
INTRODUCTION
Pancreatic cancer is the fourth leading cause of cancer-related death in developed countries, with an overall 5-year survival rate below 5%. It is generally asymptomatic until it has reached an advanced stage and is typically unresponsive to conventional radiation and chemotherapy, resulting in a mortality rate of nearly 100% within 6 months of diagnosis [1]. Therefore, novel therapeutic strategies are urgently needed.
Apolipoprotein A-I (ApoA-I), the major protein component of high-density lipoprotein (HDL), reverses cholesterol transport by extracting and transferring cholesterol and phospholipids from peripheral cells to the liver for excretion. In addition to its antiatherogenic properties, ApoA-I also exhibits anti-inflammatory and anti-oxidant properties [2]. Reconstituted HDL has been shown to reduce inflammation by inhibiting cellular expression of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 in stimulated human coronary endothelial cells and reducing CD11b expression in circulating monocytes [3]. L-4F, an ApoA-I mimetic peptide, was engineered to mimic the anti-inflammatory and anti-oxidative functionalities of ApoA-I, which have shown positive effects for cancer treatment and for decreasing inflammation [4, 5]. It has been reported that L-4F suppresses tumor progression by inhibiting the expression and activity of hypoxia-inducible factor-1α and tumor angiogenesis in human ovarian cancer [6], reduces cell viability and proliferation in ovarian cancer cells, and decreases plasma levels of lysophosphatidic acid in colon cancer [7, 8]. It has also recently been found that L-4F inhibits LPS-mediated elevation of TNF-α and IL-6 in LPS-stimulated neutrophils [9].
The therapeutic effects of L-4F on pancreatic cancer and tumor immune microenvironments remain poorly understood. In this study, we aimed to explore whether L-4F can inhibit pancreatic cancer progression and to determine its cellular and molecular mechanisms.
RESULTS
L-4F attenuates the progression of pancreatic tumors in mice
We first evaluated whether L-4F might exhibit suppressive effects on pancreatic cancer development in a mouse model. To accomplish this, C57BL/6 mice were implanted with H7 pancreatic cancer cells through direct injection into the pancreas. L-4F (10 mg/kg) or Sc-4F (10 mg/kg), a control peptide, was administered. As shown in Figure 1A–1C, compared with tumors in Sc-4F-treated mice, L-4F treatment significantly reduced both tumor size (Figure 1A, 1B) and tumor weight (0.77 g vs 0.52 g, P<0.01) (Figure 1C).
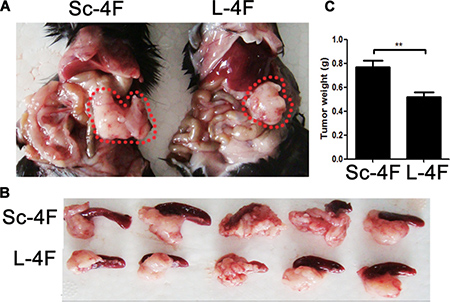
Figure 1: L-4F delays H7 tumor progression in mice. H7 cells were injected into the pancreas. Mice were euthanized after 1 wk of L-4F or Sc-4F treatment. (A, B) Representative tumors from Sc-4F- or L-4F-treated mice; pancreatic cancer is outlined using a red dashed line. (C) Final tumor weights (*P < 0.05).
L-4F did not inhibit migration, reduce proliferation or induce apoptosis of H7 cells
H7 cells were treated with vehicle or the indicated concentration of L-4F (5, 10, or 20 μg/mL) and submitted to a wound healing assay. Wound width was photographed using light microscopy at 0, 24 and 48 h after scraping. As shown in Figure 2A, there were no clear differences in wound healing for the duration of L-4F treatment. As shown in Figure 2B, the proliferative index (PI) of the L-4F-treated cells was not obviously reduced compared to the untreated or low dose-treated cells (PI = 62.74, 63.17, 62.28, and 60.22 for 0, 5, 10, and 20 μg/mL, respectively, NS). In addition, compared with the untreated cells, the populations of early apoptotic, necrotic, and late apoptotic cells were not obviously changed in the L-4F-treated cells (4.43%, 4.68%, 5.44% and 5.88%, for 0, 5, 10, and 20 μg/mL, respectively, NS) (Figure 2C).
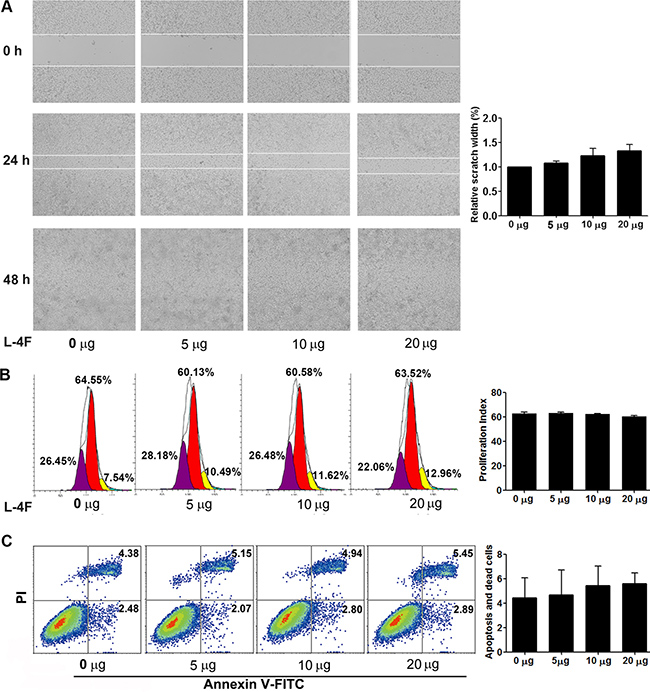
Figure 2: L-4F could not directly attenuate H7 cell invasion or proliferation and did not induce apoptosis. H7 cells were treated with L-4F (0, 5, 10, or 20 μg/mL). (A) Representative images of wound healing in a scratch assay of H7 cells treated with L-4F at 0, 24 and 48 h after wounding. The distances between wound edges in three randomly chosen regions were normalized to 100% in untreated cells at 24 h. (B) One representative result from each experiment is shown: each peak represents the population of cells with reduced CFSE content due to cell division and the proliferative index (PI) at 48 h. (C) The percentage of apoptotic cells treated with L-4F at 48 h. Cells in the lower right quadrant were scored as “early apoptotic” (Annexin+/PI-), and cells in the upper right quadrant were scored as “necrotic/late apoptotic” (Annexin+/PI+).
