
INTRODUCTION
Breast cancer is a problem for women worldwide in both developed and underdeveloped countries. According to the statistics, breast cancer is the most common cancer diagnosed and the second leading cause of cancer-related death among U.S. women. On a practical level, it makes up 1 in 3 cancers [1]. It has been reported that even with standard breast cancer treatment, up to 30% of patients still die of relapse or metastasis [2]. Accordingly, identifying the mechanisms underlying breast cancer metastasis and recurrence is the key to eradicate breast cancer.
Currently, accumulating evidence suggests that breast cancer departs from a fraction of cancer initiating cells called cancer stem cells (CSCs) [3–5]. CSCs share with normal stem cells some capacities including self-renewal and pluripotency. Hence, CSCs are considered as the cause of treatment failure and are liable for metastatic dissemination. Conventional therapies, despite target the progeny of CSCs, fail eventually owing to not killing CSCs, which results in recurrence of tumours [5–8]. Therefore, eliminating CSCs might have promise to achieve a permanent cure for breast cancer patients.
In the past few decades, mounting lines of reports have indicated that signaling pathways, including the Notch pathway, Wnt/β-catenin pathway and hedgehog pathway, are responsible for controlling the self-renewal characteristicsof CSCs [9–11]. Conversely, these pathways might contribute to stemness maintenance in CSCs when these pathways become dysregulated. Additionally, an increasing number of recent studies have revealed that non coding RNAs (ncRNAs) are related to stemness of CSCs in a manner of competing for the microRNA response elements (MREs) [12–14]. However, whether circular RNAs (circRNAs), a new type of ncRNAs, are implicated in stemness maintenance of CSCs remains poorly explored.
As a new member of ncRNAs, unlike their linear counterparts, circRNAs are able to form covalent closed circles with their 3′ heads and 5′ tails bonded together [15]. Exactly this special form confers circRNAs resistance to digestion of Rnase R and makes them ideal biomarkers for diagnosis. In addition, circRNAs are reported to be expressed in a tissue/developmental-stage-specific manner [16–18]. Significantly, several reports indicate that circRNAs harbour microRNA (miRNA) binding sites as miRNA sponges [17, 19].
Recently, circRNAs were demonstrated to be involved in abundant biological processes, including cell development, cell proliferation, cancer onset and progression [18, 20–23]. For instance, Simon J. Conn and his colleagues showed that thousands of circRNAs could be produced when cells were underwent epithelial mesenchymal transition (EMT) [21]. In addition, a recent reports revealed that fusion circRNAs generated during chromosomal translocations displayed remarkable abilities to promote cellular transformation in vitro and initiate tumours in vivo. EMT imparted cancer cells heritable phenotypic changes via epigenetic modifications. Once EMT was activated, cancer cells lost their epithelial characteristics and acquired mesenchymal features and CSC-like characteristics [24, 25]. Hence, these studies promoted us to hypothesize that circRNAs might be implicated in CSC properties. However, reports on circRNAs in CSCs were scarce. Hence, the present work aimed to establish the relationship between CSCs and circRNAs, which might be beneficial for understanding the biogenesis of CSCs.
In this paper, we examined the circRNA signature in breast cancer stem cells (BCSCs) and found 27 differentially expressed circRNAs, of which 19 circRNAs were downregulated and 8 were upregulated. In addition, some representative circRNAs were selected to testify their authenticity by Quantitative PCR (Q-PCR). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were used to investigate the possible functions of these circRNAs and a circRNA-miRNA network was constructed to illustrate the potential mechanisms. Specifically, we showed that circVRK1 could serve as a suppressor on stemness of BCSCs. Collectively, our work provides strong supports for exploring the functional roles of circRNAs in BCSCs. These findings might be beneficial for us to develop new avenues to conquer breast cancer.
RESULTS
Mammosphere cells exhibit a much higher percentage of BCSCs with CD44+CD24- phenotype
Mammosphere formation assay is a generally used method to obtain BCSCs in vitro [26]. Hence, here we conducted mammosphere assay to enrich BCSCs. Mammospheres with diameter≥ 100 μm were considered as significant (Figure 1A). In the current study, our data suggested that BCSCs with CD44+CD24- phenotye derived from mammosphere cells displayed a much higher frequency when compared to that from adherent-cultured cells (Figure 1B and 1C). These findings were in consistent with our previous report [27]. Taken together, these data validated that our platform was feasible and strengthened the notion that mammospheres were enriched CSCs.
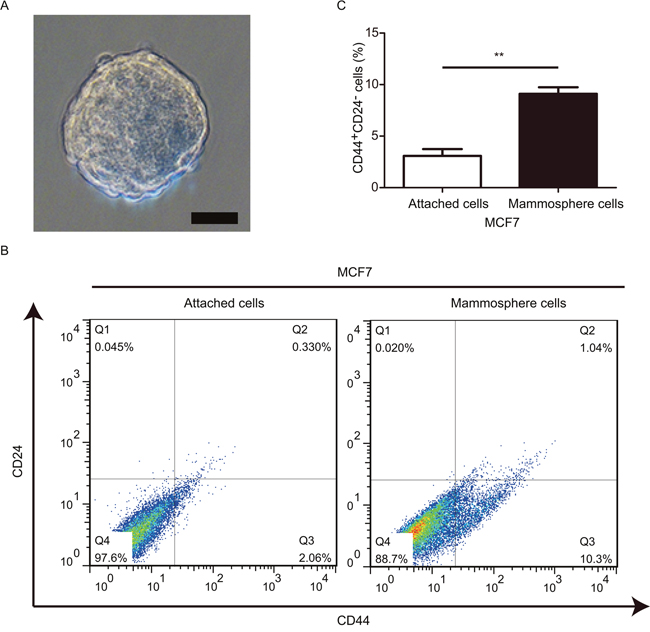
Figure 1: Mammosphere-derived cells exhibit a relative higher frequency of BCSCs. (A) Phase-contrast images of mammospheres generated by MCF-7. (B and C) FACS were used to evaluate the proportions of cells with CD44+CD24- phenotype. **P< 0.01, data are represented as mean ± SD from three different assays, Scale bar, 100 μm.
