
INTRODUCTION
The “hierarchal clustering” model of tumors posits the key role of cancer stem cells (CSC), a pool of relatively quiescent cells otherwise named tumor initiating cells (TIC), in carcinogenesis. In this context, no tumor can be cured until the CSC pool has been eradicated. As the normal counterpart, CSC display self-renewal, express telomerase and activate anti-apoptotic and multi-drug resistance pathways [1]. The availability of in vitro culture techniques since the pioneering work of Till and McCulloch and the exhaustive phenotypic characterization of myeloid progenitor cells at different differentiation levels let define the ontogenesis of normal and transformed hematopoiesis [2, 3]. Finally, identification of the causative event of CML as the BCR-ABL1 fusion protein TK let distinguish LSC from normal HSC, hence providing a host of information on signals involved in self-renewal, proliferation and life expectancy associated with leukemic transformation [4]. The most significant trait of CML LSC is BCR-ABL1 independence, which makes them autonomous from the fusion protein TK for proliferation and survival and drives their resistance to TK inhibitors IM, Nilotinib and Dasatinib, hence providing a sanctuary for the disease relapse upon drug withdrawal and a putative source of drug-resistance [5]. Pro-survival signaling pathways of BCR-ABL1+ LSC have therefore attracted great interest in view of their use as pharmacological targets.
b-catenin is a central component of BCR-ABL1+ LSC self-renewal and microenvironment protection from TK inhibitors [6–9]. β-Catenin activation in CML is mostly contingent upon mechanisms hampering its degradation, including BCR-ABL1-mediated phosphorylation at specific tyrosine residues (Y86 and Y654), which prevents the recruitment by the adenomatous polyposis coli (APC)/Axin/glycogen synthase kinase 3 β (GSK3 β) destruction complex, the overexpression of growth arrest specific 2 (GAS2), which reduces calpain-dependent degradation, and GSK3β inactivation due to the prevalence of a GSK3β mis-spliced isoform unable to phosphorylate β-catenin and/or to GSK3β de-phosphorylation by the Fas-associated phosphatase 1 (Fap1) [10–13]. The prerequisite of β-catenin transcriptional activity is nuclear import and interaction with transcription factors of the TCF/LEF1 family and additional co-factors, such as B-cell lymphoma 9 (BCL9), cyclic AMP response element binding protein (CBP) and pygopus to regulate a series of target genes [14]. β-catenin nuclear transport is mediated by direct contact with the nuclear pore and regulated by phopshorylation and binding partners, including Forkhead box M1 (FOXM1), insulin receptor substrate (IRS-1), mucin 1 (MUC-1), BCL9, androgen receptor and LEF-1 [14]. Our review will be focused on the β-catenin antagonist CBY1.
CBY is a 14.5 kDa protein highly conserved throughout evolution. It directly interacts with the C-terminal activation domain of b catenin and competes with TCF/LEF factors for b catenin binding hence repressing its transcriptional activity [15]. Moreover, it forms a stable tripartite complex with 14-3-3z and b catenin hence promoting the b catenin nuclear exclusion and cytoplasmic compartmentalization [16, 17].
CBY1 participation in the constitutive activation of β-catenin in CML was suggested by our recent study showing that a significant reduction in CBY1 expression levels is associated with BCR-ABL1 and correlates with nuclear b catenin increment [18]. The relative proximity of C22orf2 gene encoding for CBY1 to the BCR breakpoint on chromosome 22q11 as a consequence of deletions of distal BCR sequences occurring at the time of Philadelphia translocation suggested the gene haploinsufficiency eventually associated with disease worse prognosis [19]. Indeed, fluorescent in situ hybridization (FISH) did not let any evidence of C22orf2 loss associated with CML more advanced stage or worse prognosis. Conversely, we observed CBY1 down-modulation, driven by transcriptional and post-transcriptional mechanisms and evoked by the constitutive TK activity of BCR-ABL1 fusion protein. CBY1 induction in BCR-ABL1+ cell response to TK inhibitors triggers a series of events, including activation of the unfolded protein response (UPR) and autophagy eventually leading to selective leukemic cell death [20, 21]. Targeting signals involved in CBY1 down-modulation in CML may be, therefore, advanced as a complementary strategy to eradicate clonal hematopoiesis.
