
INTRODUCTION
Recently, colorectal cancer (CRC) studies have been focusing on molecule-targeted therapy [1, 2] and exploring novel chemo-preventive agents [3–6]. Protein phosphatase 2A (PP2A) is a well-established serine/threonine phosphatase [7–9], which is extremely important in regulation of mitotic progression and DNA damage responses [10]. Although PP2A was traditionally viewed as a tumor suppressor [10, 11], recent cancer studies have indicated that PP2A inhibition could inhibit cancer cells via driving senescent cancer cells into mitosis or promoting cancer cell death and apoptosis [12, 13]. Several known PP2A inhibitors were shown to inhibit cancer cell proliferation and/or to induce cancer cell apoptosis [12, 13]. LB-100 is a novel small-molecule PP2A inhibitor [12–14]. Its potential activity in human CRC cells is tested here.
One important PP2A substrate protein kinase is AMP-activated protein kinase (AMPK) [15–17], which is composed of the catalytic α and regulatory β and γ subunits. AMPK is the master energy sensor, whose activation maintains energy homeostasis under stress conditions [18–20]. Recent studies have proposed a tumor-suppressing function of activated AMPK [4, 21–24]. AMPK inhibits its substrates acetyl-CoA carboxylase (ACC) [19, 25] and mammalian target of rapamycin (mTOR) complex 1 (mTORC1) to block fatty acid and protein synthesis [26]. Activated AMPK could also induce p53 stabilization and activation, thus causing cell arrest and apoptosis [27]. Further, AMPK activation could trigger autophagic cell death via multiple mechanisms involving mTOR, ULK1, and VPS34 [28, 29]. Activation of AMPK by a number of anti-cancer agents and natural occurring compounds efficiently inhibits human CRC cells [4, 21, 22]. In the current study, we show that targeting PP2A by LB-100 and microRNA-17-92 (“miR-17-92”) activates AMPK signaling, which in turn inhibits CRC cells in vitro and in vivo.
RESULTS
LB-100 inhibits survival and proliferation of CRC cells
In order to test the potential activity of LB-100 on human CRC cells, the established HCT-116 HCC cells [5] were cultured in complete medium, and were treated with LB-100 at different concentrations. Simple cell counting assay results in Figure 1A displayed that treatment with LB-100, at 2.5 μM and 10 μM, significantly inhibited HCT-116 cell proliferation. The number of HCT-116 cells was decreased following LB-100 treatment (Figure 1A). LB-100 at 10 μM was more potent than 2.5 μM in inhibiting HCT-116 cell proliferation, displaying a concentration-dependent response (Figure 1A). BrdU incorporation is a well-utilized marker of cell proliferation. Results in Figure 1B confirmed that LB-100 treatment (2.5 μM and 10 μM) significantly decreased BrdU ELISA (enzyme-linked immunosorbent assay) optic density (OD) in HCT-116 cells, confirming its anti-proliferative activity. To test cell viability, the routine MTT tetrazolium assay was performed. As demonstrated, LB-100 treatment (2.5 μM and 10 μM, 96 hours) largely inhibited survival (“MTT OD”) of HCT-116 cells (Figure 1C). Additionally, the number of viable HCT-116 cell colonies was also significantly decreased following LB-100 treatment (2.5 μM and 10 μM, renewed every two days) (Figure 1D). Notably, the anti-survival activity by LB-100 was also concentration-dependent. At a relative low concentration (0.5 μM), LB-100 failed to decrease HCT-116 cell survival (Figure 1C and 1D).
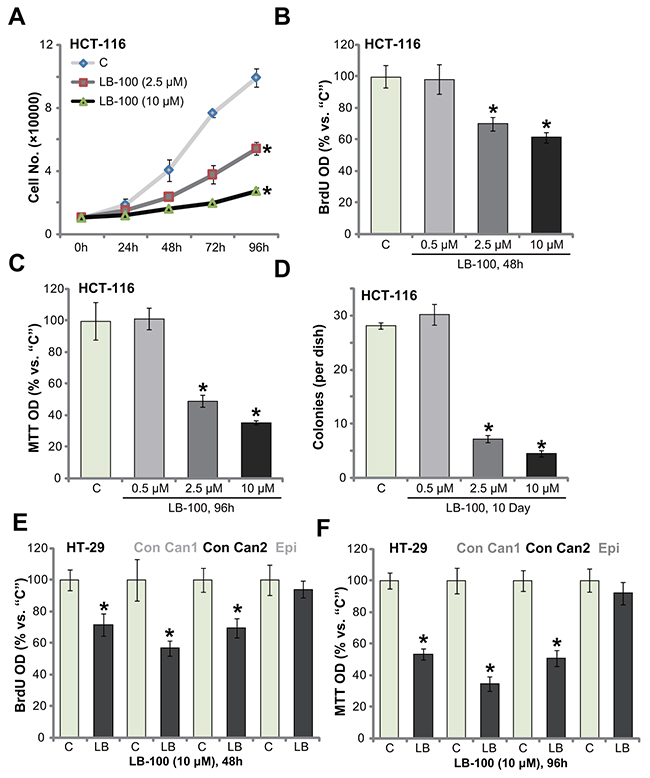
Figure 1: LB-100 inhibits survival and proliferation of CRC cells. The established human CRC cells (HCT-116 and HT-29 lines), the primary-cultured human colon cancer cells (two lines, “Con Can1/2”), or the primary-cultured colon epithelial cells (“Epi”) were treated with LB-100 at designated concentration, cells were further cultured for applied time; Cell proliferation (A, B and E) and cell survival (C, D and F) were examined by the assays mentioned in the text. Error bars indicate standard deviation (SD). “C” stands for untreated control group. *p<0.05 vs. “C” group. Experiments in this figure were repeated five times, and similar results were obtained.
We also examined the potential effect of this novel PP2A inhibitor [13, 30, 31] in other CRC cells: including the established HT-29 cells and two lines of primary human colon cancer cells (“Con Can1/2”). BrdU ELISA proliferation assay results (Figure 1E) and MTT viability assay results (Figure 1F) displayed that treatment with LB-100 (10 μM) dramatically inhibited survival and proliferation of the tested CRC cells, as the BrdU ELISA OD and MTT OD were both decreased following the LB-100 treatment (Figure 1E and 1F). Intriguingly, the very same LB-100 (10 μM) treatment failed to inhibit proliferation and survival of the primary human colon epithelial cells (“Epi”) (Figure 1E and 1F). BrdU ELISA OD and MTT OD were not significantly decreased after LB-100 treatment (Figure 1E and 1F). Collectively, the results indicate that LB-100 inhibits survival and proliferation of human CRC cells.