L-4F decreases inflammatory cell infiltration in mice with pancreatic cancer
As shown in Figure 3D, L-4F treatment obviously reduced inflammatory cell infiltration in tumor tissues collected from mice. Therefore, we further analyzed the percentages of IL-17A-, IL-6-, IFN-γ-, IL-4-, granulocyte macrophage colony stimulating factor (GM-CSF)- and IL-1β-producing cells in tumor-infiltrating cell populations from mice administered L-4F or Sc-4F.
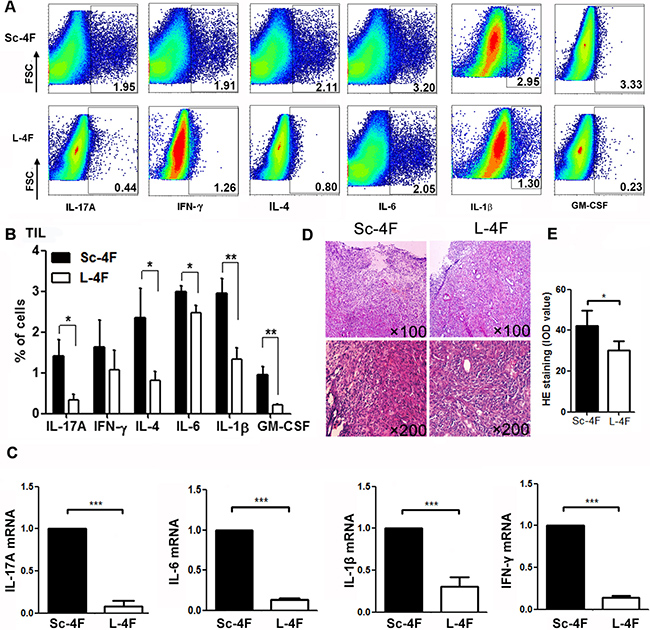
Figure 3: L-4F reduces inflammation in a mouse model of pancreatic cancer. Tumors were collected from Sc-4F- or L-4F-treated mice. Single-cell suspensions were acquired, and the cytokines were immunostained as described in the Materials and Methods section. (A) One representative result from each experiment is shown. (B) The percentages of IL-17A-, IFN-γ-, IL-4-, IL-6-, IL-1β- and GM-CSF-producing cells among tumor infiltrating cells in tumor tissues (*P < 0.05, **P < 0.01). (C) The mRNA levels of IL-17A, IFN-γ, IL-6, and IL-1β in tumor tissues. (D) H&E staining showing infiltration of inflammatory cells in tumor tissues. (E) Image-Pro Plus was used to quantify the relative IOD value of HE staining of inflammatory cells in tumor tissues (*P < 0.05).
Compared with the Sc-4F-treated group, in the mice treated with L-4F, the percentages of tumor-infiltrating cells producing IL-17A (1.41% vs 0.33%, P < 0.05), IL-4 (2.35% vs 0.81%, P < 0.05), IL-6 (2.99% vs 2.47%, P < 0.01), IL-1β (2.95% vs 1.34%, P < 0.01) and GM-CSF (0.96% vs 0.22%, P < 0.01) all significantly decreased, whereas the percentage of tumor-infiltrating cells producing IFN-γ (1.63%vs 1.07%, NS) did not exhibit significant changes (Figure 3A, 3B).
L-4F decreases mRNA levels of inflammatory cytokines in mice with pancreatic cancer
To further confirm the anti-inflammatory effects of L-4F, we analyzed mRNA levels of the inflammatory cytokines IL-17A, IFN-γ, IL-6, and IL-1β in tumor tissues from Sc-4F- or L-4F-treated tumor-bearing mice. As shown in Figure 3C, L-4F substantially decreased the mRNA levels of the inflammatory cytokines IL-17A, IFN-γ, IL-6 and IL-1β (average fold-change of 0.107 for IL-17A, 0.155 for IFN-γ, 0.069 for IL-6, 0.340 for IL-1β, P<0.001).
L-4F decreases Th17 and Th1 cell populations in mice with pancreatic cancer
We further analyzed the percentages of IL-17A-, IL-6-, IFN-γ-, IL-4- and GM-CSF-producing Th cells (T helper cells) in tumor-infiltrating lymphocytes (TILs) from H7 tumor-bearing mice treated with Sc-4F or L-4F. Compared with the Sc-4F-treated mice, in the mice treated with L-4F, the percentages of tumor-infiltrating Th cells producing IL-17A (19.65% vs 5.65%, P < 0.05) and IFN-γ (15.72% vs 7.7%, P < 0.05) significantly decreased, whereas the percentages of tumor-infiltrating Th cells producing IL-4 (11.11% vs 8.51%, NS), IL-6 (13.29% vs 12.53%, NS), and GM-CSF (13.84% vs 9.0%, NS) did not exhibit significant changes (Figure 4A, 4B).
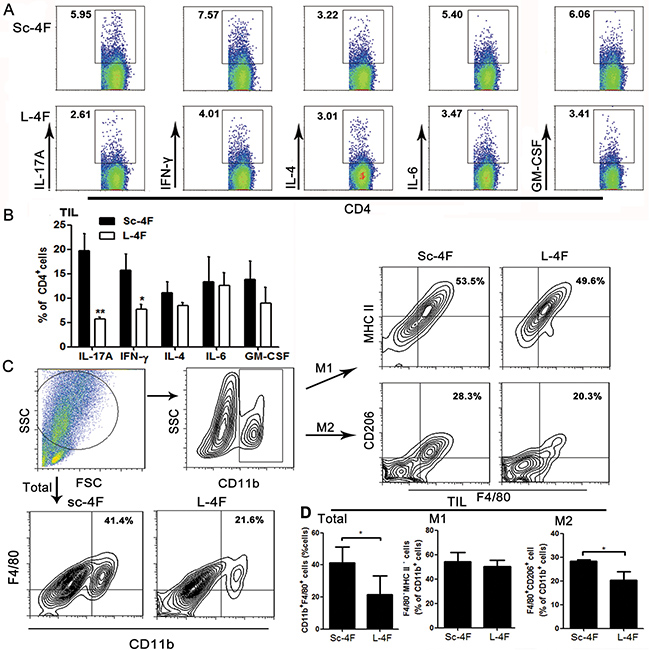
Figure 4: L-4F decreases Th17 cell, Th1 cell and TAM populations in a mouse pancreatic cancer model. Tumors were collected from Sc-4F- or L-4F-treated mice. Single-cell suspensions were acquired and stained as described in the Materials and Methods section. (A) One representative result from each experiment is shown. (B) The percentages of IL-17A-, IFN-γ-, IL-4-, IL-6- and GM-CSF-producing cells among tumor infiltrating Th cells (*P < 0.05, **P < 0.01). Th17 and Th1 cells represent two particular subsets of T helper lymphocytes characterized by production of IL-17 and IFN-γ cytokines, respectively. (C) The percentage of CD11b+F4/80+ cells among tumor infiltrating cells. The number of F4/80+ macrophages among the total CD11b+cells was quantified. (D) The blot shown was gated on CD11b+ cells, and an F4/80+MHC II+ and F4/80+CD206+ flow plot was used to identify M1 and M2 macrophages, respectively. The number of F4/80+MHC II+ and F4/80+CD206+ macrophages among the total CD11b+ cells was quantified (*P < 0.05).
