
BIOLOGY OF CHILDHOOD ETV6-RUNX1+ B-ALL
The ETV6-RUNX1 fusion gene is the most common chromosomal alteration in pediatric cancer and occurs in approximately 25% of childhood B cell precursor-acute lymphoblastic leukemia (pB-ALL) [1]. The t(12;21)(p13;q22) chromosomal translocation results in the fusion of two critical regulators of hematopoiesis, bringing together the 5′ portion of the ETV6 (TEL) gene on chromosome 12p13 and nearly the entire RUNX1 (AML1) gene on chromosome 21q22 [2–4]. The overall cure rate is excellent (approximately 90%), however, treatment is associated with severe toxic side effects, long-term sequelae and even though, 20% of children still relapse and may later succumb to their disease [5]. Preventional strategies are clearly superior to any therapy improvement. The prerequisite to develop these strategies is to discover the aetiology of ETV6-RUNX1 pB-ALL.
The ETV6-RUNX1 fusion gene is one of the most extreme examples of genotype-phenotype association. Its presence is only associated to pB-ALL. ETV6-RUNX1 pB-ALL is a clonal malignant disease that originates in a single cell and is characterized by an accumulation of immature B-cells that are phenotypically reminiscent of normal stages of B-cell differentiation. This finally leads to the suppression of normal hematopoiesis and the infiltration of many vital organs. The cells in ETV6-RUNX1 pB-ALL are generally regarded as malignant counterparts of normal B-cell precursors, but ETV6-RUNX1 pB-ALL origin is still the subject of continuing discussion, given the fact that human disease is diagnosed at late stages and cannot be monitored during its natural evolution from its cell of origin [6–8].
Pre-leukaemic clones carrying ETV6-RUNX1 oncogenic lesions are frequently found in neonatal cord blood [9, 10], but only few ETV6-RUNX1 carriers develop the disease (Figure 1). Thus, ETV6-RUNX1 gene fusion seems to confer a low risk of developing pB-ALL and represents the first event (“hit”) in the process of leukemogenesis creating a preleukemic clone, which requires secondary postnatal genetic aberrations. A better understanding of both, what causes the initial changes and which events are leading to an irreversibly transformed state can potentially allow to prevent pB-ALL development in preleukemic carriers. Thus, we can now consider childhood leukemia research in terms of seeking to prevent the occurrence of this disease. This conceptual framework requires a precise understanding of the genetic and epigenetic factors that promote pB-ALL development, and how these are influenced by environmental factors associated to human leukemia development such as infection, low-dose radiation, etc (Figure 1). However, the molecular events involved in the early genesis of ETV6-RUNX1-associated leukemia have been particularly elusive because these stages are usually not detected in children and, when they are, very little if any tissue is available for research studies. Therefore, preclinical models of ETV6-RUNX1-associated pB-ALL are an essential unmet need to prevent the occurrence of this disease.
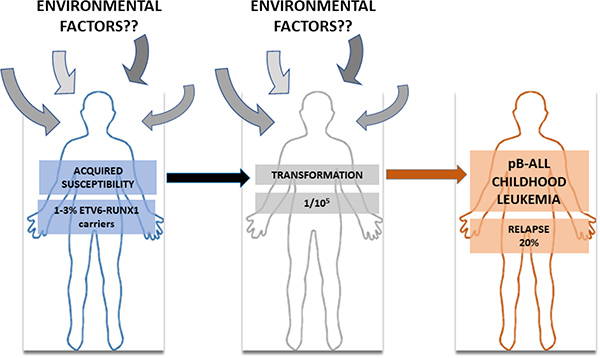
Figure 1: The natural history of ETV6-RUNX1 pB-ALL childhood leukemia. Between 1–3% of pre-leukemic clones carrying ETV6-RUNX1 translocation are frequently found in neonatal cord blood, but only few ETV6-RUNX1 carries develop pB-ALL (1/105). Many environmental factors have been pointed out as implicated in this transformation process but the lack of biological evidence has kept environmental factor under suspicion.