LB-100 activates apoptosis in CRC cells
We next studied the potential activity of LB-100 on cell apoptosis. Various apoptosis assays were performed, including the DNA end labeling (TUNEL) assay, Annexin V FACS assay and the caspase-3 activity assay. As displayed, treatment with LB-100, at 2.5 μM and 10 μM, in HCT-116 cells significantly increased the percentage of TUNEL-stained cells (Figure 2A), Annexin V ratio (Figure 2B) and the caspase-3 activity (Figure 2C). These results indicated profound apoptosis activation in LB-100-treated cells (Figure 2A-2C). LB-100-induced apoptosis was again concentration-dependent (Figure 2A-2C), and it was yet in-effective when applied at a lower concentration (0. 5 μM, Figure 2A-2C). In order to block cell apoptosis, the caspase-based inhibitors were employed. As displayed, co-treatment with the caspase-3 inhibitor zDEVDfmk or the pan caspase inhibitor zVADfmk largely attenuated LB-100-induced viability (MTT OD) reduction in HCT-116 cells (Figure 2D). The two caspase inhibitors almost blocked LB-100-induced apoptosis activation (Data not shown). These results suggest that LB-100 activates caspase-dependent apoptosis to kill the CRC cells. It should be noted that significant apoptosis activation (TUNEL assay) was also observed in LB-100 (10 μM)-treated HT-29 cells and the primary human colon cancer cells (Figure 2E). TUNEL ratio was yet unchanged in the colon epithelial cells with same LB-100 treatment, further suggesting its selective response in cancerous cells (Figure 2E). These results demonstrate that LB-100 activates apoptosis in CRC cells.
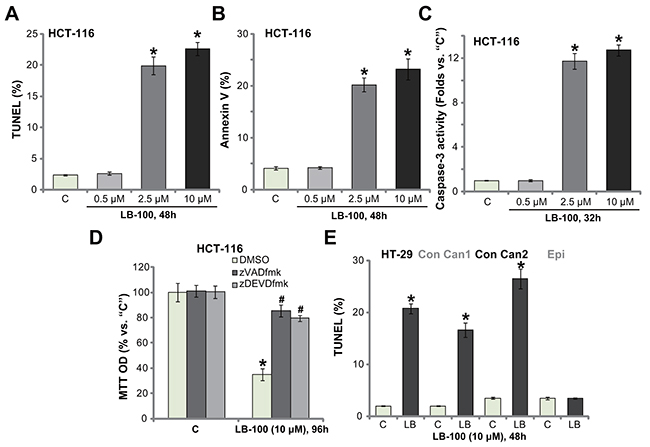
Figure 2: LB-100 activates apoptosis in CRC cells. The established human CRC cells (HCT-116 and HT-29 lines), the primary-cultured human colon cancer cells (two lines, “Con Can1/2”), or the primary-cultured colon epithelial cells (“Epi”) were treated with LB-100 at designated concentration, cells were further cultured for applied time; Cell apoptosis were tested by the assays mentioned in the text (A-C and E). For panel (D), HCT-116 cells were co-treated with 50 μM of the caspase-3 inhibitor zDEVDfmk or the pan caspase inhibitor zVADfmk, and cell viability was tested by MTT assay. Error bars indicate standard deviation (SD). “C” stands for untreated control group. *p<0.05 vs. “C” group. # p<0.05 vs. DMSO (0.1%) group (D). Experiments in this figure were repeated three times, and similar results were obtained.
LB-100 induces G1-S arrest in CRC cells
The activity of LB-100 on cell cycle progression was also tested. HCT-116 cells were treated with LB-100 (10 μM), cells were further cultured for additional 36 hours, and FACS assay was employed to test cell cycle distribution. The quantified results in Figure 3A demonstrated that, following the LB-100 treatment, the percentage of G1-phase cells was increased, but the percentages S-phase and G2-phase cells were both decreased. These results indicate G1-S arrest by LB-100 treatment in HCT-116 cells, which should favor proliferation-inhibition and apoptosis. The very similar G1-S arrest result was also obtained in the LB-100-treated primary human colon cancer cells (Figure 3B). Thus, LB-100 induces G1-S arrest in CRC cells.
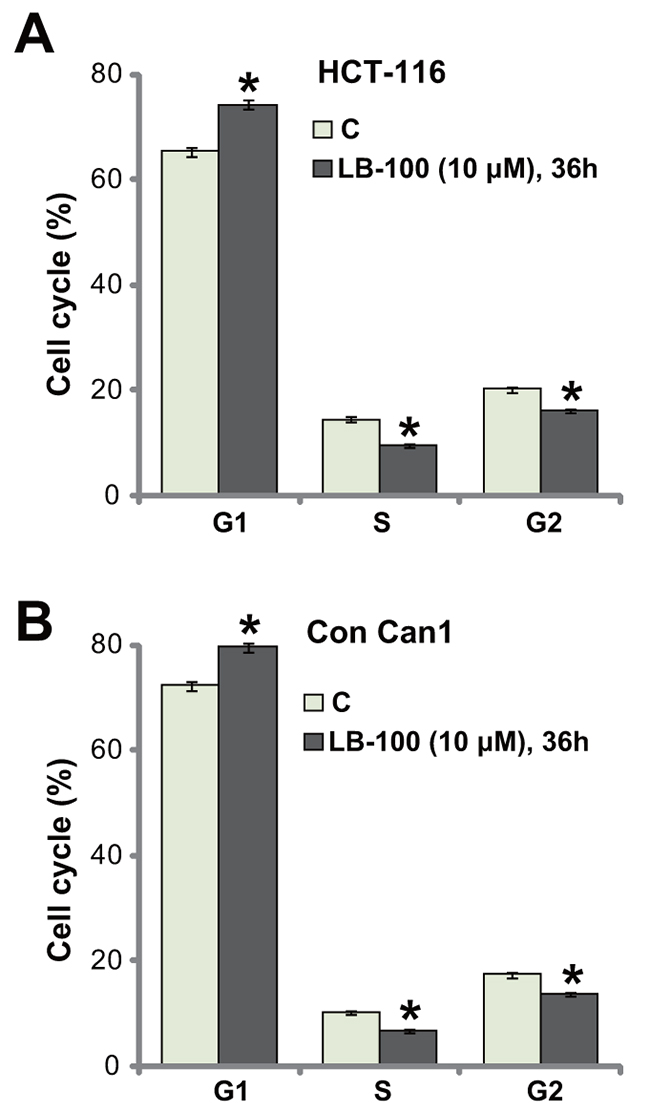
Figure 3: LB-100 induces G1-S arrest in CRC cells. HCT-116 cells (A) or the primary-cultured human colon cancer cells (“Con Can1”, (B) were treated with LB-100 at 10 μM, cells were further cultured for 36 hours; Cell cycle distribution was tested by the FACS assay, percentages of G1-, S- and G2-phase cells were quantified (A and B). Error bars indicate standard deviation (SD). “C” stands for untreated control group. *p<0.05 vs. “C” group. Experiments in this figure were repeated five times, and similar results were obtained.
LB-100 inhibits PP2A activity and activates AMPK signaling in CRC cells
LB-100 is a novel PP2A inhibitor [13, 30–32]. Its effect on PP2A activity was examined. As displayed, treatment with LB-100 (10 μM) in HCT-116 cells largely inhibited PP2A activity (Figure 4A). Expression of PP2A was unchanged by the same LB-100 treatment (Data not shown). As discussed, recent studies have focused on the function of AMPK activation in suppressing human CRC cells [4, 21, 33–35]. Several anti-cancer agents were shown to activate AMPK signaling to inhibit CRC cells [4, 21, 33–35]. Forced-activation of AMPK, via adding AMPK activators or expressing genetic modified AMPKα, was able to efficiently inhibit CRC cells [4, 36]. Recent studies have proposed PP2A as a key AMPKα phosphotase [15, 16, 37].