RNA-Sequencing reveals dysregulated circRNA signature in BCSCs
To investigate the expression pattern of circRNAs in BCSCs, we performed High-Throughput Sequencing to screen the expression profile of circRNAs in BCSCs and matched non-BCSCs. The flow chart was depicted in Figure 2A. A total of 5727 circRNA candidates were identified via high-throughput sequencing in 3 couple of samples. The distribution of genome alignment counts was showed in Supplementary Figure 1. Normalized log2 scales were used in scatter plot to evaluate the variations between two groups (Figure 2B) and the dysregulated circRNAs identified here were displayed in the volcano plot (Figure 2C). To further depict the traits of circRNAs, the distribution of circRNAs on human chromosomes were analysed (Figure 2D). In line with previous reports, the current study showed that circRNAs were derived from exons in most cases, some circRNAs generated from introns were also present and other sources of circRNAs were detected (Figure 2E). We summarized the differentially expressed circRNAs information in Table 1 by fold change. In the present study, 27 differently expressed circRNAs were identified by fold change≥ 1.8, P value < 0.05, including 8 upregulated circRNAs and 19 downregulated circRNAs (Figure 3). These data together showed that there was a difference in the expression levels of circRNAs between BCSCs and non-BCSCs.
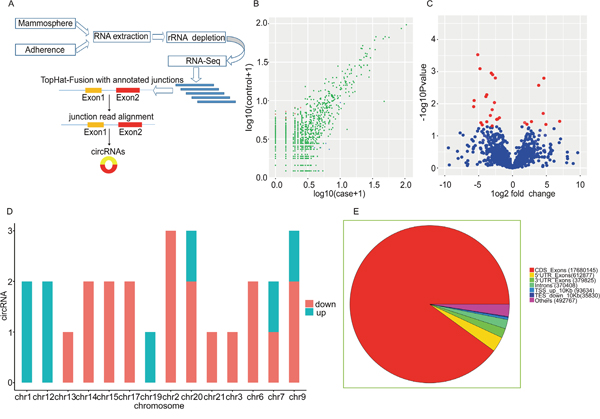
Figure 2: Discrepancy and characterizations in circRNA expression signature between BCSCs and non-BCSCs. (A) Flow chart of circRNA detection and annotation. (B) The difference in the expression of circRNAs between BCSCs and non-BCSCs was estimated with the scattered plot. The values drawn on X and Y axes were the standardized signal values of each group (log10 scaled). Red dots indicated downregulated circRNAs and blue ones represented upregulated circRNAs. (C) Volcano plots were depicted to assess the differential expression between the 2 groups. The Y axis showed a P-value of 0.05 (−log10 scaled). The horizontal line equaled 2.0 fold (log10 scaled) up and down, respectively. The red dots in the plot indicated the aberrantly expressed circRNAs with statistical significance. (D) The distribution of aberrantly expressed circRNAs in human chromosomes. (E) The pie diagram showed the circRNA category. Most of the differentially expressed circRNAs originated from the exons. Some were from introns, while a few were from other sources.
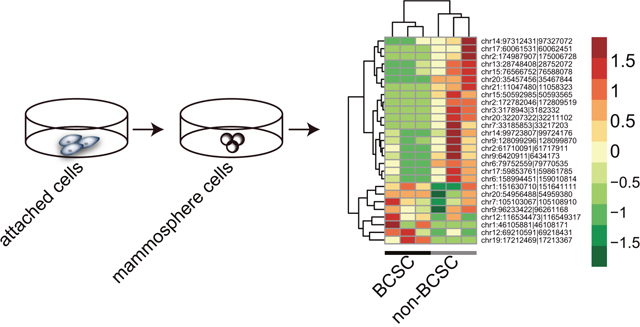
Figure 3: Hierarchical clusters of aberrantly expressed circRNAs between BCSCs and non-BCSCs.
Table 1: The list of differentially expressed circRNAs
CircID |
Isoform Name |
Uniprot Gene name |
logFC |
P.Value |
|---|---|---|---|---|
chr1:151630710|151641111 |
NM_030918 |
SNX27 |
2.133333 |
0.043621 |
chr12:116534473|116549317 |
NM_015335 |
MED13L |
4.933333 |
0.044239 |
chr12:69210591|69218431 |
NM_002392 |
MDM2 |
6.966667 |
0.035245 |
chr13:28748408|28752072 |
NM_175854 |
PAN3 |
-4.8 |
0.000812 |
chr1:46105881|46108171 |
NM_021639 |
GPBP1L1 |
1.8 |
0.03871 |
chr14:97312431|97327072 |
NM_003384 |
VRK1 |
-2.33333 |
0.030737 |
chr14:99723807|99724176 |
NM_022898 |
BCL11B |
-5.73333 |
0.012355 |
chr15:50592985|50593565 |
NM_005254 |
GABPB1 |
-2.66667 |
0.009209 |
chr15:76566752|76588078 |
NM_000126 |
ETFA |
-3.8 |
0.005233 |
chr17:59853761|59861785 |
NM_032043 |
BRIP1 |
-2.2 |
0.030266 |
chr17:60061531|60062451 |
NM_005121 |
MED13 |
-3 |
0.01532 |
chr19:17212469|17213367 |
NM_001130065 |
MYO9B |
4.533333 |
0.020082 |
chr20:32207322|32211102 |
NM_001032999 |
CBFA2T2 |
-3.93333 |
0.006012 |
chr20:35457456|35467844 |
NM_199181 |
SOGA1 |
-1.93333 |
0.0274 |
chr20:54956488|54959380 |
