Author contributions
Helei Hou and Xiaochun Zhang conceived and designed the experiments; Yanxia Jiang performed the pathological assessment; Dong liu, Chuantao Zhang, Guifang Lu and Na Zhou collected the study patients; Xiaonan Yang, Xiaoping Zhang, Zhaoyang Qian, Hongmei Zhu and Zhuokun Li analyzed the genetic data; Helei Hou and Xiaochun Zhang wrote the manuscript; All authors reviewed and approved the final version of the paper.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the Taishan Scholar foundation (No.tshw201502061), the Qingdao Key Discipline foundation, the Qingdao people's Livelihood Science and technology program (16-6-2-3-nsh) and the third batch of Qingdao entrepreneurial innovation leading talent program.
REFERENCES
1. Stintzing S. Management of colorectal cancer. F1000Prime Rep. 2014; 6:108.
2. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008; 26:1626-1634.
3. De Roock W, Lambrechts D, Tejpar S. K-ras mutations and cetuximab in colorectal cancer. N Engl J Med. 2009; 360:834-836.
4. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010; 11:753-762.
5. Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009; 69:1851-1857.
6. Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007; 97:1139-1145.
7. Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E, Canestrari E, Funel N, Schiavon G, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 2009; 27:2622-2629.
8. Seo AN, Kwak Y, Kim DW, Kang SB, Choe G, Kim WH, Lee HS. HER2 status in colorectal cancer: its clinical significance and the relationship between HER2 gene amplification and expression. Plos One. 2014; 9:e98528.
9. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Cora D, Di Nicolantonio F, Buscarino M, Petti C, Ribero D, Russolillo N, Muratore A, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011; 1:508-523.
10. Barry GS, Cheang MC, Chang HL, Kennecke HF. Genomic markers of panitumumab resistance including ERBB2/HER2 in a phase II study of KRAS wild-type (wt) metastatic colorectal cancer (mCRC). Oncotarget. 2016; 7:18953-18964. https://doi.org/10.18632/oncotarget.8006.
11. Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, Niknafs N, Nesselbush M, Lytle K, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015; 526:263-267.
12. Deeb KK, Sram JP, Gao H, Fakih MG. Multigene assays in metastatic colorectal cancer. J Natl Compr Canc Netw. 2013; 11 Suppl 4:S9-S17.
13. Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Med. 2015; 7:80.
14. Schwaederle M, Daniels GA, Piccioni DE, Fanta PT, Schwab RB, Shimabukuro KA, Parker BA, Kurzrock R. On the Road to Precision Cancer Medicine: Analysis of Genomic Biomarker Actionability in 439 Patients. Mol Cancer Ther. 2015; 14:1488-1494.
15. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013; 31:1023-1031.
16. Han SW, Kim HP, Shin JY, Jeong EG, Lee WC, Lee KH, Won JK, Kim TY, Oh DY, Im SA, Bang YJ, Jeong SY, Park KJ, et al. Targeted sequencing of cancer-related genes in colorectal cancer using next-generation sequencing. Plos One. 2013; 8:e64271.
17. Belardinilli F, Capalbo C, Buffone A, Petroni M, Colicchia V, Ferraro S, Zani M, Nicolussi A, D'Inzeo S, Coppa A, Screpanti I, Gulino A, Giannini G. Validation of the Ion Torrent PGM sequencing for the prospective routine molecular diagnostic of colorectal cancer. Clin Biochem. 2015; 48:908-910.
18. Gao J, Wu H, Wang L, Zhang H, Duan H, Lu J, Liang Z. Validation of targeted next-generation sequencing for RAS mutation detection in FFPE colorectal cancer tissues: comparison with Sanger sequencing and ARMS-Scorpion real-time PCR. BMJ Open. 2016; 6:e9532.
19. Malapelle U, Vigliar E, Sgariglia R, Bellevicine C, Colarossi L, Vitale D, Pallante P, Troncone G. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J Clin Pathol. 2015; 68:64-68.
20. Bai J, Gao J, Mao Z, Wang J, Li J, Li W, Lei Y, Li S, Wu Z, Tang C, Jones L, Ye H, Lou F, et al. Genetic mutations in human rectal cancers detected by targeted sequencing. J Hum Genet. 2015; 60:589-596.
21. D'Haene N, Le Mercier M, De Neve N, Blanchard O, Delaunoy M, El HH, Dessars B, Heimann P, Remmelink M, Demetter P, Tejpar S, Salmon I. Clinical Validation of Targeted Next Generation Sequencing for Colon and Lung Cancers. Plos One. 2015; 10:e138245.
22. Haley L, Tseng LH, Zheng G, Dudley J, Anderson DA, Azad NS, Gocke CD, Eshleman JR, Lin MT. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod Pathol. 2015; 28:1390-1399.
23. Malapelle U, Vigliar E, Sgariglia R, Bellevicine C, Colarossi L, Vitale D, Pallante P, Troncone G. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J Clin Pathol. 2015; 68:64-68.
24. Zhang L, Chen L, Sah S, Latham GJ, Patel R, Song Q, Koeppen H, Tam R, Schleifman E, Mashhedi H, Chalasani S, Fu L, Sumiyoshi T, et al. Profiling cancer gene mutations in clinical formalin-fixed, paraffin-embedded colorectal tumor specimens using targeted next-generation sequencing. Oncologist. 2014; 19:336-343.
25. Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014; 53:852-864.
26. Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, Fujisaka Y, Philips J, Shimizu T, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011; 3:86-99.
27. Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I, Martinelli E, Troiani T, Ciardiello F, Racca P, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016; 17:738-746.
28. Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C, Burris H, Bose R, Guo S, Bernaards C, Beattie MS, Stein A, Brammer M, Kurzrock R. Targeted therapy for advanced solid tumors based on molecular profiles: Early results from MyPathway, an open-label, phase IIa umbrella basket study. J Clin Oncol. 2016; 34(suppl; abstr LBA11511).
29. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003; 3:11-22.
30. Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V, Smith P, Smith I, Crino L. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013; 14:38-47.
31. Johnson DB, Puzanov I. Treatment of NRAS-mutant melanoma. Curr Treat Options Oncol. 2015; 16:15.
32. Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T, Daud AI, McCalmont TH, LeBoit PE, Ortiz-Urda S. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci U S A. 2013; 110:4015-4020.
33. Johnson DB, Smalley KS, Sosman JA. Molecular pathways: targeting NRAS in melanoma and acute myelogenous leukemia. Clin Cancer Res. 2014; 20:4186-4192.
34. Temraz S, Mukherji D, Shamseddine A. Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers. Int J Mol SCI. 2015; 16:22976-22988.
35. Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012; 28:1307-1313.