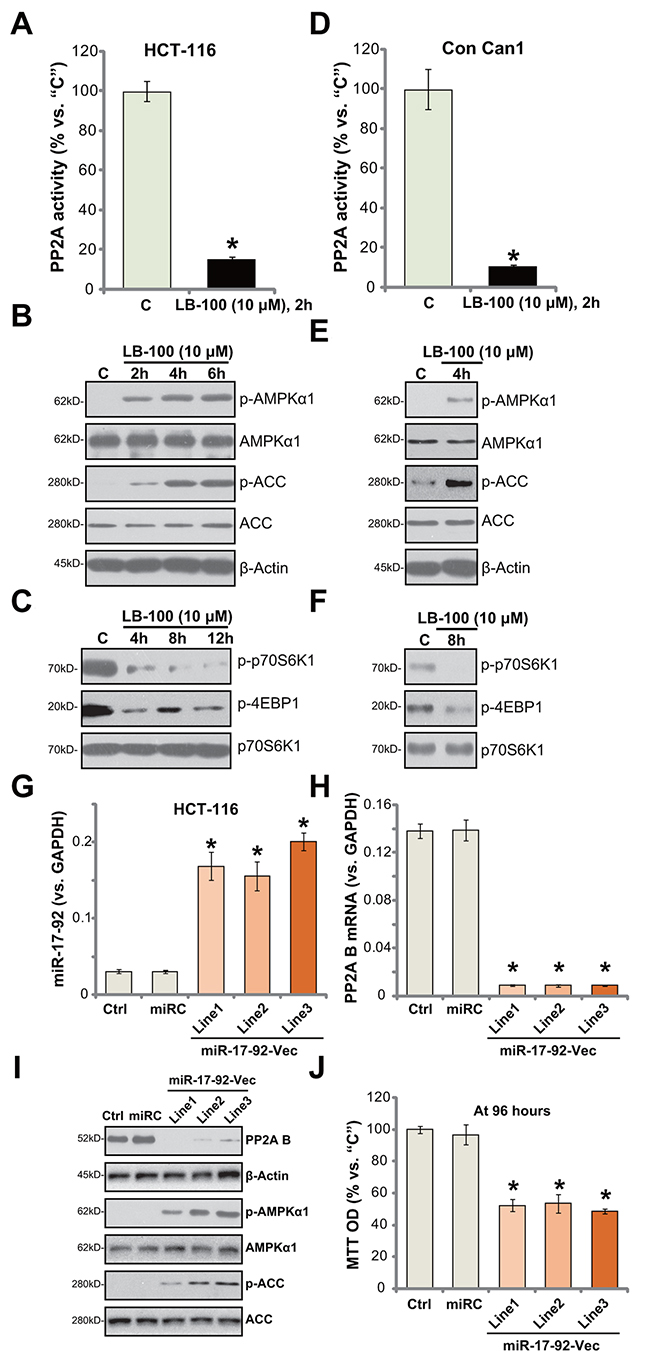
Figure 4: LB-100/miR-17-92 inhibits PP2A and activates AMPK signaling in CRC cells. HCT-116 cells (A-C) or the primary-cultured human colon cancer cells (“Con Can1”, D-F) were treated with LB-100 at 10 μM, cells were further cultured for the designated time; Relative PP2A activity (A and D) was analyzed; Expressions of listed proteins were tested by Western blotting assay (B, C, E and F). Stable HCT-116 cells, expressing miR-17-92 expression vector (“miR-17-92-Vec”, three lines, “Line1/2/3”), non-sense control microRNA (“miR”), or the parental control cells (“Ctrl”) were subjected to quantitative real-time PCR (“qRT-PCR”) assay of miR-17-92 (G) and PP2A B subunit (H) as well as the Western blotting assay of listed proteins (I); Cell survival at 96 hours was tested by MTT assay (J). Error bars indicate standard deviation (SD). “C” stands for untreated control group. *p<0.05 vs. “C” group (A and D). *p<0.05 vs. “Ctrl” cells (G, H and J). Experiments in this figure were repeated three times, and similar results were obtained.
Since LB-100 blocked PP2A activity (Figure 4A), we thus tested AMPK signaling in LB-100-treated cells. The Western blotting assay results in Figure 4B demonstrated that LB-100 (10 μM) induced profound AMPK activation in HCT-116 cells. AMPK activation was tested by phosphorylated (“p-“) AMPKα1 (at Thr-172) and its major downstream substrate protein, acetyl-CoA carboxylase (ACC, Ser-79) [19, 25]. One major consequence following AMPK activation is mTOR complex 1 (mTORC1) inhibition (See discussion below [19, 25]). Here, we demonstrate that treatment with LB-100 (10 μM) in HCT-116 cells largely inhibited phosphorylations of two primary mTORC1 substrates, including p70S6K1 and 4E-binding protein 1 (4E-BP1) [38–40], suggesting mTORC1 in-activation [41] (Figure 4C). Above-mentioned signalings were also tested in the primary human colon cancer cells. As demonstrated, LB-100 similarly blocked PP2A activation (Figure 4D), activated AMPK signaling (Figure 4E) and inhibited mTORC1 (Figure 4F) in the primary cancer cells. These results indicate that LB-100 blocks PP2A and activates AMPK signaling in HCT-116 cells.
miR17-92-mediated silence of PP2A activates AMPK and inhibits HCT-116 cell survival
miRNAs (miRs) are capable of suppressing targeted-gene expression via binding to 3’-untranslated region (UTR) of targeted-mRNAs [42–44]. Previous studies have identified an anti-PP2A miR, miR-17-92 [45]. Here, a miR-17-92-expression vector was constructed and transfected to the HCT-116 cells. Three lines with the construct were established (“Line1/Line2/Line3”). As demonstrated, forced-expression of miR-17-92-vector indeed significantly increased miR-17-92 level in all three lines (Figure 4G). Consequently, PP2A (B subunit) mRNA (Figure 4H) and protein (Figure 4I) level was sharply reduced. AMPK activation, tested by p-AMPKα1/p-ACC, was increased (Figure 4I). Additionally, HCT-116 cell survival, tested by MTT assay, was inhibited in miR-17-92-expressing HCT-116 cells (Figure 4J). Thus, in line with the LB-200’s data, miR17-92-mediated silence of PP2A also activated AMPK and inhibited HCT-116 cell survival. Notably, the nonsense microRNA control (“miRC”) failed to affect miR-17-92/PP2A expression (Figure 4G-4I) nor HCT-1116 cell survival (Figure 4J).