L-4F inhibits the augmentation of tumor-associated macrophage populations in mice with pancreatic cancer
The activation status of macrophages in tumor tissue was also assessed using flow cytometry. Populations of M1 macrophages and M2 macrophages were determined using MHC II and CD206 (also called mannose receptor C type 1, Mrc1) as markers. Compared with the Sc-4F-treated group, the percentage of CD11b+F4/80+ macrophages (41.4% vs 21.6%, P < 0.05) significantly decreased (Figure 4C, 4D) in the L-4F treated group. Similarly, L-4F effectively inhibited the augmentation of F4/80+CD206+ M2 macrophage populations (28.3% vs 20.3%, P<0.01) (Figure 4C, 4D).
L-4F suppresses M2 macrophage polarization and decreases the expression of macrophage-associated genes
To further examine the effect of L-4F in macrophages in vitro, we differentiated mouse bone marrow-derived macrophages to M1 or M2 macrophages using LPS (for M1) or IL-4 (for M2) in the presence of Sc-4F or L-4F. M1-like macrophages are defined here as CD11b+ F4/80+MHC II+ cells, while M2-like macrophages are defined as CD11b+ F4/80+CD206+ cells. As shown in Figure 5A–5C, the number of M2 macrophages significantly decreased in the group treated with L-4F compared to the Sc-4F-treated group (31.3% vs 20.1%, P<0.05). In contrast, there were no significant changes in M1 macrophage populations. These results indicate that L-4F treatment inhibits the augmentation of M2 macrophage populations in vitro.
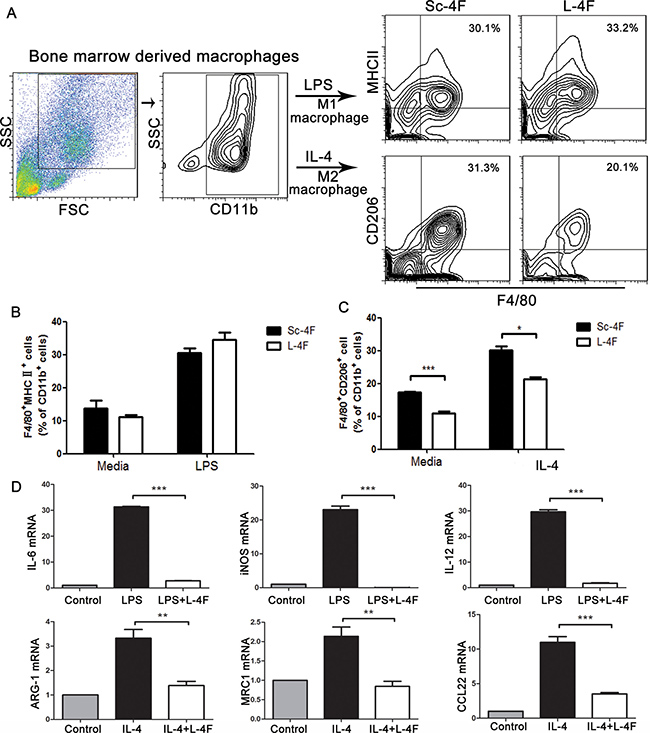
Figure 5: L-4F inhibits M2 macrophage polarization and decreases the expression of macrophage-associated genes. BMDMs were exposed to LPS or IL-4 in the presence or absence of L-4F as described in the Materials and Methods section. (A) The blot shown was gated on CD11b+ cells, and an F4/80+MHC II+ and F4/80+CD206+ flow plot was used to identify M1 and M2 macrophages, respectively. (B and C) The number of F4/80+MHC II+ and F4/80+CD206+ macrophages among the total CD11b+ cells was quantified. (D) The expression of the M1 genes IL-6, iNOS and IL-12 and the M2 genes ARG-1, MRC1, and CCL22 was analyzed using qPCR (*P < 0.05, ***P < 0.001).
The M1 and M2 phenotypes were further confirmed by gene expression analysis. To accomplish this, we measured the expression of several M1 cytokines (IL-6, iNOS, and IL-12) and M2 molecules (ARG-1, MRC1, and CCL22). Compared to undifferentiated macrophages, M1- or M2-differentiated macrophages showed significant upregulation of M1- or M2-specific inflammatory genes, and expression of these genes was substantially decreased in the L-4F-treated group (average fold-change of 0.0042 for IL-6, 0.0014 for iNOS, 0.0842 for IL-12, 0.415 for ARG-1, 0.82 for MRC1, 0.317 for CCL22, , P < 0.001) (Figure 5D).
L-4F down-regulated STAT3 and MAPK signaling pathways in lipopolysaccharide-treated RAW 264.7 cells
We further evaluated the effect of L-4F on activation of STAT3 and LPS-induced activation of the MAPK pathway in RAW 264.7 cells. As shown in Figure 5, compared to the LPS control group, L-4F treatment significantly suppressed the phosphorylation of STAT3 (Figure 6A) and inhibited the phosphorylation of JNK and ERK1/2 (Figure 6C and 6D) in LPS-treated RAW 264.7 cells in a low dose-dependent manner. However, L-4F had no obvious effect on the p38MAPK (Figure 6B) signaling pathway.