CBY1 DOWN-MODULATION ASSOCIATED WITH BCR-ABL1 TK CONTRIBUTES TO Β-CATENIN ACTIVATION IN LEUKEMIC HEMATOPOIESIS
CBY1 is a small conserved antagonist of β-catenin. It is encoded by the C22orf2 gene at chromosome 22q13.1, downstream of BCR cluster region (22q11) involved in the t [9, 22] translocation [22]. CBY1 antagonistic function on β-catenin encompasses its direct interaction with the C-terminal activation domain of β-catenin (which hinders β-catenin binding with TCF/LEF transcription factors hence repressing β-catenin transcriptional activation) and 14-3-3 scaffolding proteins (s or z, which drives CBY1 nuclear export into a stable tripartite complex with β-catenin) [15–17]. Accordingly, CBY1 “loss of function” has been involved in the pathogenesis of some types of cancers, such as colon carcinomas and pediatric ependymomas [23, 24]. Due to the relative proximity of C22orf2 [22q13.1] to the breakpoint cluster region on BCR (22q11) we first investigated whether C22orf2 haplo-insufficiency, originated by deletion(s) downstream of BCR sequences as a result of the t(9, 22) translocation, was correlated with CML prognosis [22]. However, fluorescent in situ hybridization (FISH) established that the full length C22orf2 gene follows BCR sequences in CML myeloid progenitors, and relocates to the derivative chromosome 9 (der(9q)) in patients with the typical translocation t [9, 22] [q34;q11] or to the second fusion gene in patients with variant translocations [18, 19]. Still, CBY1 expression is reduced in hematopoietic progenitors of CML patients at clinical diagnosis compared to healthy donors and further lowered in the LSC (CD34+) compartment, where β-catenin provides a key signal for proliferation and survival [8]. Restored expression of CBY1 in CML patients at the time of complete or major molecular response during treatment with TK inhibitors (when the whole or major part of hematopoiesis is the normal, BCR-ABL1- one) supports CBY1 down-modulation as a trait of leukemic clone. Lack of correlation between CBY1 expression, disease prognosis and response to TK inhibitors seems to exclude its involvement in the disease progression [18]. Further investigation let establish the dependency of CBY1 down-modulation from BCR-ABL1 TK. The fusion protein inhibition in response to IM induced, in fact, CYB1 expression, which, in turn, abolished the leukemic clone growth and survival advantage through events proceeding from β-catenin nuclear export and degradation in the cytoplasm, activation of ER stress-associated pathway known as UPR, which leads to apoptotic death, and induction of an autophagic pathway, which addresses β-catenin to proteasome-independent degradation [20, 21, 25].
CBY1 down-modulation associated with BCR-ABL1 TK is driven by multiple events, including transcriptional mechanisms, caused by the gene promoter hyper-methylation, and post-transcriptional modifications involved in the ubiquitin-mediated degradation by proteasome [15–17].
CBY1 DOWN-MODULATION ASSOCIATED WITH BCR-ABL1 IS MEDIATED BY GENE PROMOTER HYPERMETHYLATION AND PROTEIN INSTABILITY
DNA methylation consists in the attachment of methyl groups (CH3) at the 5′ carbon position of the cytosine ring. It predominantly occurs at high density CpG regions named CpG islands, which cover the transcriptional initiation sites of approximately 70% of annotated gene promoters [26]. It is promoted by a family of enzymes, the DNA methyltransferases (DNMTs, encompassing the de novo methyltransferases DNMT3a and DNMT3b and the maintenance methyltransferase DNMT1), which catalyze CH3 group transfer to establish a permissive landscape for methyl-binding (MBD) proteins, such as MeCP2, MBD1, MBD2 and MBD4, involved in transcriptional repression [27]. Indeed, DNA hyper-methylation is a critical epigenetic mechanism for transcriptional silencing of genes involved in cancer development and progression, including those controlling DNA repair, cell cycle, cell adhesion, apoptosis and angiogenesis. Accordingly, it may be considered as a second hit in the Knudson’s two-hit model of cellular transformation [28]. From a clinical perspective, such integrated view into cancer genomics might improve the therapeutic approach through DNA de-methylating agents. DNA hyper-methylation is a common event in CML and affects multiple genes [29]. We found that enhanced recruitment of DNMT1 at the C22orf2 promoter is a component of CBY1 down-modulation in CML hematopoietic progenitors and LSC, hence suggesting the putative advantage of DNA-demethylating drugs, such as 5-Aza-CdR (also referred to as decitabine), in the disease therapy [30].