AMPK activation is required for LB-100-induced cytotoxicity in HCT-116 cells
In order to further confirm that LB-100 activates AMPK signaling in CRC cells. AMPK activity was tested. As shown in Figure 5A, treatment with LB-100 (10 μM) in HCT-116 cells significantly increased AMPK activity in HCT-116 cells. The effect by LB-100 was again dose-dependent (Figure 5A). Next, genetic strategies were applied to block AMPK activation. First, a dominant negative AMPKα1 (T172A, from Dr. Wu [4]) was introduced to HCT-116 cells, which in-activated AMPKα1 (Figure 5B). Secondly, the lentiviral AMPKα1 shRNA was added to HCT-116 cells to stably knockdown AMPKα1 (Figure 5B). Third, the CRISPR/Cas9 method was utilized to knockout AMPKα1 [46] in HCT-116 cells (Figure 5B). As displayed, AMPKα1 mutation (T172A), knockdown (by targeted-shRNA) or complete knockout (by CRISPR/Cas9) almost completely blocked LB-100-induced AMPK activation, or AMPKα1/ACC phosphorylations (Figure 5B). Consequently, mTORC1 activation, tested by p-p70S6K1, was restored (Figure 5C). Remarkably, AMPKα1 mutation, shRNA knockdown or complete knockout largely attenuated LB-100 (10 μM)-induced HCT-116 cell viability (“MTT OD”) reduction (Figure 5D) and apoptosis activation (TUNEL staining, Figure 5E). These results show that genetic in-activation of AMPK largely attenuated LB-200-induced cytotoxicity against HCT-116 cells. Thus, AMPK activation is required for LB-100-induced cytotoxicity in HCT-116 cells.
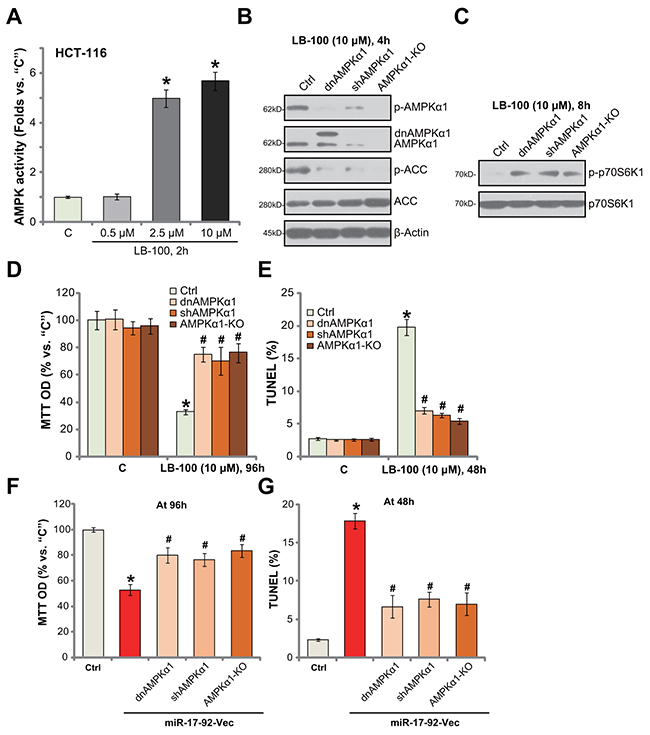
Figure 5: AMPK activation is required for LB-100/miR-17-92-induced cytotoxicity in HCT-116 cells. HCT-116 cells were treated with LB-100 (at applied concentration), cells were further cultured for designated time; Relative AMPK activity was shown (A) Stable HCT-116 cells, expressing AMPKα1 shRNA (“shAMPKα1”), dominant negative AMPKα1 (T172A, Flag-tagged, “dnAMPKα1”) or CRISPR/Cas9 AMPKα1 (“AMPKα1-KO”), as well as the control parental HCT-116 cells (“Ctrl”) were treated with LB-100 (10 μM) or transfected with miR-17-92 expression vector (“miR-17-92-Vec”), cells were further cultured for designated time; Expressions of listed proteins were tested by Western blotting assay (B and C) Cell survival (MTT assay, D and F) and apoptosis (TUNEL assay, E and G) were also tested. Error bars indicate standard deviation (SD). “C” stands for untreated control group. *p<0.05 vs. “C” group. # p<0.05 vs. “Ctrl” cells (D and E). # p<0.05 vs. miR-17-92-Vec only cells (F and G). Experiments in this figure were repeated three times, and similar results were obtained.
AMPK activation is required for miR17-92-induced cytotoxicity against HCT-116 cells
Further studies showed that forced-expression of miR17-92-induced HCT-116 cell death (MTT OD reduction, Figure 5F) and apoptosis (TUNEL increase, Figure 5G) were also largely attenuated with AMPKα mutation (T172A), silence or knockout. These results suggest that AMPK activation is also required for miR17-92-induced cytotoxicity against HCT-116 cells.
LB-100 administration activates AMPK signaling and inhibits HCT-116 tumor growth in nude mice
The LB-100’s activity on CRC growth in vivo was tested. HCT-116 cells, with or without AMPKα1, were injected s.c. to the nude mice. HCT-116 xenografts were then established. Weekly tumor growth curve results in Figure 6A displayed that daily intraperitoneally (i.p.) administration of LB-100 (5 mg/kg body weight) potently inhibited growth of HCT-116 tumors in the mice. The volume of LB-100-treated tumors was markedly lower than that of the vehicle control mice (Figure 6A). The estimated daily tumor growth (in mm3 per day), which was calculated by (tumor volume at day-35—tumor volume at day-0)/35, was also significantly decreased by LB-100 administration (Figure 6B). Additionally, the average tumor weight at day-35 was dramatically lower in the LB-100-treated group (Figure 6C). These results indicated that LB-100 i.p. administration inhibited HCT-116 tumor growth in nude mice. The mice body weights, which reflected animals’ general health condition, were not significantly changed by LB-100 administration (Figure 6D). No significant or apparent toxicities were observed in the experimental mice.
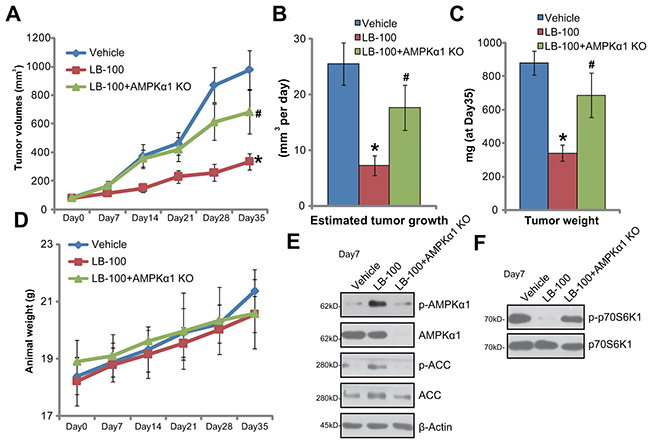
Figure 6: LB-100 administration activates AMPK signaling and inhibits HCT-116 tumor growth in nude mice. Weekly tumor growth curve of xenografts (from AMPKα1 knockout or control HCT-116 cells) (A) and mice body weight curve (D) with indicated treatment: Saline (“Vehicle”, i.p.), or LB-100 (5 mg/kg, i.p. daily, for 21 days), were shown; Estimated daily tumor growth (B) and tumor weights (at Day-35, C) were also shown; Seven days after initial LB-100 treatment, one tumor of each group was removed; Tumor tissues were subjected to Western blotting assay of listed proteins (E and F). Error bars indicate standard deviation (SD). * p < 0.05 vs. “Vehicle” group. # p < 0.05 vs. control tumors.