Zebrafish transgenic models for childhood ETV6-RUNX1+ pB-ALL
Zebrafish has been instrumental to delineate the cell-of-origin where the ETV6-RUNX1 fusion gene could facilitate the development of pB-ALL. When ETV6-RUNX1 expression was under the control of the rag2 promoter, zebrafish transgenic did not develop any leukemia, suggesting that the cell-of-origin of pB-ALL is not a lymphoid committed progenitor [11]. On the contrary, ETV6-RUNX1 expression under the control of ubiquitous promoters in zebrafish resulted in precursor B-cell malignancies, suggesting that ETV6-RUNX1-associated leukemias only might develop when ETV6-RUNX1 was expressed at the level of non-committed progenitors [11]. Like in humans, these B-cell leukemias appeared with low penetrance and long latency suggesting the need for a second hit. However, this zebrafish transgenic model has not been able to identify the molecular events involved in the early genesis of ETV6-RUNX1-associated leukemia and to define if the environmental factors associated to the human disease influence leukemia disease development in the zebrafish transgenic model.
Murine mouse models for childhood ETV6-RUNX1+ pB-ALL
Transplantation of ETV6-RUNX1-expressing human or murine B cells into immunodeficient mice has been one of the main approaches used to try to understand childhood ETV6-RUNX1+ pB-ALL development. In agreement with the zebrafish observations mentioned above, initial studies in pro-B cell lines shown that ETV6-RUNX1 by itself cannot confer cytokine factor-independent growth (Table 1) [12]. However, sporadic T- or B-cell leukemias appeared when total bone marrow cells from wild-type mice were transduced with ETV6-RUNX1, suggesting that an uncommitted progenitor could the cell-of-origin of ETV6-RUNX1+ leukemia [13]. Similarly, mice reconstituted with transduced hematopoietic progenitors with ETV6-RUNX1 did not develop leukemia, but a bias in the differentiation of the progenitors towards B cell development was observed [14]. Overall, the transplantation of ETV6-RUNX1-expressing human or murine B cells into immunodeficient mice has failed to reproduce the childhood ETV6-RUNX1+ pB-ALL [15, 16]. In addition, this technological approach presents many disadvantages. First, these models lack an immune system in contrast to children where the leukemia appears. Second, the lack of reproducibility and homogeneity is given by the fact that many parameters (injection site, sorting parameters, pre-treatment of recipients with specific antibodies, etc) influence the grafting efficiency and capacity of the injected cells in the recipient animals [6]. In addition, another main problem is that recipient mice should be irradiated with gamma radiation before the injection of transduced cells, rendering these approaches impractical for the study of the molecular events involved in the early genesis of ETV6-RUNX1-associated leukemia and the contribution of environmental factors associated to the human disease to leukemia development. Consequently, in order to accurately reproduce all the aspects intervening in B-ALL development, it is necessary to perform experiments in an in vivo setting in which the leukemia emerges in the appropriate microenvironment.
Table 1: Main experimental models of ETV6-RUNX1
Experimental design |
Phenotype |
Year |
Reference |
|---|---|---|---|
Immunoglobulin heavy chain enhancer/promoter |
The mice did not develop leukemia. |
2001 |
[12] |
Retroviral gene transfer |
T cell leukemia and myeloid leukemia |
2002 |
[13] |
Fetal liver HPCs transduced with retroviral vectors |
ETV6-RUNX1 did not induce leukemia in transplanted mice. |
2004 |
[14] |
Bone marrow transplantation model |
No leukemia development was observed. |
2005 |
[15] |
ETV6-RUNX1 expression is conditionally driven from the endogenous promoter (knock-in) |
ETV6-RUNX1 renders mice prone to T-cell malignancy after chemical mutagenesis when expressed in HSCs, but not in early lymphoid progenitors. |
2009 |
[20] |
Constitutive expression of ETV6-RUNX1 from the endogenous promoter |
No leukemia development was observed without a genetically induced random mutagenesis approach. |
2011 |
[21] |
ETV6-RUNX1 expression is driven from a CMV promoter and constitutively activated since fetal HSC stage. |
No leukemia development was observed even under low-dose radiation exposure. |
2013 |
[22] |
The expression of the fusion protein is restricted to CD19(+) B cells |
No leukemia development was observed. |
2013 |
[18] |
ETV6-RUNX1 expression is restricted to HS/PCs |
Low penetrance of pB-ALL under common infection exposure. |
2017 |
[28] |
Given the fact that the ETV6-RUNX1 fusion gene is only associated to pB-ALL, many genetically engineered mouse models have been designed to express the ETV6-RUNX1 gene in a committed B-cell (Table 1) [12–17]. These models allow bypassing the limitations and experimental variability of the xenotransplant models, but these mice did not develop hematologic disorder of any kind. Recently, a novel ETV6-RUNX1 transgenic mouse model with restricted expression of the fusion gene to CD19+ B cells was generated. This model showed that ETV6-RUNX1 induced reactive oxygen species and drove the accumulation of DNA damage in B cells [18]. However, this model did not give rise to leukemia [18]. Even more, when the CD19-ETV6-RUNX1 model [18] was crossed with a vav-Bcl2 transgenic model resulted in shortened latency to follicular lymphoma [19], but the double transgenic mice did not develop pB-ALL (Table 1). These attempts to model the childhood ETV6-RUNX1+ pB-ALL by targeting committed B-cells suggest that the cell-of-origin is an undifferentiated hematopoietic precursor cell.