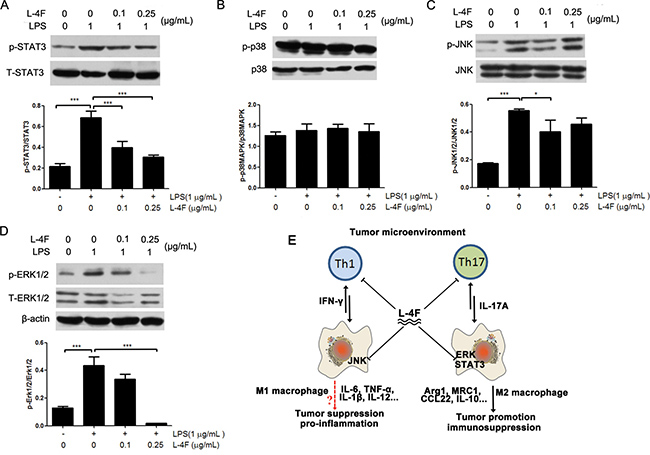
Figure 6: L-4F down-regulated STAT3 and MAPK signaling pathways in lipopolysaccharide-treated RAW 264.7 cells. RAW 264.7 macrophages were treated with the indicated concentrations of L-4F and then stimulated with LPS. The levels of pSTAT3, pp38, pJNK and pERK were analyzed by western blotting, and the results are shown in (A–D) respectively (*P < 0.05, **P < 0.01). (E) Regulation mechanism of L-4F in the tumor microenvironment. The tumor microenvironment (TME) is a highly dynamic system that adopts a variety of activation states in response to inflammation. L-4F is a negative regulator of Th1 and Th17 cells as well as macrophages in the TME. Specifically, L-4F is a crucial suppressor of M2-type macrophage polarization and inhibits IFN-γ and IL-17A production in response to the TME. Mechanistically, L-4F suppresses the STAT3, ERK and JNK signaling pathways in macrophages. These pathways are critical for tumor progression and eventually lead to fatal outcomes.
DISCUSSION
It has been reported that L-4F can suppress tumor progression in ovarian cancer and colon cancer by reducing cell viability and proliferation [7, 8]. In addition, L-4F was shown to significantly reduce cell viability in the human ovarian cancer cell lines SKOV3, OV2008, and A2780 by binding and removing lysophosphatidic acid, which is a well-known activator of proliferation in cancer cells. Despite these promising results, however, L-4F was not effective in the OVCAR5 cell line [8]. These reports indicate that L-4F can suppress cancer growth, but not all tumor cells are sensitive to L-4F. In our in vivo study, we found that L-4F substantially reduced the tumorigenicity of H7 pancreatic tumors in a mouse model. However, our in vitro results demonstrated that L-4F could not suppress the invasion (Figure 2A) or proliferation (Figure 2B) capacity of H7 cells (or Panc1 cells, Supplementary Figure 1) and did not induce cell apoptosis in these cells (Figure 2C). Therefore, we conclude that L-4F has no significant direct anti-tumor effect in the mouse pancreatic cancer H7 cell line, leaving the biological basis underlying the ability of L-4F to help mice resist pancreatic cancer unresolved.
Inflammation, such as in chronic pancreatitis, has been shown to represent a major risk factor for pancreatic cancer [10]. Inflammation plays a critical role in tumorigenesis: it can increase the proliferation and viability of malignant cells, promote angiogenesis and metastasis, and subvert adaptive immune responses [11]. Cytokines derived from immune cells or cancer cells play important roles in tumor immunoediting. In particular, IL-6 has emerged as an important factor in the modulation of cancer-associated inflammation [12], and pancreatic cancer cells highly express this cytokine [13]. Pancreatic tumor cells also release the pro-inflammatory cytokines IL-1β and TNF-α, which play a critical role in the malignancy of pancreatic ductal adenocarcinoma (PDA) [14, 15]. Serum levels of GM-CSF in pancreatic cancer patients are also significantly higher than in healthy individuals [16], and increased expression of the inflammatory cytokines IFN-γ, CXCL1 and CXCL2 were observed in a mouse model of spontaneous pancreatic cancer [17].
Several studies have determined that L-4F possesses effective anti-inflammatory properties. For example, L-4F stimulates HDL anti-inflammatory activity and inhibits LDL pro-inflammatory activity in the plasma of patients with end-stage renal disease [4]. L-4F also significantly decreased serum interleukin (IL)-6, TNF-α and IL-1β in obese mice [18]. Moreover, L-4F was shown to inhibit LPS-induced inflammatory responses by reducing the synthesis of cytokines, chemokines, and adhesion molecules [5]; reduce pro-inflammatory gene expression in activated human coronary artery endothelial cells and macrophage foam cells; and prevent reactive oxygen species formation in activated neutrophils [19]. In this study, we investigated the anti-inflammatory effect of L-4F in a mouse pancreatic cancer model. Our results show that L-4F treatment significantly inhibited tumor progression (Figure 1A–1C), decreased inflammatory cell infiltration in tumor tissues (Figure 3D), and reduced percentages of IL-17A-, IL-6-, GM-CSF- and IL-1β-producing cells to varying degrees in tumor tissues (Figure 3A, 3B). Moreover, L-4F decreased mRNA levels of the inflammatory cytokines IL-17A, IFN-γ, IL-6, and IL-1β (Figure 3C). These results indicate that L-4F suppresses pancreatic cancer progression through inhibition of inflammation by decreasing the infiltration of pro-inflammatory and inflammatory cytokine-producing cells in mice with pancreatic cancer.
Th cells, including Th1, Th2, and Th17 cells, can exert both tumor-suppressive and tumor-promoting effects [20, 21]. There is evidence that the processes of tumor promotion, progression, and metastasis are governed by select Th cell subsets, including IFN-γ-producing Th1 cells [22], IL-4-producing Th2 cells [15, 23] and IL-17-producing Th17 cells [24]. In this study, we investigated changes in Th1, Th2 and Th17 cell subpopulations by detecting the cytokines IFN-γ, GM-CSF, IL-4, IL-6 and IL-17A in TILs. We found that only the percentages of IL-17A- and IFN-γ-producing Th cells decreased (Figure 4A, 4B). Therefore, we conclude that L-4F inhibited pancreatic cancer progression by decreasing the infiltration of Th17 and Th1 inflammatory cells, but not other Th cell subpopulations, in a mouse model of pancreatic cancer.