Notably, LB-100-induced anti-tumor activity in vivo was largely compromised against tumors that were derived from AMPKα1-knockout (by CRISPR/Cas9 method) HCT-116 cells (“+AMPKα1 KO”, Figure 6A-6C). These results suggest that AMPK activation should also be required or LB-100-induced activity in vivo. Indeed, when analyzing signaling changes in tumor tissues, we demonstrate that LB-100 administration activated AMPK (AMPKα1/ACC phosphorylations) in HCT-116 tumors (Figure 6D), which was followed by mTORC1 (p-p70S6K1) inhibition (Figure 6F). Tumors-derived from AMPKα1-knockout cells showed depleted AMPKα1 (Figure 6E). LB-100-induced AMPK activation (Figure 6E) and mTORC1 inhibition (Figure 6F) were almost abolished in AMPKα1-knockout tumors. Together, we conclude that LB-100 administration activates AMPK and inhibits HCT-116 tumor growth in nude mice.
DISCUSSION
AMPKα1 phosphorylation at Thr-172 is vital for AMPK activation [20, 47, 48]. Studies have been focusing on the upstream mechanisms of phosphorylation of AMPKα1 [26]. A number of potential AMPKα1 kinases have been identified. These kinases, including LKB1 (Liver kinase B1) [48] and CaMKK (calcium/calmodulin-dependent protein kinase kinase) [49], directly phosphorylate AMPKα1 at Thr-172, leading to profound AMPK activation. On the other hand, the mechanisms of AMPKα1 de-phosphorylation are largely unknown. Several potential AMPKα1 phosphatase were proposed [50]. One key AMPKα1 phosphatase is PP2A [15, 37]. In the current study, we show that targeted inhibition of PP2A by LB-100 activated AMPK signaling in CRC cells. Notably, AMPKα in-activation, silence or complete depletion via the described methods didn’t result in total abolition of LB-100-induced cytotoxicity against HCT-116 cells (See Figure 5). These results suggest that other signalings, besides AMPK activation, could also be responsible for LB-100’s actions in CRC cells. These results are not surprising, as PP2A could de-phosphorylate substrate kinases other than AMPK [10, 51]. It should also be noted that miR-17-92-mediated silence of PP2A also activated AMPK and induced HCT-116 cell death. Such effects were again largely attenuated by AMPKα mutation, silence or complete knockout.
Dysregulation of mTOR signaling has been detected in CRC tissues [52, 53]. Sustained and over-activation of mTOR in CRC is positively involved in a number of key oncogenic behaviors, including uncontrolled cancer cell survival, proliferation and migration, as well as chemo-resistance and angiogenesis [52, 53]. Thus, mTOR over-activation represents an important oncotarget of CRC treatment [3, 4]. AMPK activation shall lead to mTORC1 in-activation, thus inhibiting CRC and other cancer cells. Tuberous sclerosis 2 (TSC2) phosphorylation and activation by activated AMPK would lead to downstream mTORC1 in-activation [54–56]. Meanwhile, AMPK is also shown to directly phosphorylate and in-activate Raptor, a key mTORC1 component, to shut down mTORC1 activation [57, 58].
In the current study, we show that AMPK activation by LB-100 also inhibited mTORC1 activation in CRC cells. Phosphorylations of mTORC1 substrates, including p70S6K1 and 4E-BP1, were dramatically inhibited in LB-100-treated cells. Notably, LB-100-induced mTORC1 inhibition was almost completely reversed with AMPKα1 mutation, shRNA knockdown or complete knockout. Thus, we propose that blockage of PP2A by LB-100 activates AMPK to inhibit mTORC1 activation in CRC cells. This mechanism could a primary reason of CRC cell inhibition. These results might also explain the in-effectiveness of LB-100 against human colon epithelial cells, as the normal cells displayed extremely low level of mTORC1 activation [59].
MATERIALS AND METHODS
Reagents
LB-100 was provided by Selleck (Beijing, China). The caspase-3 inhibitor zDEVDfmk and the pan caspase inhibitor zVADfmk were purchased from Sigma (Nanjing, China). All the antibodies of this study were purchased from Cell Signaling Technology (Beverly, MA). The tissue-culture reagents were provide by Gibco (Nanjing, China).
Culture of established cell lines
The established human CRC cell lines, HCT-116 and HT-29, were purchased from the iBS Cell Bank of Fudan University (Shanghai, China). Cells were cultured in DMEM medium, plus 6% FBS. DNA fingerprinting and profiling were performed to distinguish the cell line from possible cross-contamination.
Primary human colon cancer cells and colon epithelial cells
The protocols of isolation and culture of human colon cancer cells as well as the primary colon epithelial cells were described in detail previously [60, 61]. In brief, the colon cancer issues and the surrounding normal colon epithelial tissues from patients (two male, 49 and 59 years old) were obtained, washed, and digested. The single cell suspensions were pelleted, and resuspended in the described medium plus growth factors for the primary human cells [61]. A total of two primary human colon cancer cell lines and one colon epithelial cell line were established. Written-informed consent was obtained from each patient. The protocols were approved by the IRB (Institutional Review Board) and Ethics Committee of all authors’ institutions, and experiments were conducted according to the principles of Declaration of Helsinki.
Cell viability assay
The cell survival was always tested by the 3-[4,5-dimethylthylthiazol-2-yl]-2,5 diphenyltetrazolium bromide (MTT) (Sigma, Nanjing, China) assay according to the attached protocol [61]. The MTT OD of treatment cells was always expressed as the percentage of that of untreated control cells.
Colony formation assay
HCT-116 cells were initially plated at 5 × 103 cells per 10-cm tissue culture dish. LB-100-containing medium was renewed every two days. After 10 days of incubation, the remaining HCT-116 colonies were stained and manually counted.
Cell proliferation assay
The simple cell counting assay and BrdU incorporation ELISA assay were performed to test cell proliferation. The detailed protocols were described previously [4, 62].
Annexin V FACS analysis
The Annexin V In Situ Cell Apoptosis Detection Kit (Roche, Indianapolis, IN) [61, 63] was employed to test cell apoptosis. In brief, following the applied LB-100 treatment, cells were washed, fixed and stained with Annexin V (2.5 μg/mL) and propidium iodide (PI) (2.5 μg/mL). The cell apoptosis ratio was reflected by Annexin V+/+/PI-/- plus Annexin V+/+/PI+/+ percentage detected by fluorescence-activated cell sorting (FACS) counter (Beckman Coulter, Shanghai, China).
Apoptosis assays
Other apoptosis assays, including TUNEL assay and caspase-3 activity, were performed as previously described [4].
Cell cycle analysis
Following the applied LB-100 treatment, CRC cells were labeled with 10 μM EdU (Biyuntian, Nanjing, China) for 1 hour and were then fixed. Cells were then analyzed by flow cytometry (Beckman Coulter, Shanghai, China). Cell cycle distribution was analyzed with Kaluza software (Beckman Coulter).
Western blotting assay
Cells and tumor tissues were lysed by the commercial available RIPA lysis buffer (Biyuntian) plus Complete Protease Inhibitor Cocktail (Roche) and phosphatase inhibitors (Roche). Quantified lysate proteins (40 μg of each condition) were separated by the 7.5-10 % SDS-PAGE gels (Biyuntian), and were transferred onto the polyvinylidene fluoride (PVDF) membrane. The blot was then blocked, and were probed overnight with designated primary antibody. The corresponding horseradish peroxidase (HRP)-conjugated secondary antibody was then added. Super-Signal West Pico Chemiluminescent Substrates (Thermo Scientific, Shanghai, China) were added to visualize the targeted protein band (based on the molecular weight), under the X-ray film development.