The next-generation ETV6-RUNX1 mouse models were based on conditional gene targeting of the Runx1 gene into the endogenous Etv6 locus of the mouse by which the ETV6-RUNX1 gene is activated in a temporal- and tissue-specific manner following expression of Cre recombinase [20]. Schindler et al. observed malignancies in the T-lymphoid lineage following ENU mutagenesis when the ETV6-RUNX1 protein is expressed in HSCs [20]. A similar constitutive knock-in approach was employed by van der Weyden et al. [21]. These mice do not develop pB-ALL spontaneously. However, when an insertional mutagenesis screen was performed by intercrossing these mice with those carrying a Sleeping Beauty transposon array, 20% of the offspring developed pB-ALL, but even more often T cell leukemia and myeloid leukemias appeared (Table 1) [21]. Similarly, constitutive activation of ETV6-RUNX1 in fetal HSCs gave not rise to leukemia development even when the mice were exposed to low-dose radiation [22]. Thus far, no experimental model of translocation t(12;21)(p13;q22) associated leukemia has recapitulated the full disease phenotype of pB-ALL, irrespective of the mode and timing of expression or the source of target cells. The inability to generate spontaneous pB-ALL may reflect the requirement for specific secondary molecular hits, which are not induced efficiently by the nonspecific environmental mutagenesis strategies used.
The infective theory of ETV6-RUNX1 leukemia
Infection was the first suggested causal exposure for childhood ETV6-RUNX1 pB-ALL and remains the strongest candidate [17–22]. However, its role in the conversion of the ETV6-RUNX1 preleukemic clone into pB-ALL remains unknown. We recently showed that an inherited susceptibility to childhood pB-ALL (Pax5+/−) linked to a B-cell deficiency and followed by infection exposure may lead to pB-ALL [23]. However, it is unclear if a somatically acquired gene fusion which occurs in humans prenatally during early steps in haematopoiesis, like in the pB-ALL-associated ETV6-RUNX1-translocation, act in a similar manner as an inherited Pax5SNP in the germline because ETV6-RUNX1 [24, 25] and Pax5 [26, 27] are fundamentally different in terms of inheritance and mode of cellular action. Furthermore, inherited Pax5 mutations are rare, hence we aimed to prove the theory of exposure to infection in pB-ALL development in a common subtype of pediatric pB-ALL such as ETV6-RUNX1 positive leukemia. Nevertheless, the lack of genetically engineered human-like ETV6-RUNX1 pB-ALL models has hampered our better understanding of the pathogenesis of this disease
Limiting ETV6-RUNX1 expression to stem cells induces childhood pB-ALL development under common infection exposure
Preleukemic clones carrying ETV6-RUNX1 oncogenic lesions are frequently found in neonatal cord blood [9, 10], where majority of preleukemic carriers do not convert into pB-ALL. These findings suggest that ETV6-RUNX1 might promote leukemogenesis by creating an aberrant progenitor compartment that is susceptible to malignant transformation through accumulation of additional secondary hits that will be acting as drivers of leukemogenesis. Thus, ETV6-RUNX1 does not seem to be a dominant oncogene within the natural cellular hematopoietic stem cell compartment where the ETV6-RUNX1 oncogenic lesion takes place. The existence of this process is, however, difficult to demonstrate using available models of transgenic-driven ETV6-RUNX1, because current available models do not mimic the human scenario where majority of preleukemic carriers do not convert into pB-ALL, and the initiating ETV6-RUNX1 oncogenic lesion that would create the susceptibility is genetically preserved in the cancer-initiating cells, but also through the different pathological cellular intermediates, until the final tumour differentiated cells, and thus making it difficult to ascertain their molecular role at the different stages of tumour biology. Thus, we engineered mice to restrict ETV6-RUNX1 gene expression to hematopoietic stem/progenitor cells (HS/PCs) in order to unmask this unknown