Th cells respond to inflammation to maximize cytotoxicity against cancer cells in association with macrophages [25]. In addition, macrophages play a key role in the immune response and are the main component of inflammatory cell populations in solid tumors. As such, they are key regulators of the link between inflammation and various types of cancer. Macrophage depletion has been shown to successfully limit tumor growth, metastasis and angiogenesis, thereby leading to a better response to conventional therapy in metastatic breast cancer and colitis-associated cancer [26, 27]. Macrophages are categorized into “classically activated” M1 and “alternatively activated” M2 subtypes. M1 macrophages express high levels of pro-inflammatory cytokines (TNF-α, IL-6, and IL-12) and are capable of killing pathogens and priming anti-tumor immune responses. By contrast, M2 or “alternatively” activated macrophages show increased expression of the anti-inflammatory cytokine IL-10 as well as scavenger receptor A and arginase. M2-polarized macrophages have been shown to be significantly more abundant in primary PDA samples [28], and the presence of M2-polarized macrophages in the stroma is strongly correlated with tumors located in the tail and body of the pancreas [29]. Most tumor-associated macrophages (TAMs) are considered to have an M2 phenotype and to promote tumor angiogenesis and tissue remodeling [30]. TAMs are also an important source of cytokines [29]. Functionally, TAMs promote tumor growth and may be obligatory for promoting angiogenesis, enhancing tumor cell migration and invasion, and suppressing anti-tumor immune responses [8]. TAMs are also immunosuppressive, preventing tumor cells from being attacked by natural killer and T cells during tumor progression and after recovery from chemo- or immunotherapy [31]. The balance between pro-inflammatory and anti-inflammatory cytokines in the tumor microenvironment finely regulates macrophage polarization and activity [32]. M1 macrophages are activated by IFN-γ and LPS, whereas M2 macrophages arise in response to IL-4, IL-10 and IL-17A [33, 34]. Furthermore, higher numbers of M2-polarized TAMs are associated with an increased risk of lymph node metastasis, neural invasion, and chemoresistance in PDA and hence a worse prognosis and reduced survival [35, 36].
Therefore, targeting TAMs provides an opportunity for prevention and treatment of pancreatic cancer [37]. In the present study, we elucidated the effect of L-4F on TAMs using a pancreatic mouse model. Our results show (Figure 4C, 4D) that L-4F treatment augmented CD11b+F4/80+ macrophage populations and significantly decreased CD11b+F4/80+CD206+ M2-macrophage populations. This finding was further demonstrated in the in vitro, where IL-4-induced M2 polarization was directly reversed by L-4F (Figure 5A). Furthermore, L-4F led to down-regulation of IL-17A and IL-4 (Figure 3), suggesting that, in addition to its direct anti-inflammatory effect, L-4F can change the phenotypes of TAMs in pancreatic tumors by altering the microenvironment.
Increasing evidence indicates that signal transducer and activator of transcription 3 (STAT3) and mitogen-activated protein kinase (MAPK) signaling pathways are closely associated with inflammatory processes and play major roles in inflammation and cancer [38, 39]. Moreover, the major pathways of macrophage polarization have been outlined and reviewed [40]. In brief, M1 macrophage polarization results from exposure to LPS and activation of NF-κB and STAT1. In contrast, STAT3 and MAPK activation results in M2 macrophage polarization, which is associated with immune suppression and tumor progression [41]. The MAPK family of proteins includes ERK, p38, and JNK [42]. Some studies have reported that ApoA-I can inhibit the LPS-induced activation of JNK in mouse macrophage RAW264.7 cells [43] and can inhibit the production of IFN-γ and IL-6 in mouse serum [44]. In addition, ApoA-I inhibited STAT3, which plays a major role in suppressing LPS-induced production of the inflammatory cytokine IL-6, in a mouse model of colitis-associated carcinogenesis [45]. Here, our results demonstrated that L-4F could not inhibited the IFN-γ producing cells in tumor tissues and L-4F inhibited phosphorylation of LPS-induced JNK in mouse RAW264.7 macrophages, but not inhibit p38MAPK. On the other hand, L-4F inhibited both the cytokines (IL-4 and IL-17A) producing cells in tumor tissues and the important signaling molecules (phosphorylation of ERK and STAT3). These results indicated that L-4F could inhibit all of the key regulators of M2 macrophage, but not M1 macrophage. STAT3 signaling in macrophage cells is the potential mechanism that L-4F regulation.
In conclusion, we have demonstrated that L-4F substantially inhibits pancreatic cancer progression mostly by exerting anti-inflammatory activity. Additionally, macrophage targeting is a key component of the anti-inflammatory and anti-tumor properties of L-4F. Notably, L-4F prevents M2 macrophage differentiation through inhibition of the STAT3 and MAPK pathways in macrophages. Thus, L-4F plays a critical role in regulating the tumor microenvironment (Figure 6E). The current results strengthen the rationale for future validation of ApoA-I-mimetic peptides (such as L-4F), which may serve as adjuvant therapeutic agents for the treatment of pancreatic cancer.
MATERIALS AND METHODS
Cell lines
The highly metastatic mouse pancreatic cancer H7 cell line was established using an in vivo selection method described by Wang et al. [46]. H7 and RAW 264.7 macrophages were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 100 mg/mL penicillin and 100 mg/mL streptomycin (Gibco) in a humidified atmosphere of 5% CO2 at 37°C.
Animals and tumor model
Six- to eight-week-old female C57BL/6 mice were purchased from the Academy of Military Medical Science of China and were acclimated for 7 days in the laboratory before experimentation. All of the experiments were performed in accordance with university guidelines for animal care and were approved by the Animal Ethics Committee of Tianjin Medical University.
Mouse abdomens were prepped with betadine solution, and an approximately 1-cm-wide incision was made in the left upper quadrant of each abdomen. The tip of the pancreatic tail was grasped gently, and the pancreas and spleen were externalized in a lateral direction to fully expose them. A needle was inserted into the tail of the pancreas and positioned in the pancreatic head region. A 27-gauge needle was then used to slowly inject 1×106 H7 cells suspended in 50 mL of PBS. The spleen was then returned to the appropriate position in the abdomen, and the skin and peritoneum were closed with 3–0 vicryl sutures. Subgroups of mice received either vehicle (50 mM ammonium bicarbonate, pH = 7.0, containing 0.1 mg/mL Tween-20) containing L-4F (the peptide Ac-D-W-F-K-A-F-Y-D-K-V-A-E-K-F-K-E-A-F-NH2 was synthesized from all L-amino acids, 10 mg/kg, n = 8) or vehicle containing scrambled peptide (Sc-4F) (containing the same amino acids as the 4F peptide but arranged in the sequence Ac-D-W-F-A-K-D-Y-F-K-K-A-F-V-E-E-F-A-K-NH2, 10 mg/kg, n = 8) by daily intraperitoneal injection beginning on day 3. All of the mice were euthanized after one week of treatment, and the tumor tissues were collected for further study.