PP2A phosphatase activity assay
After the designated LB-100 treatment, CRC cells were washed and lysed in the described RIPA lysis buffer. The supernatants of 50 μg of total cellular proteins were assayed with the PP2A Phosphatase Assay Kit (Millipore) by the attached protocol. The PP2A activity data were presented as percentage of relative PP2A activity compared with control.
AMPK activity assay
Following the LB-100 treatment, cell lysates were achieved. AMPK was firstly immunoprecipitated with anti-pan-AMPKα antibody. The AMPK activity was determined in kinase assay buffer (see previous study [64]) plus AMP-[γ-32P] ATP mixture, and SAMS peptide (HMRSAMSGLHLVKRR) [64]. The reaction was terminated by spotting the reaction mixture on phosphocellulose paper (P81), which was extensively washed with 150 mM of phosphoric acid. The radioactivity was measured with scintillation counter. AMPK activity in the treatment group was always normalized to that of the untreated control group.
AMPKα1 shRNA
The lentiviral particles, with human AMPKα1 shRNA or scramble non-sense control shRNA, were obtained from Santa Cruz Biotech (Nanjing, China). The lentivirus was added to HCT-116 cells for 24 hours. Puromycin (3.0 μg/mL, Sigma) was then added, which helped to select the stable cells.
AMPKα1 mutation
The dominant negative AMPKα1 (dn-AMPK-α1, T172A, Flag-tagged) construct was from Dr. Wu [4]. The transfection of mutant AMPKα1 by Lipofectamine 2000 was described previously [4]. Neomycin (1 μg/mL) was added to select the stable cells. The mutant AMPKα1 expression was confirmed by Western blotting assay.
CRISPR/Cas9-mediated AMPKα1 knockout
The small guide RNA (sgRNA) targeting human AMPKα1 was through the Optimized Crispr Design application from Dr. Zhang’s lab (http://crispr.mit.edu/), which was inserted into the lentiCRISPR plasmid (Addgene plasmid 49535, Shanghai, China). The plasmid was transfected to HCT-116 cells using the described protocol [46]. AMPKα1 expression in the stable cells was confirmed by Western blotting assay.
Forced miR-17-92 expression
The pSuper-GPF-puro-miR-17-92 expression vector, encoding miR-17-92 (based on the sequence reported previously [65, 66]), was designed, synthesized and sequence-verified by Shanghai Genepharm Co. (Shanghai, China). The HCT-116 cells were transfected with miR-17-92 expression vector or the scramble miRNA control (“miR-C”) via the Lipofectamine 2000 reagent (Invitrogen, Nanjing, China). Forty-eight hours after transfection of miR, stable cells were selected by puromycin (0.5 μg/mL, Sigma).
RNA isolation and RT-PCR
Total cellular RNA was extracted by the RNeasy Mini Kit (Qiagen, Wuxi, China). A total of 600 ng RNA per treatment was reverse-transcribed by commercial SYBR green kit (TOYOBO, Japan). Quantitative real-time PCR (“qRT-PCR”) was performed through the ABI-7500 fast PCR system (Applied Biosystems, Shanghai, China) [67, 68]. PP2A regulatory (B subunit) mRNA primers were reported early [45, 69]. For miR analysis, miR was converted to cDNA from 600 ng of total RNA using the First-Strand Synthesis Kit (SABiosciences, Frederick, MD). miR-17-92 analysis was performed through qRT-PCR assay using the miR-17-92 primers (SABiosciences) [65, 66]. miR-17-92 level was also normalized to GAPDH.
Xenograft tumor assay
BALB/c nude mice (17.8-18.5 grams, 4-5 week old, all female) were injected subcutaneously (s.c.) in the right flanks with 1 × 106 HCT-116 cells (per mouse). Animals were housed in temperature- and humidity-controlled cages, with free access to water and rodent food on a 12-h light/dark cycle. After a tumor volume of 80 mm3 was reached, LB-100 were injected intraperitoneally (i.p.) daily for a total of 21 days. Control mice were injected with equal quantity of vehicle. Tumor volumes were recorded weekly, calculated via the following formula: π/6 × larger diameter × (smaller diameter)2. All animal studies were performed in accordance with the standards of ERB and IACUC of all authors’ institutions.
Statistical analysis
The results were expressed as the mean ± standard deviation (SD). Ordinary one-way ANOVA test was employed for comparison between groups. p < 0.05 was considered as statistically significant.
CONCLUSION
The previous cancer studies have suggested that PP2A inhibition is likely to be most effective for cancer therapy when combined with traditional cytotoxic agents [14, 31, 32]. The results of this study show that PP2A inhibition by LB-100 or miR-17-92 may have significant anti-CRC cell activity in vitro and in vivo. LB-100 or miR-17-92 could be further tested as promising anti-CRC agents.
Author contributions
All authors carried out the experiments, participated in the design of the study and performed the statistical analysis, participated in its design and coordination and helped to draft the manuscript.
CONFLICTS OF INTEREST
The listed authors have no conflicts of interest.
FUNDING
This study was supported in part by the “533 Talents Project” research project in 2011 of Huaian City (Hygiene category 78), by the Medical Technology Development Project of Health Department of Jiangsu Province (J200912), by the Social Development Fund of Technology Project, in Huaian City, Jiangsu Province, China (HAS2009002-3) and by the Science and Technology Development Project, in Huaian City, Jiangsu Province, China (HAS201605 and HAS2009002-3).
REFERENCES
1. McCarthy N. Colorectal cancer: Editing an invasion. Nat Rev Cancer. 2014; 14: 297. https://doi.org/10.1038/nrc3735.
2. Kuipers EJ, Rosch T, Bretthauer M. Colorectal cancer screening: optimizing current strategies and new directions. Nat Rev Clin Oncol. 2013; 10: 130-42. https://doi.org/10.1038/nrclinonc.2013.12.
3. Lu XS, Qiao YB, Li Y, Yang B, Chen MB, Xing CG. Preclinical study of cinobufagin as a promising anti-colorectal cancer agent. Oncotarget. 2017; 8: 988-98. https://doi.org/10.18632/oncotarget.13519.
4. Lu PH, Chen MB, Ji C, Li WT, Wei MX, Wu MH. Aqueous Oldenlandia diffusa extracts inhibits colorectal cancer cells via activating AMP-activated protein kinase signalings. Oncotarget. 2016; 7: 45889-900. https://doi.org/10.18632/oncotarget.9969.
5. Li JP, Huang ZJ, Lu XS, Zhou YC, Shao Y, He XP, Chen SR, Wang DD, Qin LS, Sun WH. Pre-clinical characterization of PKC412, a multi-kinase inhibitor, against colorectal cancer cells. Oncotarget. 2016; 7: 77815-24. https://doi.org/10.18632/oncotarget.12802.
6. Wang L, Zhao Z, Feng W, Ye Z, Dai W, Zhang C, Peng J, Wu K. Long non-coding RNA TUG1 promotes colorectal cancer metastasis via EMT pathway. Oncotarget. 2016; 7: 51713-9. https://doi.org/10.18632/oncotarget.10563.
7. Cunningham CE, Li S, Vizeacoumar FS, Bhanumathy KK, Lee JS, Parameswaran S, Furber L, Abuhussein O, Paul JM, McDonald M, Templeton SD, Shukla H, El Zawily AM, et al. Therapeutic relevance of the protein phosphatase 2A in cancer. Oncotarget. 2016; 7: 61544-61. https://doi.org/10.18632/oncotarget.11399.