potential mode of action of ETV6-RUNX1 (Sca1-ETV6-RUNX1 mice). These Sca1-ETV6-RUNX1 mice developed exclusively pB-ALL at a low disease penetrance only when they were exposed to common pathogens [28] (Figure 2). This limited expression of ETV6-RUNX1 during early hematopoietic development mimicked human ETV6-RUNX1 preleukemic biology, where ETV6-RUNX1 expression is not detected in progenitor B-cells of healthy children carrying preleukemic clone [29, 30], and conferred a low risk of developing pB-ALL after exposure to common pathogens corroborating the low incidence in humans and facilitated the identification of genetic basis of the clonal evolution of an ETV6-RUNX1 preleukemic clone to pB-ALL. The infection-driven pB-ALLs that developed in Sca1-ETV6-RUNX1 mice closely resemble the human disease both in low penetrance and in pathology and genomic lesions (Figure 2). Moreover, these results show that the control of oncogenesis is a function of both the target-cell and the genetic oncogenic alteration(s), being the molecular mechanisms of action of ETV6-RUNX1 at the HSC level different from those acting at later stages of tumoral cell differentiation [31]. We have shown that the Sca1-ETV6-RUNX1 model mimicked human ETV6-RUNX1 preleukemic biology [9, 10] and provided a means to evaluate the potential for oncogenic environments that contribute to pB-ALL development.
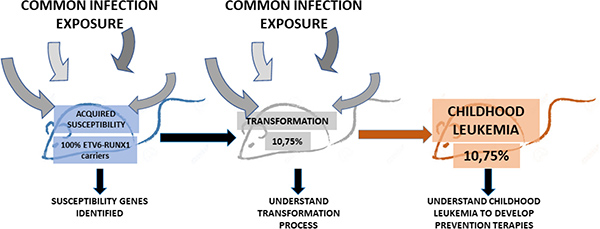
Figure 2: The natural history of ETV6-RUNX1 pB-ALL childhood in the mouse. The Sca1-ETV6-RUNX1 mouse model restricts the oncogen expression to the HS/PCs. The pB-ALL development in this mouse model is driven by the exposure of the mice to common infections and has a low penetrance (10,75%), as in humans. This mouse model is valuable because it allows us to identify susceptible genes and understand the transformation process and hopefully to develop new prevention therapies for pB-ALL childhood leukemia.
Importance and implications of these findings in childhood pB-ALL development and prevention
This preclinical model of ETV6-RUNX1-associated pB-ALL can be essential tool to prevent the occurrence of this disease in preleukemic carriers for the following reasons: 1) pB-ALL develops de novo in the context of the native tissue environment and intact immune system, 2) pB-ALL arises from genetic and/or environmental factors that are relevant for the childhood pB-ALL, 3) the model mimics multistage progression, including preleukemic lesions that have the potential to progress to malignant ones, 4) the murine phenotypes display histological and biological features in common with their human counterpart, 5) murine leukemias present similar molecular pathways that are dysregulated in the human pB-ALL, 6) murine pB-ALL originate in appropriate cells of origin, 7) murine pB-ALL display heterogeneity, as occurs in childhood pB-ALL, and 8) the mice also model genetic diversity, as happens in human populations (Figure 3). So, how could this Sca1-ETV6-RUNX1 model be fully integrated into childhood pB-ALL prevention? In our view analyses of the Sca1-ETV6-RUNX1 model offers unique opportunities to interrogate aspects of childhood pB-ALL prevention that are difficult to study in preleukemic carriers such as accessing preleukemia and/or early-stage pB-ALL with known progressive potential. In addition, it will facilitate the discovery of how relevant environmental factors promote leukemogenesis in genetically defined preleukemic contexts. Realizing the benefits of early pB-ALL detection will require improved means of distinguishing, at the earliest possible stages, ‘true’ pB-ALL that require intervention, from the majority of non-malignant ones. In addition, identification of early-stage leukemic lesions (true pB-ALL) as early as possible would probably enable us to prevent or delay progression to clinically relevant disease. In recognition of these challenges, the identification of extrinsic and intrinsic risk factors in healthy preleukemic carriers will allow the goal of minimizing their exposure and thereby reducing leukemia incidence.