NM_003600 |
AURKA |
3.866667 |
0.002675 |
chr21:11047480|11058323 |
NM_182481 |
NA |
-2.53333 |
0.001577 |
chr2:172782046|172809519 |
NM_003642 |
HAT1 |
-2.93333 |
0.001248 |
chr2:174987907|175006728 |
NM_001011708 |
OLA1 |
-3.13333 |
0.001115 |
chr2:61710091|61717911 |
NM_003400 |
XPO1 |
-4.46667 |
0.045581 |
chr3:3178943|3182332 |
NM_182916 |
TRNT1 |
-5.13333 |
0.000298 |
chr6:158994451|159010814 |
NM_020823 |
TMEM181 |
-5.66667 |
0.007929 |
chr6:79752559|79770535 |
NM_017934 |
PHIP |
-3 |
0.049127 |
chr7:105103067|105108910 |
NM_019042 |
PUS7 |
2.8 |
0.036061 |
chr7:33185853|33217203 |
NM_001033604 |
BBS9 |
-3.26667 |
0.02315 |
chr9:128099296|128099870 |
NM_015635 |
GAPVD1 |
-4.66667 |
0.038967 |
chr9:6420911|6434173 |
NM_152896 |
UHRF2 |
-3.8 |
0.036463 |
chr9:96233422|96261168 |
NM_014612 |
FAM120A |
4.633333 |
0.001599 |
Q-PCR results of six randomly selected circRNAs validate the sequencing data’s reliability
Six circRNAs were randomly selected to validate the RNA-Sequencing results via Q-PCR. Our results demonstrated that the 6 downregulated circRNAs identified by sequencing were truly downregulated in mammosphere cells compared to attached cells (Figure 4A). In addition, the log2 fold-changes suggested that the Q-PCR results were in agreement with RNA-Sequencing data (Figure 4B). Specifically, one of selected circRNAs, circVRK1 were further confirmed with sanger sequencing. Our data showed that only cDNA templates possessed the potential to amplify circVRK1, however, both cDNA templates and gDNA templates were able to obtain linear VRK1 (Figure 5A and 5B). These results showed that RNA-Sequecing analysis was in line with Q-PCR results and indicated that these circRNAs were significantly differentially expressed.
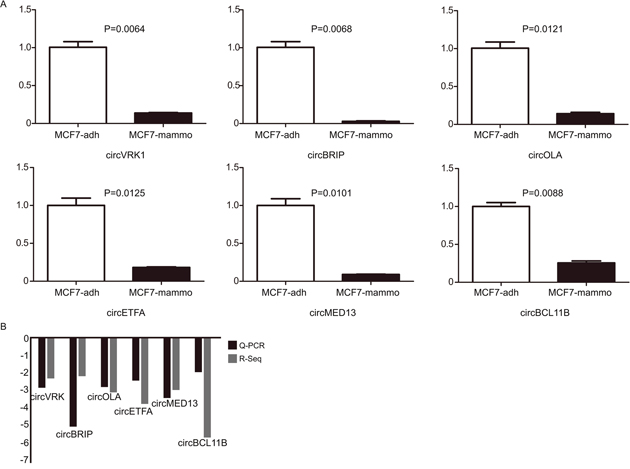
Figure 4: Q-PCR validates the expression of 6 selected circRNAs. (A) The expression levels of 6 circRNAs were validated by Q-PCR in adherent cells and floating mammosphere cells. (B) The comparison between RNA-Seq data and Q-PCR results. The Y axis showed the average of fold change (log2 transformed) of each circRNAs measured by Q-PCR and sequencing respectively. *P<0.05, **P<0.01 and ***P<0.001, data are shown as mean ± SEM of triplicated experiments.
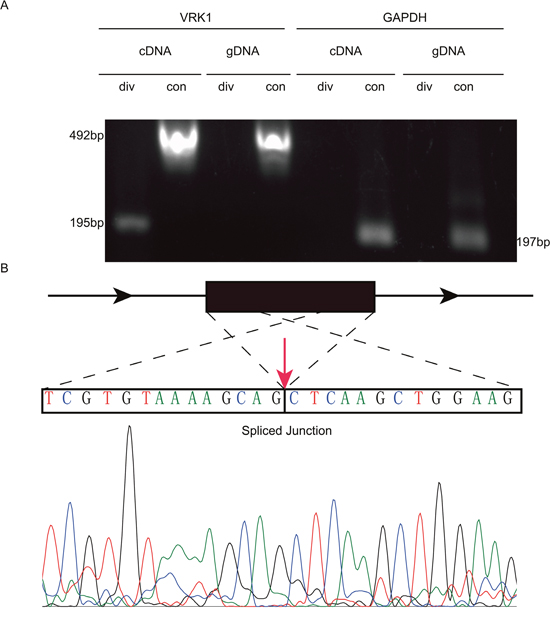
Figure 5: PCR and Sanger sequencing validates the existence of circVRK1. (A) Gel electrophoresis, (B) Sanger sequencing results.
The potential functionalities of identified circRNAs are predicted via GO and KEGG pathway analysis
To investigate the functional roles of identified circRNAs, GO enrichment analysis and KEGG pathway analysis were employed to analyse the functional effects of dysregulated circRNAs. GO analysis indicated that the differentially expressed circRNAs were present predominantly in the organelle of the endomembrane system, such as the ribosomes, and lumen of the Golgi (Figure 6A). Further, GO analysis showed that biological processes including stem cell proliferation, differentiation, and development were correlated with the identified dysregulated circRNAs, which suggested that the differentially expressed circRNAs identified here were possible to be involved in biogenesis of BCSCs (Figure 6B and 6C). KEGG pathway analysis for the circRNAs showed strong enrichment for stem-cell-characteristic-related pathways including the Hippo signaling pathway, Notch signaling pathway, and Wnt signaling pathway. To date, abundance of reports have revealed that these pathways were strongly linked with stem cell characteristics [28–30]. The particulars of functional analyses were summarized in Figure 6D. These results taken together provided new viewpoints on the functionalities of circRNAs on stemness of BCSCs.