8. Lai TY, Yen CJ, Tsai HW, Yang YS, Hong WF, Chiang CW. The B56gamma3 regulatory subunit-containing protein phosphatase 2A outcompetes Akt to regulate p27KIP1 subcellular localization by selectively dephosphorylating phospho-Thr157 of p27KIP1. Oncotarget. 2016; 7: 4542-58. https://doi.org/10.18632/oncotarget.6609.
9. Zhang W, Chen H, Chen Y, Liu J, Wang X, Yu X, Chen JJ, Zhao W. Cancerous inhibitor of protein phosphatase 2A contributes to human papillomavirus oncoprotein E7-induced cell proliferation via E2F1. Oncotarget. 2015; 6: 5253-62. https://doi.org/10.18632/oncotarget.2867.
10. Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta. 2009; 1795: 1-15. https://doi.org/10.1016/j.bbcan.2008.05.005.
11. Khanna A, Pimanda JE, Westermarck J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res. 2013; 73: 6548-53. https://doi.org/10.1158/0008-5472.CAN-13-1994.
12. Fu QH, Zhang Q, Zhang JY, Sun X, Lou Y, Li GG, Chen ZL, Bai XL, Liang TB. LB-100 sensitizes hepatocellular carcinoma cells to the effects of sorafenib during hypoxia by activation of Smad3 phosphorylation. Tumour Biol. 2016; 37: 7277-86. https://doi.org/10.1007/s13277-015-4560-2.
13. Chung V, Mansfield AS, Braiteh F, Richards D, Durivage H, Ungerleider RS, Johnson F, Kovach JS. Safety, tolerability, and preliminary activity of LB-100, an inhibitor of protein phosphatase 2A, in patients with relapsed solid tumors: an open-label, dose escalation, first-in-human, phase I trial. Clin Cancer Res. 2017; 23: 3277-84. https://doi.org/10.1158/1078-0432.CCR-16-2299.
14. Bai XL, Zhang Q, Ye LY, Hu QD, Fu QH, Zhi X, Su W, Su RG, Ma T, Chen W, Xie SZ, Chen CL, Liang TB. Inhibition of protein phosphatase 2A enhances cytotoxicity and accessibility of chemotherapeutic drugs to hepatocellular carcinomas. Mol Cancer Ther. 2014; 13: 2062-72. https://doi.org/10.1158/1535-7163.MCT-13-0800.
15. Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007; 282: 9777-88. https://doi.org/10.1074/jbc.M608310200.
16. Guo S, Chen C, Ji F, Mao L, Xie Y. PP2A catalytic subunit silence by microRNA-429 activates AMPK and protects osteoblastic cells from dexamethasone. Biochem Biophys Res Commun. 2017; 487: 660-5. https://doi.org/10.1016/j.bbrc.2017.04.111.
17. Deepa SS, Zhou L, Ryu J, Wang C, Mao X, Li C, Zhang N, Musi N, DeFronzo RA, Liu F, Dong LQ. APPL1 mediates adiponectin-induced LKB1 cytosolic localization through the PP2A-PKCzeta signaling pathway. Mol Endocrinol. 2011; 25: 1773-85. https://doi.org/10.1210/me.2011-0082.
18. O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013; 493: 346-55. https://doi.org/10.1038/nature.11862.
19. Hardie DG, Schaffer BE, Brunet A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016; 26: 190-201. https://doi.org/10.1016/j.tcb.2015.10.013.
20. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012; 13: 251-62. https://doi.org/10.1038/nrm3311.
21. Zhao Z, Feng L, Wang J, Cheng D, Liu M, Ling M, Xu W, Sun K. NPC-26 kills human colorectal cancer cells via activating AMPK signaling. Oncotarget. 2017; 8: 18312-21. https://doi.org/10.18632/oncotarget.15436.
22. Din FV, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K, Alessi DR, Dunlop MG. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012; 142: 1504-15 e3. https://doi.org/10.1053/j.gastro.2012.02.050.
23. Plews RL, Mohd Yusof A, Wang C, Saji M, Zhang X, Chen CS, Ringel MD, Phay JE. A novel dual AMPK activator/mTOR inhibitor inhibits thyroid cancer cell growth. J Clin Endocrinol Metab. 2015; 100: E748-56. https://doi.org/10.1210/jc.2014-1777.
24. Pineda CT, Potts PR. Oncogenic MAGEA-TRIM28 ubiquitin ligase downregulates autophagy by ubiquitinating and degrading AMPK in cancer. Autophagy. 2015; 11: 844-6. https://doi.org/10.1080/15548627.2015.1034420.
25. Garcia D, Shaw RJ. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell. 2017; 66: 789-800. https://doi.org/10.1016/j.molcel.2017.05.032.
26. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007; 8: 774-85. https://doi.org/10.1038/nrm2249.
27. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005; 18: 283-93. https://doi.org/10.1016/j.molcel.2005.03.027.
28. Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013; 152: 290-303. https://doi.org/10.1016/j.cell.2012.12.016.
29. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13: 132-41. https://doi.org/10.1038/ncb2152.
30. Gordon IK, Lu J, Graves CA, Huntoon K, Frerich JM, Hanson RH, Wang X, Hong CS, Ho W, Feldman MJ, Ikejiri B, Bisht K, Chen XS, et al. Protein Phosphatase 2A Inhibition with LB100 Enhances Radiation-Induced Mitotic Catastrophe and Tumor Growth Delay in Glioblastoma. Mol Cancer Ther. 2015; 14: 1540-7. https://doi.org/10.1158/1535-7163.MCT-14-0614.
31. Chang KE, Wei BR, Madigan JP, Hall MD, Simpson RM, Zhuang Z, Gottesman MM. The protein phosphatase 2A inhibitor LB100 sensitizes ovarian carcinoma cells to cisplatin-mediated cytotoxicity. Mol Cancer Ther. 2015; 14: 90-100. https://doi.org/10.1158/1535-7163.MCT-14-0496.
32. Bai X, Zhi X, Zhang Q, Liang F, Chen W, Liang C, Hu Q, Sun X, Zhuang Z, Liang T. Inhibition of protein phosphatase 2A sensitizes pancreatic cancer to chemotherapy by increasing drug perfusion via HIF-1alpha-VEGF mediated angiogenesis. Cancer Lett. 2014; 355: 281-7. https://doi.org/10.1016/j.canlet.2014.09.048.
33. Kang MR, Park SK, Lee CW, Cho IJ, Jo YN, Yang JW, Kim JA, Yun J, Lee KH, Kwon HJ, Kim BW, Lee K, Kang JS, et al. Widdrol induces apoptosis via activation of AMP-activated protein kinase in colon cancer cells. Oncol Rep. 2012; 27: 1407-12. https://doi.org/10.3892/or.2012.1644.
34. Chen MB, Zhang Y, Wei MX, Shen W, Wu XY, Yao C, Lu PH. Activation of AMP-activated protein kinase (AMPK) mediates plumbagin-induced apoptosis and growth inhibition in cultured human colon cancer cells. Cell Signal. 2013; 25: 1993-2002. https://doi.org/10.1016/j.cellsig.2013.05.026.