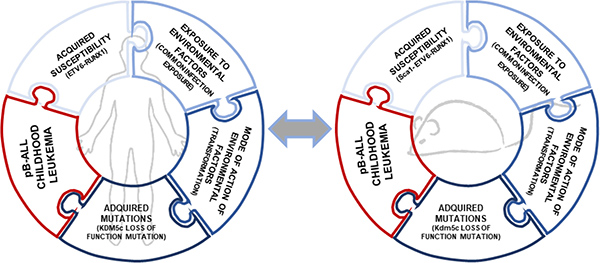
Figure 3: Parallelisms in clonal evolution of ETV6-RUNX1 preleukemic clones to pB-ALL childhoood leukemia in mouse and humans. The ETV6-RUNX1 fusion gene is linked with the most common subtype of childhood leukemia. In humans, only few ETV6-RUNX1 carriers develop pB-ALL so the essential genetic basis for development of full-blown leukemia remains to be recognized. The infective theory linked to the development of leukemia was postulated 100 years ago and has been validated experimentally in the last two years [23, 28]. Thereby, the clonal evolution of an ETV6-RUNX1 preleukemic clone to pB-ALL is a multistep process in which a preleukemic clone (ETV6-RUNX1+) is transformed only when is exposed to common infections [18]. Similar secondary genetic alterations were found in both human and mice, like the loss of function mutation in KDM5c (p.408Y>C mutation in humans and p.804R>* mutation in mice).
Author contributions
All authors contributed to the preparation of the review.
ACKNOWLEDGMENTS AND FUNDING
We are indebted to all members of our groups for useful discussions and for their critical reading of the manuscript. J.H. has been supported by the German Children’s Cancer Foundation, DJCLS 02R/2016, KKS A2016/07 and from the “Forschungskommission” of the medical faculty of the Heinrich Heine University. A.B. has been supported by the German Children’s Cancer Foundation and the Federal Ministry of Education and Research, Bonn, Germany. Funding to S.N.C. from Salus Sanguinis, Fondation contre le cancer Belgium; Interuniversity Attraction Poles IAP and ARC 10/15-027 is acknowledged. Research in ISG group is partially supported by FEDER and by MINECO (SAF2012-32810, SAF2015-64420-R and Red de Excelencia Consolider OncoBIO SAF2014-57791-REDC), Instituto de Salud Carlos III (PIE14/00066), ISCIII- Plan de Ayudas IBSAL 2015 Proyectos Integrados (IBY15/00003), by Junta de Castilla y León (BIO/SA51/15, CSI001U14, UIC-017, and CSI001U16), and by the Fundacion Inocente Inocente. ISG lab is a member of the EuroSyStem and the DECIDE Network funded by the European Union under the FP7 program. A.B. and ISG have been supported by the German Carreras Foundation (DJCLS R13/26). Research in CVD group is partially supported by a “Miguel Servet” Grant (CP14/00082 - AES 2013-2016 - FEDER) from the Instituto de Salud Carlos III (Ministerio de Economía y Competitividad) and by the Lady Tata International Award for Research in Leukaemia 2016–2017.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006; 6:193–203. https://doi.org/10.1038/nrc1816.
2. Golub TR, Barker GF, Bohlander SK, Hiebert SW, Ward DC, Bray-Ward P, Morgan E, Raimondi SC, Rowley JD, Gilliland DG. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 1995; 92:4917–21.