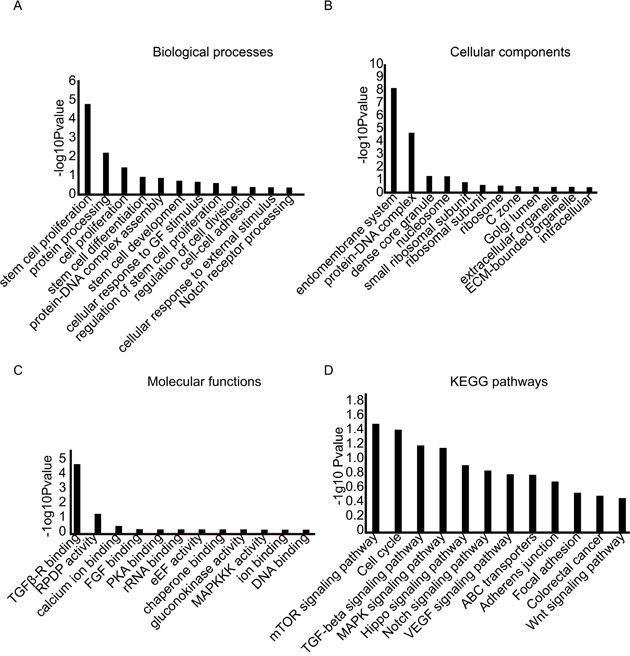
Figure 6: GO and KEGG pathway analysis. The GO database includes 3 parts. (A) biological process, (B) cellular component, (C) molecular function, (D) KEGG pathway analysis.
CircRNA/miRNA interaction network shows that the screened circRNAs are possible to serve as miRNA sponges
Reports suggested that circRNAs might binding with miRNAs thus regulating gene expression as miRNA sponges [31]. Hence, miRANDA and TargetScan were using to predict miRNA targets per conserved seed-matching sequences. In the present data, a total of 2712 miRNA candidates were identified (data not shown). To better delineate the interactions, the top 103 target miRNAs and their counterpart circRNAs were chose to construct the interaction network using cytoscape (Tot. Score≥ 90, Tot. Energy≤ –17) (Figure 7). Of these, we found that mir-153-5p was one of the predicted miRNA targets of circVRK1, which was one of the downregulated circRNAs in BCSCs. Interestingly, a previous study revealed that mir-153 was involved in stemness maintenance of triple-negative breast cancer via reducing the expression of KLF5 [13]. Combined these results with our data, these findings prompted us to hypothesize that circVRK1 was possible to negatively correlated with stemness of BCSCs.
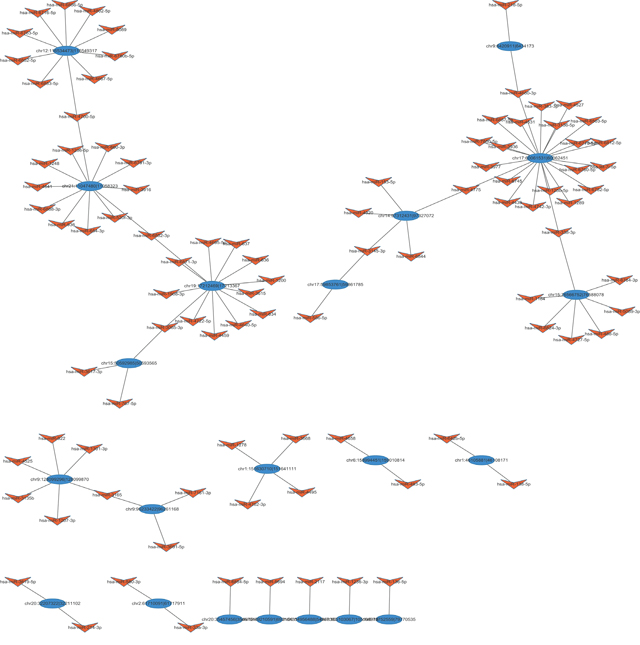
Figure 7: The circRNA/miRNA network analysis. The full view network consists of the differentially expressed circRNAs (blue) and their target miRNAs (orange).
CircVRK1 displays an inhibiting role in stemness of BCSCs
To testify our hypothesis, we designed siRNAs to silence circVRK1 and explored its influence on self-renewal capacity, BCSCs’ expansion and expression of stemness-related markers. We found that breast cancer cells showed an enhanced capacities to form mammospheres and colonies after loss of circVRK1 (Figure 8A–8D). Additionally, the proportion of BCSCs with CD44+CD24- phenotype were significantly increased when reduced circVRK1 (Figure 8E and 8F). Similarly results were also observed in the global level of stemness-related factors (Figure 8G and 8H). Taken together, these data indicated that circVRK1 was negatively correlated with maintenance of BCSC characteristics.
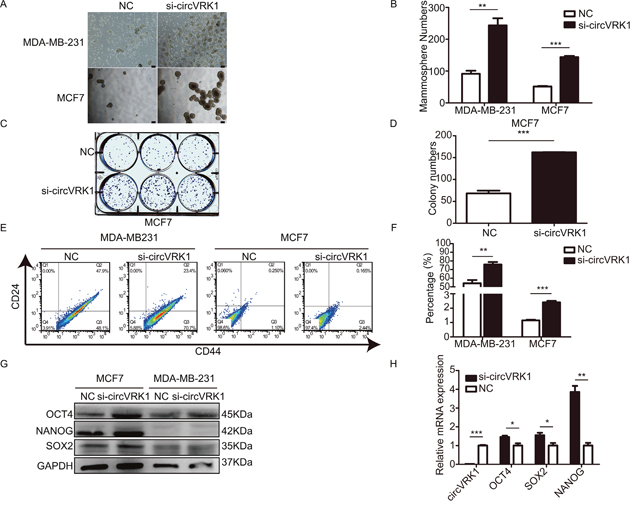
Figure 8: CircVRK1 displays a negative regulatory effect on self-renewal capacity, BCSCs’ expansion and expression levels of stemness-related markers in vitro. (A and B) Size and numbers of mammospheres derived from breast cancer cells treated or not with si-circVRK1. (C and D) Size and numbers of colonies cultured from breast cancer cells treated or not with si-circVRK1. (E and F) Proportions of CD44+CD24- in breast cancer cells were increased when treated with si-circVRK1 compared to NC. (G and H) The global abundance of stemness-related markers were markedly increased when treated with si-circVRK1 not only at mRNA level but also protein level. All the results were obtained from three independent experiments and the data are reported as mean ± SD. *P<0.05, **P<0.01 and ***P<0.001, Scale bar, 100 μm.