The reduction of protein stability is a further mechanism driving CBY1 down-modulation in CML. As CBY1 reduced transcription, the protein instability is mediated by BCR-ABL1 TK activity through events affecting CBY1 binding with 14-3-3. Those events includes phosphorylation by AKT at a critical residue of CBY1 (serine 20) for 14-3-3 binding and the impaired phosphorylation at a 14-3-3 residue (serine 186) by c-Jun N-terminal kinase (JNK) [17, 20]. The enhanced degradation of CBY1 is driven by the ubiquitin proteasome system (UPS) through a complex and tightly controlled process encompassing the covalent attachment of K48-linked polyubiquitin chain to flag target proteins for degradation through the 26S proteasome [31]. In particular, a post-translational modification which utilizes small ubiquitin-like modifier (SUMO) groups to covalently attach target substrates and promote their ubiquitination and degradation has been involved in CBY1 increased degradation associated with BCR-ABL1 [17, 32]. These findings suggest that inhibitors of AKT/mTOR axis, 14-3-3 binding or proteasome have the potential to attenuate β-catenin signalling and may be, therefore, tested for clinical use. Notably, pilot studies support the synergistic effects of proteasome inhibitors and TK inhibitors on BCR-ABL1+ cells, including LSC [33, 34].
CBY1 INDUCTION IN BCR-ABL1+ CELLS ACTIVATES UPR AND TRIGGERS THE EXECUTION PHASE OF APOPTOSIS
β-catenin nuclear exclusion and transcriptional inactivation is the major consequence of CBY1 enforced expression following C22orf2 stable transfection and CBY1 induction in response to IM in BCR-ABL1+ cells [18]. In such a cell context, as in other cell types, CBY1-driven cytoplasmic re-location of β-catenin activates UPR, which, in turn, induced the BCL2-interacting mediator of cell death (BIM), hence contributing to the execution phase of apoptotic death [20].
UPR is a homeostatic signaling network that transduces information about the protein-folding status in the ER lumen to buffer fluctuations in the unfolded protein load and let the recovery of ER function. Under physiological conditions it acts as an adaptive mechanism to promote cell survival, while under high-level or chronic ER stress, it becomes overshadowed by alternative signals which commit cells to degeneration and culminate in apoptosis [35]. UPR occurs via three mechanisms: i) reduced translation of misfolded proteins, ii) enhanced translation of ER chaperones and iii) ER-associated degradation (ERAD) of misfolded proteins, which are transferred from the ER to the cytoplasm for subsequent ubiquitination and degradation by the 26S proteasome. UPR is triggered by the activation of three trans-membrane ER proteins: pancreatic ER kinase (PERK), inositol-requiring enzyme 1a (IRE1a) and activating transcription factor 6 (ATF6), whose oligomerization at the ER luminal domain activates downstream activities, to transduce life or death signals [36]. We proved that CBY1 enforced expression as well as its induction in response to IM activate PERK and IRE1a, which, in turn, trigger specific transcription factors to up-regulate their target genes. Activated PERK phosphorylates the eukaryotic translation initiator factor 2a (eIF2a) to slow down mRNA translation and protein synthesis [37]. Moreover, it allows selective translation of the activating translation factor 4 (ATF4) to induce transcription of the C/EBP-homologous protein (CHOP, otherwise termed GADD153), which inhibits expression of the anti-apoptotic BCL-2 to hasten cell death [38]. More importantly, CHOP induces transcription of BIM, a crucial tumor suppressor gene for CML response to IM [39, 40]. BIM participates in the death signal transmission from ER to mitochondria, hence contributing to the execution phase of apoptosis through the activation of ER resident caspase 12 [40]. Finally, CHOP induction inhibits β-catenin/TCF-dependent transcriptional activation and may, therefore, contribute to cyclin D1 reduction to promote BCR-ABL1+ cell growth arrest [41]. Still, the role of UPR in the survival of CML progenitors and LSC is elusive. Three recently published studies established the pro-survival effects of UPR on BCR-ABL1+ cells, hence raising the question of the fusion protein impact on individual UPR branches [42–44]. Further investigation is required, in particular, to elucidate the misfolded nature of BCR-ABL1 protein, whether and how its expression and phosphorylation levels are involved in UPR activation, and the participation of BCR-ABL1 downstream targets, such as JNK and AKT, in the induction of UPR sensors and effectors [36].