35. Chen MB, Wei MX, Han JY, Wu XY, Li C, Wang J, Shen W, Lu PH. MicroRNA-451 regulates AMPK/mTORC1 signaling and fascin1 expression in HT-29 colorectal cancer. Cell Signal. 2014; 26: 102-9. https://doi.org/10.1016/j.cellsig.2013.07.017.
36. Chen MB, Jiang Q, Liu YY, Zhang Y, He BS, Wei MX, Lu JW, Ji Y, Lu PH. C6 ceramide dramatically increases vincristine sensitivity both in vivo and in vitro, involving AMP-activated protein kinase-p53 signaling. Carcinogenesis. 2015; 36: 1061-70. https://doi.org/10.1093/carcin/bgv094.
37. Perera ND, Sheean RK, Scott JW, Kemp BE, Horne MK, Turner BJ. Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models. PLoS One. 2014; 9: e95549. https://doi.org/10.1371/journal.pone.0095549.
38. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017; 168: 960-76. https://doi.org/10.1016/j.cell.2017.02.004.
39. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012; 149: 274-93. https://doi.org/10.1016/j.cell.2012.03.017.
40. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006; 6: 729-34. https://doi.org/10.1038/nrc1974.
41. Chen GQ, Tang CF, Shi XK, Lin CY, Fatima S, Pan XH, Yang DJ, Zhang G, Lu AP, Lin SH, Bian ZX. Halofuginone inhibits colorectal cancer growth through suppression of Akt/mTORC1 signaling and glucose metabolism. Oncotarget. 2015; 6: 24148-62. https://doi.org/10.18632/oncotarget.4376.
42. Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016; 17: 272-83. https://doi.org/10.1038/nrg.2016.20.
43. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015; 16: 421-33. https://doi.org/10.1038/nrg3965.
44. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012; 13: 271-82. https://doi.org/10.1038/nrg3162.
45. Jablonska E, Gorniak P, Szydlowski M, Sewastianik T, Bialopiotrowicz E, Polak A, Warzocha K, Juszczynski P. MiR-17-92 represses PTPROt and PP2A phosphatases and amplifies tonic BCR signaling in DLBCL cells. Exp Hematol. 2017; 46: 56-61. https://doi.org/10.1016/j.exphem.2016.09.011.
46. Sujobert P, Poulain L, Paubelle E, Zylbersztejn F, Grenier A, Lambert M, Townsend EC, Brusq JM, Nicodeme E, Decrooqc J, Nepstad I, Green AS, Mondesir J, et al. Co-activation of AMPK and mTORC1 Induces Cytotoxicity in Acute Myeloid Leukemia. Cell Rep. 2015; 11: 1446-57. https://doi.org/10.1016/j.celrep.2015.04.063.
47. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011; 13: 1016-23. https://doi.org/10.1038/ncb2329.
48. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004; 101: 3329-35. https://doi.org/10.1073/pnas.0308061100.
49. Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JF, Richter EA. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab. 2007; 292: E1308-17. https://doi.org/10.1152/ajpendo.00456.2006.
50. Chen MB, Liu YY, Cheng LB, Lu JW, Zeng P, Lu PH. AMPKalpha phosphatase Ppm1E upregulation in human gastric cancer is required for cell proliferation. Oncotarget. 2017; 8: 31288-96. https://doi.org/10.18632/oncotarget.16126.
51. Kaur A, Westermarck J. Regulation of protein phosphatase 2A (PP2A) tumor suppressor function by PME-1. Biochem Soc Trans. 2016; 44: 1683-93. https://doi.org/10.1042/BST20160161.
52. Pandurangan AK. Potential targets for prevention of colorectal cancer: a focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pac J Cancer Prev. 2013; 14: 2201-5.
53. Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget. 2014; 5: 49-66. https://doi.org/10.18632/oncotarget.1548.
54. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003; 115: 577-90.
55. Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006; 126: 955-68. https://doi.org/10.1016/j.cell.2006.06.055.
56. Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015; 356: 165-70. https://doi.org/10.1016/j.canlet.2014.01.018.
57. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008; 30: 214-26. https://doi.org/10.1016/j.molcel.2008.03.003.
58. Hardie DG. AMPK and Raptor: matching cell growth to energy supply. Mol Cell. 2008; 30: 263-5. https://doi.org/10.1016/j.molcel.2008.04.012.
59. Wu L, Zhang J, Wu H, Han E. DNA-PKcs interference sensitizes colorectal cancer cells to a mTOR kinase inhibitor WAY-600. Biochem Biophys Res Commun. 2015; 466: 547-53. https://doi.org/10.1016/j.bbrc.2015.09.068.
60. Li C, Cui JF, Chen MB, Liu CY, Liu F, Zhang QD, Zou J, Lu PH. The preclinical evaluation of the dual mTORC1/2 inhibitor INK-128 as a potential anti-colorectal cancer agent. Cancer Biol Ther. 2015; 16: 34-42. https://doi.org/10.4161/15384047.2014.972274.
61. Zhijun H, Shusheng W, Han M, Jianping L, Li-Sen Q, Dechun L. Pre-clinical characterization of 4SC-202, a novel class I HDAC inhibitor, against colorectal cancer cells. Tumour Biol. 2016. https://doi.org/10.1007/s13277-016-4868-6.
62. Li ZW, Cai S, Liu Y, Yang CL, Tian Y, Chen G, Cao C. Over-expression of Galphai3 in human glioma is required for Akt-mTOR activation and cell growth. Oncotarget. 2016. https://doi.org/10.18632/oncotarget.10995. [Epub ahead of print].
63. Qin LS, Jia PF, Zhang ZQ, Zhang SM. ROS-p53-cyclophilin-D signaling mediates salinomycin-induced glioma cell necrosis. J Exp Clin Cancer Res. 2015; 34: 57. https://doi.org/10.1186/s13046-015-0174-1.
64. Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003; 278: 39653-61. https://doi.org/10.1074/jbc.M306104200.
65. Dal Bo M, Bomben R, Hernandez L, Gattei V. The MYC/miR-17-92 axis in lymphoproliferative disorders: A common pathway with therapeutic potential. Oncotarget. 2015; 6: 19381-92. https://doi.org/10.18632/oncotarget.4574.
66. Jo DH, Kim JH, Cho CS, Cho YL, Jun HO, Yu YS, Min JK. STAT3 inhibition suppresses proliferation of retinoblastoma through down-regulation of positive feedback loop of STAT3/miR-17-92 clusters. Oncotarget. 2014; 5: 11513-25. https://doi.org/10.18632/oncotarget.2546.
67. Li KR, Yang SQ, Gong YQ, Yang H, Li XM, Zhao YX, Yao J, Jiang Q, Cao C. 3H-1,2-dithiole-3-thione protects retinal pigment epithelium cells against Ultra-violet radiation via activation of Akt-mTORC1-dependent Nrf2-HO-1 signaling. Sci Rep. 2016; 6: 25525. https://doi.org/10.1038/srep25525.
68. Gong YQ, Huang W, Li KR, Liu YY, Cao GF, Cao C, Jiang Q. SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget. 2016; 7: 60123-32. https://doi.org/10.18632/oncotarget.11164.
69. Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol. 2001; 168: 402-12. https://doi.org/10.1006/exnr.2001.7630.