DISCUSSION
CircRNAs, an enigmatic type of RNAs, were first identified as aberrantly spliced transcripts in eukaryotes. Since its first discovery in 20 years ago, only a handful of circRNAs were detected, and they were frequently considered as noises or artifacts of abnormal splicing with little function [32–35]. Recently, with the development of bioinformatic approaches and sequencing technology, the abundance of circRNAs were found widespread in organisms [17, 31, 36, 37]. Although an increasing number of circRNAs were found continuously, the functional roles of circRNAs in CSCs were poorly studied.
In the present study, we were the first to report on the circRNA repertoire in BCSCs. There have been some lines of research into circRNA profile in cancer [22, 23, 38, 39], however, to the best of our knowledge, circRNA signature has never been explored in CSCs. Our results revealed that 27 circRNAs were differentially expressed, of which 19 circRNAs were downregulated and 8 were upregulated relative to the non-BCSCs (Figure 3). Moreover, all 27 identified circRNAs were aberrantly expressed with fold change> 1.8, which lent strong supports that these circRNAs were correlated with BCSCs. To testify the reliability of RNA-Sequencing results, six circRNAs, circVRK1, circBRIP, circOLA, circETFA, circMED13 and circBCL11B, were randomly selected to make the validation via Q-PCR. Our results indicated that all the six circRNAs were downregulated in mammosphere-derived cells compared with adherent cells in vitro, which meant that Q-PCR results were consistent with the RNA-Sequencing data (Figure 4).
Recently, a large body of evidence has revealed that circRNAs were able to regulate the expression of their parent genes [39–41]. Hence, the functions of mRNAs were possible to mirror the roles of corresponding circRNAs. Therefore, GO and KEGG pathway analysis were conducted to evaluate functional roles of circRNAs in BCSCs. GO analysis for the differential expressed circRNAs revealed that some terms under the biological process and molecular function categories were associated with the stem cell characteristics (Figure 6A–6C). These findings indicated that circRNAs revealed by RNA-Sequncing were mostly expressed in the cytoplasm. A recent report have reported that plenty of circRNAs appeared to be specifically present in developmental stage or cell-specific manner and confirmed that CDR1as was highly expressed in cytoplasm [17]. Hence, our work was partially in accordance with the report. Additionally, KEGG pathway analysis identified plenty of pathways associated with the stemness of stem cells including Hippo pathway [28, 42], Notch pathway [29], Wnt pathway [30], and TGF-β pathway [43] (Figure 6D). Combined with the current work, it was easy to conclude that the differentially expressed circRNAs might be implicated in sustaining the self-renewal and multipotent capacities of BCSCs. Hence, determining the function of the detected circRNAs could be beneficial for radical elimination of BCSCs.
In addition, circRNA/miRNA interplay network revealed here provided the first reported evidence to investigate the functional roles of circRNAs in BCSCs. The present work demonstrated that most of 27 aberrantly expressed circRNAs could interact with one or more miRNAs via bioinformatics analysis. Specifically, we found that mir-153 was one of the target miRNAs of circVRK1 (Figure 7). Interestingly, a recent published study revealed that mifepristone could inhibited the expression of KLF via inducing the expression of mir-153, thus leading to suppression on stemness of triple negtive breast CSCs [13]. These results indicated that circVRK1 might be implicated in inhibiting stemness of BCSCs through adsorbing mir-153. Previous studies revealed that circRNAs were able to serve as Competitive endogenous RNAs (ceRNAs) or miRNA sponges, which controlled the expression levels of the target genes via competing with miRNAs for binding sites [17, 18]. For instance, cerebellar degeneration related 1 antisense transcript (CDR1as), a well-studied circRNA, was proven to harbour more than 60 conserved miR-7 binding sites which were common in humans and other species [17, 18]. CDR1as was dominantly expressed in cytoplasm and might bind up to 20,000 miR-7 molecules per cell due to CDR1as and miR-7 shared specific expression domains [17]. Once CDR1as binding with miR-7, the power of miR-7 to regulate its downstream target genes was drastically attenuated. Besides, testis-specific circRNA, sex-determining region Y (Sry) was also found to harbour 16 miRNA binding sites for its target miR-138 in mice, although only one target site was found to have a human homologue [18]. Additionally, recent studies have shown that circ-ITCH could serve as a miRNA sponge by binding with miR-7 and miR-124. In this fashion, circ-ITCH could upregulate the expression of its parent gene ITCH [39]. In principle, any RNA transcripts with MREs could act as ceRNAs to sequester miRNAs. Disrupting the equilibrium among ceRNAs was found to be capable of triggering malignant biological events. However, it was noteworthy that there has been no convincing evidence supporting the conclusion that all known circRNAs could act as miRNA sponges. On the contrary, most human and mouse circRNAs seemed to have very few miRNA target sites, suggesting they might not function as miRNA sponges [44]. Hence, our results remained further investigation.