CBY1-INDUCED AUTOPHAGY PARTICIPATES IN BCR-ABL1+ CELL COMMITMENT TO APOPTOTIC DEATH
Autophagy is a further consequence of CBY1 enforced expression and induction in response to IM in BCR-ABL1+ cells [21]. Autophagy is a self-catabolic process wherein bulk cytoplasmatic components such as aggregated/misfolded proteins and organelles are sequestered within double- or multi-membrane vesicles (autophagosomes) and then delivered to lysosomes for degradation. It may either serve as a cell death mechanism (otherwise named type II programmed cell death) or play a pro-survival role as part of an adaptive and detoxifying process in response to sub-lethal stress such as starvation, hypoxia, heat shock and microbial pathogens, and contingent upon the cell context and oncogenic status [45, 46]. In BCR-ABL1+ cells autophagy has been regarded as a complementary pathway to apoptotic cell death response to IM and other TK inhibitors proceeding from ER stress and ER Ca2+ store mobilization [47, 48]. More recently, autophagy has been implicated in normal and cancer stem cell self-renewal and survival in the low-oxygen “niche” of bone marrow microenvironment as well as in their quiescence through the m-TOR complex 1 (m-TORC1)-mitochondria-reactive oxygen species (ROS) axis. Moreover, it protects BCR-ABL1+ LSC from the lethal effects of TK inhibitors, hence contributing to the disease persistence. Indeed, cells expressing the BCR-ABL1 rearranged gene of CML exhibit low basal levels of autophagy, but are highly dependent on autophagy for response to stress and leukemia induction in allograft models [49–52]. In a recently published study we showed that the autophagic phenotype originated in BCR-ABL1+ cells by the elevation of free Ca2+ release from ER stores in response to IM generates a 28 kDa cleaved calpain fragment which, in turn, promotes the cleavage of the ER-resident caspase 12 into a 42 kDa fragment corresponding to its activated isoform hence contributing to apoptosis commitment shown in a previously published study [21]. Notably, β-catenin is a calpain target [53]. Accordingly, its decrease following CBY1 induction in response to IM may be partly mediated by calpain activation and autolysosomal clearance upon autophagy induction [54]. It is worth to mention the calpain-mediated cleavage of β-catenin accumulated within the cytoplasmic compartment into a 75 kDa fragment still owning TCF-dependent transcriptional activity [55, 56]. Such β-catenin fragment may be regarded as the putative mediator of autophagy pro-survival effects in BCR-ABL1+ cells following IM treatment. Further investigation is required to identify signals directing autophagy impact on cell decision between life and death in a BCR-ABL1+ cell context.
CONCLUSIONS
Here we report of a new component of β-catenin network in CML (Figure 1). CBY1 down-modulation associated with BCR-ABL1 TK promotes β-catenin retention within the nuclear compartment and transcriptional activation. It is driven by transcriptional and post-transcriptional events involving DNA hyper-methylation and protein enhanced degradation. Through its effects on β-catenin sub-cellular partitioning, CBY1 impacts UPR and autophagy in clonal hematopoietic progenitors and, more importantly, in LSC. The activation of UPR and autophagy may have a role in the balance between life and death of BCR-ABL1+ cells in response to TK inhibitors.
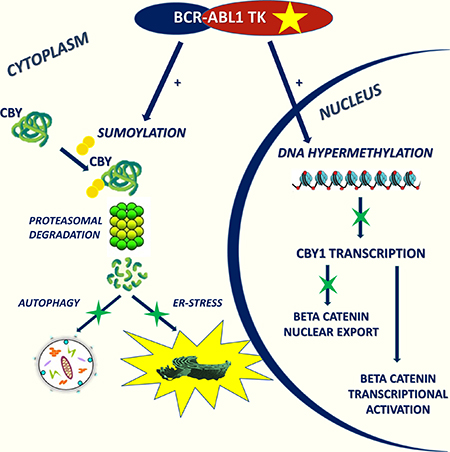
Figure 1: CBY1 down-modulation associated with BCR-ABL1 TK promotes β-catenin retention in the nucleus and its transcriptional activation. It is driven by transcriptional and post-transcriptional events involving DNA hyper-methylation and protein enhanced degradation. Through its effects on β-catenin sub-cellular localization, CBY1 impacts UPR and autophagy in clonal hematopoietic progenitors.