Histological staining
Tumor tissues were embedded in paraffin after being fixed in 4% (weight/volume) paraformaldehyde and were cut into 8-μm-thick sections. Paraffin sections were stained with hematoxylin and eosin (H&E).
Isolation of immunocytes from tumor tissues
The tumor tissues were minced into small pieces and digested with 0.05 mg/mL each of type-IV collagenase, hyaluronidase and DNase I (Sigma) for 30 min at 37°C. Single-cell suspensions were obtained by grinding the digested tissues and filtering them through a 40-μm cell strainer (BD Biosciences). Immunocytes were isolated using Ficoll density gradient centrifugation [47].
Generation of macrophages and in vitro assays
Macrophages was generated as previously described [27]. Briefly, bone marrow cells were isolated from the femurs and tibias of C57BL/6 mice and then seeded in cell culture dishes and cultured with complete DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and 20 ng/mL recombinant mouse M-CSF (PeproTech) at 37°C in a CO2 incubator for 5 days to differentiate into macrophages. The cells were washed twice with phosphate-buffered saline (PBS) every other day, and fresh medium was added. On day 6, nonadherent cells were removed and adherent cells were cultured with 20 ng/mL mouse recombinant IL-4 (PeproTech) or 1 μg/mL LPS (Sigma) for 24 h, either with or without L-4F (0.25 μg/mL). The differentiated, adherent, live macrophage population was detached from the plate with a solution containing trypsin (Gibco), and the cells were processed for phenotypic characterization. Based on the specific expression of a number of surface markers, including CD11b, F4/80, MHC II and CD206, the cells were sorted using flow cytometry.
Flow cytometric analysis
The freshly isolated immunocytes were stained with antibodies for 30 min at 4°C, washed and analyzed on a BD FACSCalibur flow cytometer (BD Biosciences). The following monoclonal anti-mouse antibodies were used: anti-CD11b-FITC (or anti-CD11b-PE), anti-F4/80-APC, anti-MHC II-PE and anti-CD206-FITC (eBioscience).
For intracellular cytokine staining, the freshly isolated immunocytes were resuspended in complete RPMI 1640 medium (Gibco) supplemented with 2 mM L-glutamine, 100 IU penicillin, 100 mg/mL streptomycin, and 4.5 × 10–5 M 2-mercaptoethanol. The cells were stimulated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA), 1 μg/mL ionomycin (Enzo Life Sciences, Farmingdale, USA) and 3 μg/mL brefeldin A (eBioscience) for 5 h. Then, the cells were fixed and permeabilized with Cytofix/Cytoperm buffers for 20 min at 4°C and washed with permeabilization wash buffer (BD Biosciences). Fc receptors were blocked with 2% rat serum and 10% bovine serum albumin prior to intracellular cytokine staining. Next, the cells were stained with rat anti-mouse IFN-γ-PE, IL-17A-PE, IL-6-FITC, IL-4-FITC, GM-CSF-PE and IL-1β-Percp (eBioscience). Finally, the cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Controls for nonspecific staining were monitored using isotype-matched mAbs, and nonspecific staining was always subtracted from the specific staining results. The acquired data were analyzed using CellQuest and WinMDI 2.9 software.
Quantitative real-time PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). The RNA was converted into cDNA with random hexamers and M-MLV reverse transcriptase (Invitrogen). GAPDH was used as an internal control. The sequences of the primers used for qPCR are as follows: IL-17A, (F) 5′-CTACCTCAACCGTTCCAC-3′ and (R) 5′-CACCCACCAGCATCTTCT-3′); IFN-γ, (F) 5′-ATGAACGCTACACACTGCATC-3′ and (R) 5′-CCATCCTTTTGCCAGTTCCTC-3′); IL-6, (F) 5′-TA GTCCTTCCTACCCCAATTTCC-3′ and (R) 5′-TTGGTC CTTAGCCACTCCTTC-3′); IL-1β, (F) 5′-GCAACTGT TCCTGAACTCAACT-3′ and (R) 5′-ATCTTTTGGGGT CCGTCAACT-3′); Argl, (F) 5′-CTCCAAGCCAAAGT CCTTAGAG-3′ and (R) 5′-AGGAGCTGTCATTAGGG ACATC-3′); MRC1, (F) 5′-CTCTGTTCAGCTATTGG ACGC-3′ and (R) 5′-CGGAATTTCTGGGATTCAGC TTC-3′); CCL22, (F) 5′-CTCTGCCATCACGTTTAGTGA A-3′ and (R) 5′-GACGGTTATCAAAACAACGCC-3′); iNOS, (F) 5′-GTTCTCAGCCCAACAATACAAGA-3′ and (R) 5′-GTGGACGGGTCGATGTCAC-3′); IL-12, (F) 5′-ACTCTGCGCCAGAAACCTC-3′ and (R) 5′-CACCCTGTTGATGGTCACGAC-3′); and GAPDH, (F) 5′-AGGTCGGTGTGAACGGATTTG-3′ and (R) 5′-TGTAGACCATGTAGTTGAGGTCA-3′). Quantitative real-time PCR was performed with SYBR Green mix (Newbio), and the data are displayed as 2-ΔCt values and are representative of at least three independent experiments.
Western blotting
RAW 264.7 macrophages were plated in 6-well plates (4 × 105/well) and cultured in 2 mL of DMEM for 4 h. The cultures were washed to remove non-adherent cells and then incubated with 2 mL of complete DMEM for 20 h. The culture medium was replaced with DMEM for 30 min to allow the cells to adjust. (i) To induce an inflammation model, 1 μg/mL LPS (Sigma) was added. After 24 h of stimulation with LPS, the cells were treated with the indicated concentrations of L-4F (0, 0.1, or 0.25 μg/mL) for 12 h, and the levels of pp38, pJNK and pERK were analyzed. (ii) Cells were stimulated by LPS and treated with the indicated concentrations of L-4F (0, 0.1, or 0.25 μg/mL) for 1 h, and the levels of pSTAT3 were analyzed.