According to the BCSCs theory, BCSCs were defined by their properties, including increasing self-renewal, multilineage differentiation, and resistance to chemotherapy, all of which were considered as contributing to the breast cancer progression, metastasis and recurrence [45, 46]. The first line of evidence for the existence of BCSCs demonstrated that a celluar population with CD44+CD24- phenotype displayed the capacity to initiate new tumours in NOD/SCID Mice [47]. In addition, BCSCs exhibited the ability to form mammospheres, which was commonly used method to enrich BCSCs in vitro. Hence, inhibiting these characteristics was an effective way to kill BCSCs. Therefore, to investigate the functionality of circVRK1 on BCSCs, we silenced circVRK1 with siRNA targeting circVRK1 and observed its implication in BCSC behaviors. We found that the breast cancer cells displayed an increased capacity of self-renewal and expanded BCSCs’ expansion when loss of circVRK1 (Figure 8A–8F). In addition, an increasing global levels of stemness-related markers including NANOG, SOX2 and OCT4 were observed when circVRK1 was silenced (Figure 8G and 8H). these data taken together provided strong supports that circVRK1 were implicated in suppressing BCSCs. Honestly, the current work remains further investigation and future studies should focus on the dysregulation of the circVRK1/miR-153 axis in BCSCs.
In conclusion, the current work was the first to indicate the circRNA signature of BCSCs. We determined the circRNA/miRNA network and analysed the potential functional roles of dysregulated circRNAs. Furthermore, we revealed that circVRK1 was capable to negatively regulate the stemness of BCSCs. Our findings strengthen the possibility that circVRK1 is able to serve as a potential target for BCSCs. Additionally, the current data also provide supports for further study on circRNAs in stemness maintenance of BCSCs.
MATERIALS AND METHODS
Antibodies
Rabbit monoclonal anti-NANOG (CAT: 4903S), anti-SOX2 (CAT: 3579S), anti-OCT4 (CAT: 2750S) and anti-GAPDH (CAT: 5174S) used for western blot assays were purchased from Cell Signaling Technology (CST, US). Goat anti-rabbit IgG-HRP was obtained from Santa Cruz Biotechnology (Santa Cruz, US). Antibodies to FITC-conjugated CD44 (CAT: 130-095-195) and PE-conjugated CD24 (CAT: 130-095-953) used for Fluorescence-activated cell sorting (FACS) were ordered from Miltenyi Biotec (US).
Cell culture
The human breast cancer cell line MCF-7 and MDA-MB-231 were obtained from American Type Culture Collection (ATCC) and were maintained in recommended media according to the ATCC’s instructions. Basically, they were cultured in DMEM high glucose media supplemented with 10% fetal bovine serum (FBS) (Hyclone, US), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, US) at 37 °C.
Mammosphere formation assay
Mammosphere formation assay was performed as described previously [48]. In brief, single-cell suspensions at a density of 1×104/ml were inoculated in a ultra-low attachment six well plate (corning, US) and maintained in serum-free DMEM/F-12 (Hyclone, US) supplemented with 50×B27 (Invitrogen, US), 20 ng/ml human epidermal growth factor (EGF) (Gibco, US), 20 ng/ml human basic fibroblast growth factor (h-basic-FGF) (Gibco, US) and 5 μg/ml human insulin (Sigma-Aldrich, US), then cultured in an incubator at 37°C with 5% CO2. The medium was replaced every 3 days and mammospheres were collected at day 7.
Colony formation assay
Breast cancer cells were digested and resuspended at a density of 104/ml. Thereafter, 600 cells were plated into a six well plate, and were maintained in medium and monitored for colony formation. At day 14, the clones were confirmed with microscope and clones more than 50 cells were considered as significant.
FACS analysis
The mammosphere-cells were detached by trypsin and resuspended in 1×PBS supplemented with 2.5% FBS, then incubated with FITC mouse anti-human CD44 and PE mouse anti-human CD24 in staining buffer including 1% bovine serum albumin and 2 mM ethylene diamine tetraacetic acid for 10 min at 4°C. Following, stained cells were subjected to sorting and divided to 2 groups: CD44+CD24- cells (BCSCs) and non- CD44+CD24- cells (non-BCSCs). Data were analysed with a FACS Aria (BD Biosciences).
Total RNA extraction and Rnase R digestion
Total RNAs were extracted from BCSCs and non-BCSCs in concordance with the manufacturers’ protocol. RNA quantity and quality was evaluated by Nano Drop ND-1000 spectrophotometer (Thermo Fisher Scientific, US). In addition, the RNAs were subjected to agarose gel electrophoresis to estimate its integrity. Then the RNAs were subjected to ribosomal RNA (rRNA) depletion with ribo-zero-magnetic-kit according to the instructions. RNase R treatment was carried out as established previously [40]. Simply, processed RNAs were incubated using RNase R (Epicenter) for 3 h at 37°C to remove the linear RNAs.
Detection of circRNAs
CIRCexplore was carried out to detect circRNAs in BCSCs and non-BCSCs samples as described previously [37]. Briefly, 2-step mapping strategy was performed. Firstly, TopHat was used to perform the multiple mapping to the sequence reads of all the samples. Then unmapped reads were screened and mapped to reference genome (GRChr37/hg19) using TopHat-Fusion [49]. These reads were separated and mapped onto the relevant reference genome while non-linear candidate positions were considered possible back-splice junction reads. Next the candidate reads were further matched to the existing gene annotation to confirm the exact splice location, the donor and acceptor positions. Notably, reads that aligned on different genes or non-canonical splice sites were mostly considered as artifacts of trans-splicing or PCR errors, these reads were not used as candidates.
Characterization of circRNAs
The subsequent analysis was performed to fully delineate the characteristics of identified circRNAs. Firstly, the distribution of genome alignment counts in chromosomes was analysed. Secondly, the difference of circRNAs between BCSCs and non-BCSCs was evaluated. Thirdly, the distribution of dysregulated circRNAs in chromosomes was determined and the sources of aberrantly circRNAs between BCSCs and non-BCSCs were analysed. In addition, Hierarchical cluster analysis was applied to depict the differentially expressed circRNAs.
Construction of circRNA/miRNA interplay network
Studies have shown that circRNAs could act as miRNA sponges [17, 18]. Hence, experiments were performed to determine whether the aberrantly expressed circRNAs identified here had the potential to interact with miRNAs. Possible miRNA targets were predicted using miRanda and TargetScan database. To construct circRNA-miRNA network, circRNAs possessing MREs were screened in Tot. Score≥ 90 and Tot. Energy≤ –17, then the miRNAs were selected in accordance with seed match sequences. The circRNA/miRNA interaction network was depicted using Cytoscape software.