ACKNOWLEDGMENTS
We thank all the member of Prof. G. Martinelli clinical and research divisions. We also thank Prof. Michele Cavo and Prof. Ken-Ichi Takemaru for insightful discussions.
CONFLICTS OF INTEREST
Authors have no conflicts of interests to declare with relation to this manuscript.
FUNDING
University of Bologna [RFO funds], Ministero della Pubblica Istruzione e della Ricerca [PRIN funds], Umberto Veronesi Foundation, AIRC, FP7 NGS-PTL project, Progetto Regione-Università 2010–12 [L. Bolondi] are acknowledged for financial support.
REFERENCES
1. Abbaszadegan MR, Bagheri V, Razavi MS, Momtazi AA, Sahebkar A, Gholamin M. Isolation, identification, and characterization of cancer stem cells: A review. J Cell Physiol. 2017; 232:2008–2018.
2. Till JE, McCulloch EA, Siminovitch L. A stochastic model of stem cell proliferation based on the growth of spleen clonony-forming cells. Proc Natl Acad Sci USA. 1964; 51:29–36.
3. Jiang X, Forrest D, Nicolini F, Turhan A, Guilhot J, Yip C, Holyoake T, Jorgensen H, Lambie K, Saw KM, Pang E, Vukovic R, Lehn P, et al. Properties of CD34+ CML stem/progenitor cells that correlate with different clinical responses to imatinib mesylate. Blood. 2010; 116:2112–2121.
4. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myeloigenous leukemia in mice by the P210 bcr/abl gene of the Philadelphia chromososme. Science. 1990; 247:824–830.
5. Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Muller-Tidow C, Bhatia R, Brunton VG, Koschmieder S, Holyoake TL. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012; 119: 1501–1510.
6. Zhao J, Blum A, Chen HY, Kwon SH, Jung JM, Cook A, Lagoo T, Reya.T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007; 12: 528–541.
7. Minami Y, Stuart SA, Ikawa T, Jiang Y, Banno SH, Huntons IC, Young DJ, Naoe T, Murre C, Jamieson CH, Wang JY. BCR-ABL-transformed GMP as myeloid leukemic stem cells. Proc Natl Acad Sci USA. 2008; 105:17967–17972.
8. Hu, Y. Chen, L. Douglas, S. Li S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia. 2009; 23: 109–116.
9. Zhang B, Li M, McDonald T, Holoyake TL, Moon RT, Campana D, Shultz L, Bhatia R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-b-catenin signaling. Blood. 2013; 121: 1824–1838.
10. Coluccia AM, Vacca A, Duñach M, Mologni L, Redaelli S, Bustos VH, Benati D, Pinna LA, Gambacorti-Passerini C. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 2007; 26: 1456–1466.
11. Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton JG, Giles FJ, Durocher J, Creusot RS, Karimi M, Jones C, Zehnder JL, Keating A, et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci USA. 2009; 106:3925–3929.2010,30,4575.
12. Huang W, Zhou W, Saberwal G, Konieczna I, Horvath E, Katsoulidis E, Platanias LC, Eklund EA. Interferon consensus sequence binding protein (ICSBP) decreases beta-catenin activity in myeloid cells by repressing GAS2 transcription. Mol Cell Biol. 2010; 30:4575–4594.
13. Huang W, Bei L,. Eklund EA. Fas-associated phosphatase 1 (Fap1) influences bcatenin activity in myeloid progenitor cells expressing the Bcr-Abl oncogene. J Biol Chem. 2013; 288:12766–12776.
14. Jamieson C, Sharma M, Henderson BR. Targeting the b-catenin nuclear transport in cancer. Semin Cancer Biol. 2014; 27:20–29.
15. Takemaru, K Yamaguchi S, Lee YS, Zhang Y, Carthew RW, Moon RT. Chibby, a nuclear beta-catenin-associated antagonist of the Wnt/Wingless pathway. Nature. 2003; 422: 905–909.
16. Li FQ, Mofunanya A, Harris K, Takemaru K. Chibby cooperates with 14-3-3 to regulate beta-catenin subcellular distribution and signalling activity. J Cell Biol. 2008; 181:1141–1154.
17. Takemaru K, Fischer V, Li FQ. Fine tuning of nuclear-catenin by Chibby and 14-3-3. Cell Cycle. 2009; 8:210–213.
18. Leo E, Mancini M, Aluigi M, Luatti S, Castagnetti F, Testoni N, Soverini S, Santucci MA, Martinelli G. BCR-ABL1-associated reduction of beta catenin antagonist Chibby1 in chronic myeloid leukemia. PLoS One. 2013; 8:e81425.