3. Romana SP, Mauchauffe M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, Bernard OA. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood. 1995; 85:3662–70.
4. Shurtleff SA, Buijs A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, Chan GC, Pui CH, Grosveld G, Downing JR. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. 1995; 9:1985–9.
5. Eckert C, Hagedorn N, Sramkova L, Mann G, Panzer-Grumayer R, Peters C, Bourquin JP, Klingebiel T, Borkhardt A, Cario G, Alten J, Escherich G, Astrahantseff K, et al. Monitoring minimal residual disease in children with high-risk relapses of acute lymphoblastic leukemia: prognostic relevance of early and late assessment. Leukemia. 2015; 29:1648–55. https://doi.org/10.1038/leu.2015.59.
6. Cobaleda C, Sanchez-Garcia I. B-cell acute lymphoblastic leukaemia: towards understanding its cellular origin. Bioessays. 2009; 31:600–9. https://doi.org/10.1002/bies.200800234.
7. Castor A, Nilsson L, Astrand-Grundstrom I, Buitenhuis M, Ramirez C, Anderson K, Strombeck B, Garwicz S, Bekassy AN, Schmiegelow K, Lausen B, Hokland P, Lehmann S, et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med. 2005; 11:630–7. https://doi.org/10.1038/nm1253.
8. Fuka G, Kauer M, Kofler R, Haas OA, Panzer-Grumayer R. The leukemia-specific fusion gene ETV6/RUNX1 perturbs distinct key biological functions primarily by gene repression. PLoS One. 2011; 6:e26348. https://doi.org/10.1371/journal.pone.0026348.
9. Cazzaniga G, van Delft FW, Lo Nigro L, Ford AM, Score J, Iacobucci I, Mirabile E, Taj M, Colman SM, Biondi A, Greaves M. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood. 2011; 118:5559–64. https://doi.org/10.1182/blood-2011-07-366542.
10. Kosik P, Skorvaga M, Durdik M, Jakl L, Nikitina E, Markova E, Kozics K, Horvathova E, Belyaev I. Low numbers of pre-leukemic fusion genes are frequently present in umbilical cord blood without affecting DNA damage response. Oncotarget. 2017; 8:35824–34. https://doi.org/10.18632/oncotarget.16211.
11. Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD. TEL-AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2006; 103:15166–71. https://doi.org/10.1073/pnas.0603349103.
12. Andreasson P, Schwaller J, Anastasiadou E, Aster J, Gilliland DG. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genet Cytogenet. 2001; 130:93–104. https://doi.org/S0165-4608(01)00518-0.
13. Bernardin F, Yang Y, Cleaves R, Zahurak M, Cheng L, Civin CI, Friedman AD. TEL-AML1, expressed from t(12;21) in human acute lymphocytic leukemia, induces acute leukemia in mice. Cancer Res. 2002; 62:3904–8.
14. Morrow M, Horton S, Kioussis D, Brady HJ, Williams O. TEL-AML1 promotes development of specific hematopoietic lineages consistent with preleukemic activity. Blood. 2004; 103:3890–6. https://doi.org/10.1182/blood-2003-10-3695.
15. Fischer M, Schwieger M, Horn S, Niebuhr B, Ford A, Roscher S, Bergholz U, Greaves M, Lohler J, Stocking C. Defining the oncogenic function of the TEL/AML1 (ETV6/RUNX1) fusion protein in a mouse model. Oncogene. 2005; 24:7579–91. https://doi.org/10.1038/sj.onc.1208931.
16. Tsuzuki S, Seto M, Greaves M, Enver T. Modeling first-hit functions of the t(12;21) TEL-AML1 translocation in mice. Proc Natl Acad Sci U S A. 2004; 101:8443–8. https://doi.org/10.1073/pnas.0402063101.
17. Ford AM, Palmi C, Bueno C, Hong D, Cardus P, Knight D, Cazzaniga G, Enver T, Greaves M. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J Clin Invest. 2009; 119:826–36. https://doi.org/10.1172/JCI36428.
18. Kantner HP, Warsch W, Delogu A, Bauer E, Esterbauer H, Casanova E, Sexl V, Stoiber D. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia. 2013; 15:1292–300.