Western blot procedure: Briefly, cells were lysed in radio-immunoprecipitation assay buffer containing the phosphatase and protease inhibitors phenylmethanesulfonyl fluoride and aprotinin (Sigma), and protein was collected. Then, the protein was separated by SDS-PAGE, transferred to PVDF membranes (Roche) and probed with the indicated primary antibodies (phosphorylated STAT3, phosphorylated p38, phosphorylated JNK and phosphorylated ERK; Cell Signaling Technology). The antibody-antigen complexes were detected using a Chemiluminescent HRP substrate kit (Millipore) according to the manufacturer’s protocols.
Wound healing assay
H7 cells were seeded into 6-well plates and cultured in complete DMEM. Upon reaching approximately 70% confluence, the medium was replaced with serum-free medium. Following an overnight incubation, the cells reached at least 95% confluence, forming a confluent monolayer. A linear scratch was made using a 10-μL micropipette tip. The cells were further cultured in DMEM containing 2% FBS and the indicated concentrations of L-4F (0, 5, 10, or 20 μg/mL). Wound width was photographed using light microscopy (40×) at 0, 24 and 48 h. For evaluation of wound closure, three randomly selected points along each wound were marked. Each experiment was performed in triplicate. The measurements were obtained by measuring the distance between the wound edges using Image J software.
Cell proliferation assay
Cells were starved overnight, and cell proliferation (cell division) was assayed using carboxyfluorescein succinimidyl ester (CFSE) labeling (Invitrogen). Briefly, the cells were trypsinized, washed twice in sterile PBS and resuspended in 2 mL of DMEM. CFSE was applied at a final concentration of 5 μM, and the cells were incubated at 37°C for 10 min in the dark. To quench the labeling, the cells were washed twice with complete DMEM. The labeled cells were treated with vehicle or the indicated concentrations of L-4F (5, 10, or 20 μg/mL) for 48 h in complete DMEM. Next, the cells were trypsinized and washed in PBS. Finally, the cells were assessed for labeling using a FACSCalibur flow cytometer. The acquired data were analyzed using CellQuest and Modfit software (BD Biosciences).
Cellular apoptosis assay
Cells were treated with vehicle or the indicated concentrations of L-4F (5, 10, or 20 μg/mL) in complete medium for 48 h. After the indicated treatments, apoptosis was assessed using an Annexin V/PI assay kit (Invitrogen) according to the manufacturer’s protocol. The acquired data were analyzed using CellQuest software.
Statistical analysis
All of the results were derived from at least three independent experiments. The data represent the mean values ± the standard deviation (SD). Comparisons between two groups were performed using Student’s unpaired t-test or one-way analysis of variance for comparison of three or more groups as indicated. Statistical analysis was performed using SPSS 10.0 software. P < 0.05 was considered statistically significant, and P ≥ 0.05 was not statistically significant (NS).
Abbreviations
ApoA-I : Apolipoprotein A-I; HDL: high-density lipoprotein; DMEM: Dulbecco’s modified Eagle’s medium; FBS: fetal bovine serum; H&E: hematoxylin and eosin; PBS: phosphate-buffered saline; PMA: phorbol 12-myristate 13-acetate; GM-CSF: granulocyte macrophage colony stimulating factor; Th cells: T helper cells; TILs: tumor-infiltrating lymphocytes; Mrc1: mannose receptor C type 1; PDA: pancreatic ductal adenocarcinoma; TAMs: tumor-associated macrophages; STAT3: signal transducer and activator of transcription 3; MAPK: mitogen-activated protein kinase;
ACKNOWLEDGMENTS
Not applicable.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
GRANT SUPPORT
This work was supported by the National Natural Science Foundation of China through grant nos. 81272317, 81602496 and 81502469 and the Science Foundation of Shangdong Province through grants no. ZR2015HL061, funding from the public school teachers domestic visits program of Weifang Medical University, Province Health Department through grants no. 2013WS0287.
REFERENCES
1. Almhanna K, Philip PA. Defining new paradigms for the treatment of pancreatic cancer. Curr Treat Options Oncol. 2011; 12:111–125.
2. Tardif JC, Heinonen T, Noble S. High-density lipoprotein/apolipoprotein A-I infusion therapy. Curr Atheroscler Rep. 2009; 11:58–63.
3. Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye KA, Chin-Dusting J, Hoang A, Sviridov D, Celermajer DS, Kingwell BA. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol. 2009; 53:962–971.
4. Vaziri ND, Moradi H, Pahl MV, Fogelman AM, Navab M. In vitro stimulation of HDL anti-inflammatory activity and inhibition of LDL pro-inflammatory activity in the plasma of patients with end-stage renal disease by an apoA-1 mimetic peptide. Kidney Int. 2009; 76:437–444.
5. Gupta H, Dai L, Datta G, Garber DW, Grenett H, Li Y, Mishra V, Palgunachari MN, Handattu S, Gianturco SH, Bradley WA, Anantharamaiah GM, White CR. Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein AI mimetic peptide. Circ Res. 2005; 97:236–243.
6. Gao F, Chattopadhyay A, Navab M, Grijalva V, Su F, Fogelman AM, Reddy ST, Farias-Eisner R. Apolipoprotein A-I mimetic peptides inhibit expression and activity of hypoxia-inducible factor-1alpha in human ovarian cancer cell lines and a mouse ovarian cancer model. J Pharmacol Exp Ther. 2012; 342:255–262.
7. Su F, Grijalva V, Navab K, Ganapathy E, Meriwether D, Imaizumi S, Navab M, Fogelman AM, Reddy ST, Farias-Eisner R. HDL mimetics inhibit tumor development in both induced and spontaneous mouse models of colon cancer. Mol Cancer Ther. 2012; 11:1311–1319.
8. Su F, Kozak KR, Imaizumi S, Gao F, Amneus MW, Grijalva V, Ng C, Wagner A, Hough G, Farias-Eisner G, Anantharamaiah GM, Van Lenten BJ, Navab M, et al. Apolipoprotein A-I (apoA-I) and apoA-I mimetic peptides inhibit tumor development in a mouse model of ovarian cancer. Proc Natl Acad Sci U S A. 2010; 107:19997–20002.
9. Sharifov OF, Xu X, Gaggar A, Tabengwa EM, White CR, Palgunachari MN, Anantharamaiah GM, Gupta H. L-4F inhibits lipopolysaccharide-mediated activation of primary human neutrophils. Inflammation. 2014; 37:1401–1412.
10. Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andrén-Sandberg A, Domellöf L, and International Pancreatitis Study Group. Pancreatitis and the risk of pancreatic cancer. N Engl J Med. 1993; 328:1433–37.
11. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454:436–444.