19. Sinclair PB, Nacheva EP, Leversha M, Telford N, Chang J, Reid A, Bench A, Champion K, Huntly B, Green AR. Large deletions at the t(9;22) breakpoint are common and may identify a poor-prognosis subgroup of patients with chronic myeloid leukemia. Blood. 2000; 95:738–743.
20. Mancini M, Leo E, Takemaru K, Campi V, Borsi E, Castagnetti F, Gugliotta G, Santucci MA, Martinelli G. Chibby drives b catenin cytoplasmic accumulation leading to activation of the unfolded protein response in BCR-ABL1+ cells. Cell Signal. 2013; 25:1820–1827.
21. Mancini M, Leo E, Campi V, Castagnetti F, Zazzeroni L, Gugliotta G, Santucci MA, Martinelli G. A calpain-cleaved fragment of b-catenin promotes BCRABL1+ cell serviva evoked by autophagy induction in response to imatinib. Cell Signal. 2014; 26:1690–1697.
22. Timmons MS, Witte ON. Structural characterization of the BCR gene product. Oncogene. 1989; 4:559–567.
23. Schuierer MM, Graf E, Takemaru K, Dietmaier W, Bosserhoff AK. Reduced expression of beta-catenin inhibitor Chibby in colon carcinoma cell lines. World J Gastroenterol. 2006; 12:1529–1535.
24. Karakoula K, Suarez-Merino B, Ward S, Phipps KP, Harkness R, Thompson D, Jacques TS, Harding B, Beck J, Thomas DG, Warr TJ. Real-time quantitative PCR analysis of pedriatric ependymomas identifies novel candidate genes including TPR at 1q25 and CHIBBY at 22q12-q13. Genes Chromosomes Cancer. 2008; 47:1005–1022.
25. Mancini M, Leo E, Takemaru K, Campi V, Castagnetti F, Soverini S, De Benedittis C, Rosti G, Cavo M, Santucci MA, Martinelli G. 14-3-3 binding and somoylation concur to the down-modulation of b-catenin antagonist CHIBBY 1 in chronic myeloid leukemia. PLoS One. 2015; 10:e0131074.
26. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011; 25:1010–1022.
27. Bogdanovi O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009; 118:549–564.
28. Knudson AG. Hereditary cancer: two hits revised. J Cancer Res Clin Oncol. 1996; 122:135–140.
29. Leo E, Martinelli G. DNA Methylation in Chronic Myeloid Leukemia. J Mol Genet Med. 2014; 8:118. https://doi.org/10.4172/1747–0862.1000118.
30. Leo EW, Mancini M, Castagnetti F, Gugliotta G, Santucci MA, Martinelli G. DNA methyltransferase 1 drives transcriptional down-modulation of b-catenin antagonist Chibby 1 associated with the BCR-ABL1 gene of chronic myeloid leukemia. J Cell Biochem. 2015; 116:589–597.
31. Inobe T, Matouschek A. Paradigms of protein degradation by the proteasome. Curr Opin Struct Biol. 2014; 24:156–164.
32. Wilkinson KA, Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J. 2010; 428: 133–145.
33. Hu Z, Pan XF, Wu FQ, Ma LY, Liu DP, Feng TT, Meng FY, Liu XL, Jiang QL, Chen XQ, Liu JL, Liu P, Chen Z, et al. Synergy between proteasome inhibitors and imatinib mesylate in chronic myeloid leukemia. PLos One. 2009; 4:e6257.
34. Crawford LJ, Chan ET, Aujay M, Holyoake TL, Melo JV, Jorgenses HG, Suresh S, Walker B, Irvine AE. Synergistic effects of proteasome inhibitor carfilzomib in combination with tyrosine kinase inhibitors in imatinib-sensitive and-resistant chronic myeloid leukemia models. Oncogenesis. 2014; 3:e90.
35. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012; 13:89–102.
36. Maly DJ, Papa FR. Druggable sensors of the unfolded protein response. Nat Chem Biol. 2014; 10:892–901.
37. Su Q, Wang S, Gao HQ, Kazemi S, Harding HP, Ron D, Koromilas AE. Modulation of the eukaryotic initiation factor 2 alpha subunit PERK by tyrosine phosphorylation. J. Biol. Chem. 2008; 283: 469–475.
38. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001; 21:1249–1259.
39. Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, Roberts AW, Strasser A. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance to their loss is overcome by BH3 mimetic. Proc Natl Acad Sci USA. 2006; 103:14907–12.
40. Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E. Translocation of Bim to the endoplasmic reticulum [ER] mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J Biol Chem. 2004; 279:50375–50381.
41. Raab MS, Breitkreutz I, Tonon G, Zhang J, Hayden PJ, Nguyen T, Freuhauf JH, Lin BK, Chauhan D, Hideshima T, Munshi NC, Anderson KC, Podar K. Targeting PKC: a novel role for beta catenin in ER stress and apoptotic signaling. Blood. 2009; 113: 1513–1521.
42. Tanimura A, Yujiri T, Tanaka Y, Hatanaka M, Mitani N, Nakamura Y, Mori K, Tanizawa Y. The anti-apoptotic role of the unfolded protein response in Bcr-Abl-positive leukemia cells. Leuk. Res. 2009; 33: 924–928.
43. Li B, Yi P, Zhang B, Xu C, Liu Q, Pi Z, Xu X, Chevet E, Liu J. Differences in endoplasmic reticulum stress signaling kinetics determine cell survival ourcome through activation of MKP-1. Cell Signal. 2011; 23: 35–45.
44. Kusio-Kobialka M, Podszywalow-Bartnicka P, Peidis P, Glodkowska-Mrowka E, Wolanin K, Leszak G, Seferynska I, Stoklosa T, Koromilas AE, Piwocka K.The PERK-eIF2b phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle. 2012; 11: 4069–4078.
45. Baehrecke EH. Autophagy: dual roles in life and death. Nat Rev Mol Cell Biol. 2005; 6:505–508.
46. He C, Klionsky DJ. Regulation mechanisms and signaling of autophagy. Annu Rev Genet. 2009; 43:67–93.
47. Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, Duyster J, Elsasser HP, Schaltzl HM. The anticancer drug imatinib induces cellular autophagy. Leukemia. 2007; 21:936–942.
48. Kamitsuji J, Kuroda S, Kimura S, Toyokuni K, Watanabe E, Ashihara H, Tanaka Y, Yui H, Matsubara Y, Mizushima Y, Hiraumi E, Kawata T, Yoshikawa, et al. The Bcr-Abl kinase inhibitor INNO-406 induces autophagy and different modes of cell death execution in Bcr-Abl-positive leukemias. Cell Death Differ. 2008; 15: 1712–1722.
49. Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R, Van Etten RA, Donato N, Hunter A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009; 119: 1109–1123Y.
50. Altman BJ, Jacobs SR, Mason EF, Michaleck EF, MacIntyre AN, Coloff JL, Ilkayeva O, Jia W, He YW, Rathmell JC. Autophagy is essential to suppress cell death and to allow BCR-Abl-mediated leukemogenesis. Oncogene. 2011; 30: 1855–1867.
51. Crowley LC, Elzinga BM, O’Sullivan GC, McKenna SL. Autophagy induction by Bcr-Abl-expressing cells facilitates their recovery from targeted and nontargeted treatment. Am J Hematol. 2011; 86:38–47.
52. Helgason GV, Mukhopadhyay A, Karvela M, Salomoni P, Calabretta B, Holyoake TL. Autophagy in chronic meloid leukemia: stem cell survival and implication in therapy. Curr Cancer Drug Targets. 2013; 13: 724–734.
53. Benetti R, Copetti T, Dell’Orso S, Melloni E, Brancolini C, Monte M, Schneider C. The calpain system is involved in the constitutive regulation of beta-catenin signaling functions. J Biol Chem. 2005; 280:22070–22080.
54. Petherick KJ, Williams AC, Lane JD, Ordonez-Moran P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik K, Paraskeva C, Greenhough A. Autolysosomal b-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013; 32:1903–1916.
55. Rios-Doria J, Kuefer R, Ethier SP, Day ML. Cleavage of beta-catenin by calpain in prostate and mammary tumor cells. Cancer Res. 2004; 64: 7237–7240.
56. Lade A, Ranganathan S, Luo J, Monga SPS. Calpain induces N-terminal truncation of b-catenin in normal murine liver development: diagnostic implications in hepatopblastomas. J Biol Chem. 2012; 287: 22789–22798.