Bioinformatics analysis
To investigate the potential functions of the differentially expressed genes, GO enrichment analysis was consulted to describe gene and gene products attributes (http://www.geneontology.org). Distinct GO subtypes were considered significantly enriched when P-values< 0.05. KEGG pathway analysis was conducted to determine aberrantly genes in different biological processes (http://www.kegg.jp). The P-value here represents the significance of the pathways. The lower the P-value, the more significant the pathway.
Q-PCR
Reverse transcription for circRNAs was implemented using Super-Script II (Takara, Japan) as per the manufacturers’ instructions. Q-PCR was performed with a 1:10 dilution of the cDNAs with FastStart Universal SYBR Green Master Kit (Roche, Switzerland) and an ABI PRISM 7900HT sequence detection system was applied to execute the program (Applied Biosystems, U.S.). Two pairs of primers (convergent primers and divergent primers) were designed for each selected circRNA by primer 5 and are given in Supplementary Table 1. Theoretically, convergent primers were supposed to amplify the linear transcripts, while divergent primers were capable of achieving the circular amplification products. CDNAs transcribed from total RNAs served as templates, and genomic DNAs (gDNAs) were selected as a control. The RT-PCR protocol was as follows: first, 2 min at 50°C, followed by 10 min at 95°C, next to 40 cycles of PCR followed standard conditions with 15 s denaturation at 95°C, elongation at 60°C for 1 min, then at 95°C for 15 s, and 1 min for 60°C. The relative abundance was normalized to GAPDH, and fold change was analysed with the 2-ΔΔCt method [50]. Error bars represent standard deviations. All the experiments were performed in triplicate.
Western blotting
Breast cancer cells were harvested and lysed in RIPA lysis and extraction buffer (Thermo Fisher Scientific, US), Western blotting was carried out as previously described [27]. Briefly, equal amounts of proteins were loaded into SDS-PAGE gels and transferred onto polyvinylidene difluoride membrane (Millipore, US). Then the membranes were blocked with 5% non-fat milk for 1 h at room temperature, and incubated with primary antibodies overnight at 4°C. Then membranes were washed with 1×PBST and incubated with horseradish peroxidase-conjugated secondary antibodies (Santa cruz, US) for 1 h. Images were obtained through a LAS-3000 Imager (Fuji film).
Sanger sequencing
PCR was carried out for 40 cycles according to the protocol. Detailed, denaturation at 95°C for 30 s, followed by annealing at 60°C for 30 s, then elongation at 72°C for 1 min. PCR products were resolved and size separated on 1.5% agarose gel supplemented with yealRed Nucleic Acid Gel stain (Yeasen, China) and confirmed by Sanger sequencing with standard methods (Genewiz, China).
Silencing of circVRK1
Breast cancer cells were transfected with a siRNA target circVRK1 or Negative Control (NC) (GenePharma, China) using Lipofectamine 2000 (Invitrogen, US), after treated for 48 h, cells were collected for further research. The siRNA sequence for circVRK1 and NC were as follows:
CircVRK1: Sense: 5’ - GCAGUUGGAGAGAUA AUAATT,
Antisense: 5’ -UUAUUAUCUCUCCAACUGCTT.
NC: sense: 5’ – UUCUCCGAACGUGUCACGUTT,
Antisense: 5’ – ACGUGACACGUUCGGAGAATT.
Statistical analysis
All data were analysed using Prism 5 unless where indicated (GraphPad Software, Inc.). Student’s t-test was used for the comparison in the aberrant levels of expression of circRNAs between distinct groups. Fisher’s exact test was performed in GO and KEGG pathway analysis. The mean ± SEM was used to evaluate the values. All experiments were performed in triplicate, and P-values< 0.05 were considered statistically significant.
Abbreviations
BCSCs: breast cancer stem cells, CSCs: cancer stem cells, ncRNAs: non coding RNAs, MRE: micro RNA response element, circRNAs: circular RNAs, EMT: epithelial mesenchymal transition, Q-PCR: Quantitative PCR, miRNA: microRNA: ATCC: American Type Culture Collection, EGF: human epidermal growth factor, h-basic-FGF: human basic fibroblast growth factor, rRNA: ribosomal RNA, GO: Gene Ontology, KEGG: Kyoto Encyclopedia of Genes and Genomes, NC: negative control, ceRNAs: Competitive endogenous RNAs, CDR1as: cerebellar degeneration related 1 antisense transcript, Sry: sex-determining region Y.
CONFLICTS OF INTEREST
All authors declare no competing interests. All authors have given consent for the publication of the manuscript.
FUNDING
This work was supported by the National High Technology Research and Development Program of China (81172522, 81301858) and Suzhou science and technology project (SYS201508, SYS201404, SYS201308).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66: 7-30. https://doi.org/10.3322/caac.21332.
2. Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007; 608: 1-22.
3. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006; 66: 9339-44. https://doi.org/10.1158/0008-5472.can-06-3126.
4. Wicha MS. Cancer stem cells and metastasis: lethal seeds. Clin Cancer Res. 2006; 12: 5606-7. https://doi.org/10.1158/1078-0432.ccr-06-1537.
5. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012; 10: 717-28. https://doi.org/10.1016/j.stem.2012.05.007.
6. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005; 5: 275-84. https://doi.org/10.1038/nrc1590.
7. Bomken S, Fiser K, Heidenreich O, Vormoor J. Understanding the cancer stem cell. Br J Cancer. 2010; 103: 439-45. https://doi.org/10.1038/sj.bjc.6605821.
8. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014; 13: 497-512. https://doi.org/10.1038/nrd4253.
9. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX, Ivy SP. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015; 12: 445-64. https://doi.org/10.1038/nrclinonc.2015.61.
10. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012; 12: 133-43. https://doi.org/10.1038/nrc3184.
11. Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Beachy PA, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009; 458: 776-9. https://doi.org/10.1038/nature07737.