12. Guthrie GJ, Roxburgh CS, Horgan PG, McMillan DC. Does interleukin-6 link explain the link between tumour necrosis, local and systemic inflammatory responses and outcome in patients with colorectal cancer? Cancer Treat Rev. 2013; 39:89–96.
13. Feurino LW, Zhang Y, Bharadwaj U, Zhang R, Li F, Fisher WE, Brunicardi FC, Chen C, Yao Q, Min L. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol Ther. 2007; 6:1096–1100.
14. Egberts JH, Cloosters V, Noack A, Schniewind B, Thon L, Klose S, Kettler B, von Forstner C, Kneitz C, Tepel J, Adam D, Wajant H, Kalthoff H, et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008; 68:1443–1450.
15. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011; 208:469–478.
16. Vasiliades G, Kopanakis N, Vasiloglou M, Zografos G, Margaris H, Masselou K, Kokosi E, Liakakos T. Role of the hematopoietic cytokines SCF, IL-3, GM-CSF and M-CSF in the diagnosis of pancreatic and ampullary cancer. Int J Biol Markers. 2012; 27:e186-e194.
17. Rachagani S, Torres MP, Kumar S, Haridas D, Baine M, Macha MA, Kaur S, Ponnusamy MP, Dey P, Seshacharyulu P, Johansson SL, Jain M, Wagner KU, et al. Mucin (Muc) expression during pancreatic cancer progression in spontaneous mouse model: potential implications for diagnosis and therapy. J Hematol Oncol. 2012; 5:68.
18. Peterson SJ, Drummond G, Kim DH, Li M, Kruger AL, Ikehara S, Abraham NG. L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res. 2008; 49:1658–1669.
19. Nguyen SD, Maaninka K, Lappalainen J, Nurmi K, Metso J, Oorni K, Navab M, Fogelman AM, Jauhiainen M, Lee-Rueckert M, Kovanen PT. Carboxyl-Terminal Cleavage of Apolipoprotein A-I by Human Mast Cell Chymase Impairs Its Anti-Inflammatory Properties. Arterioscler Thromb Vasc Biol. 2016; 36:274–284.
20. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007; 450:903–907.
21. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007; 117:1137–1146.
22. Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, Minoda Y, Sanada T, Yoshioka T, Mimata H, Kato S, Yoshimura A. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006; 203:1391–1397.
23. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009; 16:91–102.
24. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009; 206:1457–1464.
25. Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016; 44:450–462.
26. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011; 475:222–225.
27. Wu T, Dai Y, Wang W, Teng G, Jiao H, Shuai X, Zhang R, Zhao P, Qiao L. Macrophage targeting contributes to the inhibitory effects of embelin on colitis-associated cancer. Oncotarget. 2016; 7:19548–58. https://doi.org/10.18632/oncotarget.6969.
28. Hu H, Hang JJ, Han T, Zhuo M, Jiao F, Wang LW. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol. 2016; 37:8657–8664.
29. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883–899.
30. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008; 267:204–215.
31. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013; 339:286–291.
32. Rhee I. Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res. 2016; 39:1588–1596.
33. Li Q, Liu L, Zhang Q, Liu S, Ge D, You Z. Interleukin-17 Indirectly Promotes M2 Macrophage Differentiation through Stimulation of COX-2/PGE2 Pathway in the Cancer Cells. Cancer Res Treat. 2014; 46:297–306.
34. Nishikawa K, Seo N, Torii M, Ma N, Muraoka D, Tawara I, Masuya M, Tanaka K, Takei Y, Shiku H, Katayama N, Kato T. Interleukin-17 induces an atypical M2-like macrophage subpopulation that regulates intestinal inflammation. PLoS One. 2014; 9:e108494.
35. Meng F, Li C, Li W, Gao Z, Guo K, Song S. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life. 2014; 66:835–846.
36. Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011; 167:e211–19.
37. Cui R, Yue W, Lattime EC, Stein MN, Xu Q, Tan XL. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget. 2016; 7:50735–54. https://doi.org/10.18632/oncotarget.9383.
38. Kashyap D, Kumar G, Sharma A, Sak K, Tuli HS, Mukherjee TK. Mechanistic insight into carnosol-mediated pharmacological effects: Recent trends and advancements. Life Sci. 2017; 169:27–36.
39. Steele CW, Kaur GN, Jamieson NB, Carter CR. Targeting inflammation in pancreatic cancer: Clinical translation. World J Gastrointest Oncol. 2016; 8:380–388.
40. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012; 122:787–795.
41. Agoro R, Mura C. Inflammation-induced up-regulation of hepcidin and down-regulation of ferroportin transcription are dependent on macrophage polarization. Blood Cells Mol Dis. 2016; 61:16–25.
42. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003; 21:335–376.
43. Wang Y, Lu S, Li X, Du N, Sun Y, Xing J, Pan X, Chen B, Miao Z. The lipid-bound apolipoprotein A-I cysteine mutant (N74C) inhibits the activation of NF-kappaB, JNK and p38 in endotoxemic mice and RAW264.7 cells. PLoS One. 2012; 7:e51327.
44. Sharifov OF, Xu X, Gaggar A, Grizzle WE, Mishra VK, Honavar J, Litovsky SH, Palgunachari MN, White CR, Anantharamaiah GM, Gupta H. Anti-inflammatory mechanisms of apolipoprotein A-I mimetic peptide in acute respiratory distress syndrome secondary to sepsis. PLoS One. 2013; 8:e64486.
45. Gkouskou KK, Ioannou M, Pavlopoulos GA, Georgila K, Siganou A, Nikolaidis G, Kanellis DC, Moore S, Papadakis KA, Kardassis D, Iliopoulos I, McDyer FA, Drakos E, et al. Apolipoprotein A-I inhibits experimental colitis and colitis-propelled carcinogenesis. Oncogene. 2016; 35:2496–2505.
46. Wang B, Shi Q, Abbruzzese J, Xiong Q, Le X, Xie K. A Novel, Clinically Relevant Animal Model of Metastatic Pancreatic Adenocarcinoma Biology and Therapy. Int J Gastrointest Cancer. 2001; 29:37–46.
47. Peng M, Huang B, Zhang Q, Fu S, Wang D, Cheng X, Wu X, Xue Z, Zhang L, Zhang D, Da Y, Dai Y, Yang Q, et al. Embelin inhibits pancreatic cancer progression by directly inducing cancer cell apoptosis and indirectly restricting IL-6 associated inflammatory and immune suppressive cells. Cancer Lett. 2014; 354:407–416.