19. Bauer E, Schlederer M, Scheicher R, Horvath J, Aigner P, Schiefer AI, Kain R, Regele H, Hoermann G, Steiner G, Kenner L, Sexl V, Villunger A, et al. Cooperation of ETV6/RUNX1 and BCL2 enhances immunoglobulin production and accelerates glomerulonephritis in transgenic mice. Oncotarget. 2016; 7:12191–205. https://doi.org/10.18632/oncotarget.7687.
20. Schindler JW, Van Buren D, Foudi A, Krejci O, Qin J, Orkin SH, Hock H. TEL-AML1 corrupts hematopoietic stem cells to persist in the bone marrow and initiate leukemia. Cell Stem Cell. 2009; 5:43–53. https://doi.org/10.1016/j.stem.2009.04.019.
21. van der Weyden L, Giotopoulos G, Wong K, Rust AG, Robles-Espinoza CD, Osaki H, Huntly BJ, Adams DJ. Somatic drivers of B-ALL in a model of ETV6-RUNX1; Pax5(+/-) leukemia. BMC Cancer. 2015; 15:585. https://doi.org/10.1186/s12885-015-1586-1.
22. Li M, Jones L, Gaillard C, Binnewies M, Ochoa R, Garcia E, Lam V, Wei G, Yang W, Lobe C, Hermiston M, Passegue E, Kogan SC. Initially disadvantaged, TEL-AML1 cells expand and initiate leukemia in response to irradiation and cooperating mutations. Leukemia. 2013; 27:1570–3. https://doi.org/10.1038/leu.2013.15.
23. Martin-Lorenzo A, Hauer J, Vicente-Duenas C, Auer F, Gonzalez-Herrero I, Garcia-Ramirez I, Ginzel S, Thiele R, Constantinescu SN, Bartenhagen C, Dugas M, Gombert M, Schafer D, et al. Infection Exposure is a Causal Factor in B-cell Precursor Acute Lymphoblastic Leukemia as a Result of Pax5-Inherited Susceptibility. Cancer Discov. 2015; 5:1328–43. https://doi.org/10.1158/2159-8290.CD-15-0892.
24. Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003; 3:639–49. https://doi.org/10.1038/nrc1164.
25. Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, Saha V, Biondi A, Greaves MF. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999; 354:1499–503. https://doi.org/S0140673699094039.
26. Auer F, Ruschendorf F, Gombert M, Husemann P, Ginzel S, Izraeli S, Harit M, Weintraub M, Weinstein OY, Lerer I, Stepensky P, Borkhardt A, Hauer J. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia. 2014; 28:1136–8. https://doi.org/10.1038/leu.2013.363.
27. Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, Wechsler J, Yang J, Hayes J, Klein RJ, Zhang J, Wei L, Wu G, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013; 45:1226–31. https://doi.org/10.1038/ng.2754.
28. Rodriguez-Hernandez G, Hauer J, Martin-Lorenzo A, Schafer D, Bartenhagen C, Garcia-Ramirez I, Auer F, Gonzalez-Herrero I, Ruiz-Roca L, Gombert M, Okpanyi V, Fischer U, Chen C, et al. Infection Exposure Promotes ETV6-RUNX1 Precursor B-cell Leukemia via Impaired H3K4 Demethylases. Cancer Res. 2017; 77:4365–77. https://doi.org/10.1158/0008-5472.CAN-17-0701.
29. Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, Tsuzuki S, Greaves M, Enver T. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008; 319:336–9. https://doi.org/10.1126/science.1150648.
30. Lausten-Thomsen U, Madsen HO, Vestergaard TR, Hjalgrim H, Nersting J, Schmiegelow K. Prevalence of t(12;21)[ETV6-RUNX1]-positive cells in healthy neonates. Blood. 2011; 117:186–9. https://doi.org/10.1182/blood-2010-05-282764.
31. Vicente-Duenas C, Romero-Camarero I, Cobaleda C, Sanchez-Garcia I. Function of oncogenes in cancer development: a changing paradigm. Embo J. 2013; 32:1502–13. https://doi.org/10.1038/emboj.2013.97.