12. Zhou M, Hou Y, Yang G, Zhang H, Tu G, Du YE, Wen S, Xu L, Tang X, Tang S, Yang L, Cui X, Liu M. LncRNA-Hh Strengthen Cancer Stem Cells Generation in Twist-Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem Cells. 2016; 34: 55-66. https://doi.org/10.1002/stem.2219.
13. Liu R, Shi P, Nie Z, Liang H, Zhou Z, Chen W, Chen H, Dong C, Yang R, Liu S, Chen C. Mifepristone Suppresses Basal Triple-Negative Breast Cancer Stem Cells by Down-regulating KLF5 Expression. Theranostics. 2016; 6: 533-44. https://doi.org/10.7150/thno.14315.
14. Dimri M, Kang M, Dimri GP. A miR-200c/141-BMI1 autoregulatory loop regulates oncogenic activity of BMI1 in cancer cells. Oncotarget. 2016; 7: 36220-34. https://doi.org/10.18632/oncotarget.8811.
15. Danan M, Schwartz S, Edelheit S, Sorek R. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2012; 40: 3131-42. https://doi.org/10.1093/nar/gkr1009.
16. Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016; 17: 205-11. https://doi.org/10.1038/nrm.2015.32.
17. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495: 333-8. https://doi.org/10.1038/nature11928.
18. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013; 495: 384-8. https://doi.org/10.1038/nature11993.
19. Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, Liang L, Gu J, He X, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016; 7: 11215. https://doi.org/10.1038/ncomms11215.
20. Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR-7 in cancer. Cancer Res. 2013; 73: 5609-12. https://doi.org/10.1158/0008-5472.can-13-1568.
21. Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015; 160: 1125-34. https://doi.org/10.1016/j.cell.2015.02.014.
22. Yang W, Du WW, Li X, Yee AJ, Yang BB. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene. 2016; 35: 3919-31. https://doi.org/10.1038/onc.2015.460.
23. Guarnerio J, Bezzi M, Jeong JC, Paffenholz SV, Berry K, Naldini MM, Lo-Coco F, Tay Y, Beck AH, Pandolfi PP. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell. 2016; 166: 1055-6. https://doi.org/10.1016/j.cell.2016.07.035.
24. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133: 704-15. https://doi.org/10.1016/j.cell.2008.03.027.
25. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017. https://doi.org/10.1038/nrclinonc.2017.44.
26. Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005; 65: 5506-11. https://doi.org/10.1158/0008-5472.can-05-0626.
27. Xu L, Zhang L, Hu C, Liang S, Fei X, Yan N, Zhang Y, Zhang F. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int J Oncol. 2016; 48: 1175-86. https://doi.org/10.3892/ijo.2016.3337.
28. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, Daidone MG, Dupont S, Basso G, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011; 147: 759-72. https://doi.org/10.1016/j.cell.2011.09.048.
29. Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR, Lindeman GJ, Visvader JE. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell. 2008; 3: 429-41. https://doi.org/10.1016/j.stem.2008.08.001.
30. Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014; 346: 1248012. https://doi.org/10.1126/science.1248012.
31. Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013; 19: 141-57. https://doi.org/10.1261/rna.035667.112.
32. Cocquerelle C, Daubersies P, Majerus MA, Kerckaert JP, Bailleul B. Splicing with inverted order of exons occurs proximal to large introns. EMBO J. 1992; 11: 1095-8.
33. Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B. Scrambled exons. Cell. 1991; 64: 607-13.
34. Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell-Badge R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell. 1993; 73: 1019-30.
35. Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976; 73: 3852-6.
36. Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012; 7: e30733. https://doi.org/10.1371/journal.pone.0030733.
37. Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014; 159: 134-47. https://doi.org/10.1016/j.cell.2014.09.001.
38. Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D. Correlation of circular RNA abundance with proliferation--exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015; 5: 8057. https://doi.org/10.1038/srep08057.
39. Li F, Zhang L, Li W, Deng J, Zheng J, An M, Lu J, Zhou Y. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/beta-catenin pathway. Oncotarget. 2015; 6: 6001-13. https://doi.org/10.18632/oncotarget.3469.
40. Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. Circular intronic long noncoding RNAs. Mol Cell. 2013; 51: 792-806. https://doi.org/10.1016/j.molcel.2013.08.017.
41. Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, Zhu P, Chang Z, Wu Q, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015; 22: 256-64. https://doi.org/10.1038/nsmb.2959.
42. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014; 94: 1287-312. https://doi.org/10.1152/physrev.00005.2014.
43. Han D, Wu G, Chang C, Zhu F, Xiao Y, Li Q, Zhang T, Zhang L. Disulfiram inhibits TGF-beta-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-kappaB/Snail pathway. Oncotarget. 2015; 6: 40907-19. https://doi.org/10.18632/oncotarget.5723.
44. Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014; 15: 409. https://doi.org/10.1186/s13059-014-0409-z.
45. Geng SQ, Alexandrou AT, Li JJ. Breast cancer stem cells: Multiple capacities in tumor metastasis. Cancer Lett. 2014; 349: 1-7. https://doi.org/10.1016/j.canlet.2014.03.036.
46. De Los Angeles A, Ferrari F, Xi R, Fujiwara Y, Benvenisty N, Deng H, Hochedlinger K, Jaenisch R, Lee S, Leitch HG, Lensch MW, Lujan E, Pei D, et al. Hallmarks of pluripotency. Nature. 2015; 525: 469-78. https://doi.org/10.1038/nature15515.
47. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003; 100: 3983-8. https://doi.org/10.1073/pnas.0530291100.
48. Wang H, Huang M, Zhang DY, Zhang F. Global profiling of signaling networks: study of breast cancer stem cells and potential regulation. Oncologist. 2011; 16: 966-79. https://doi.org/10.1634/theoncologist.2010-0230.
49. Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011; 12: R72. https://doi.org/10.1186/gb-2011-12-8-r72.
50. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25: 402-8. https://doi.org/10.1006/meth.2001.1262.