
INTRODUCTION
Small cell lung cancer (SCLC) is a clinically aggressive subtype of lung cancer, which accounts for 10~15% of all lung cancers [1]. SCLC patients, who are diagnosed with extensive disease (i.e. spreading of cancer beyond the supraclavicular areas, or with distant metastasis) are treated with standard platinum-based chemotherapy plus etoposide. Although the initial response rate is quite high, almost all patients are bound to relapse within 3-6 months [2]. SCLC patients with extensive diseases have a median survival of less than 1 year [3]. Unlike non-small cell lung cancer (NSCLC), there is no targeted therapy being clinically approved for the treatment of SCLC.
The rare occurrences of druggable oncogenes or kinases make SCLC difficult to be harnessed for precision treatment. This is in contrast with non-small lung cancer (NSCLC), with which multiple druggable kinases are readily drugged in specific subgroups of NSCLC patients in a personalized manner. This includes Epidermal Growth Factor Receptor (EGFR) inhibitors for EGFR-mutated NSCLC and Anaplastic Lymphoma Kinase (ALK) inhibitors for ALK-rearranged NSCLC [4, 5]. In SCLC, the most frequently mutated genes are the non-druggable tumor suppressors, such as TP53 and RB1[6]. Yet, amplification or overexpression of non-kinase oncogenes such as MYC family genes and Bcl-2 are very frequent events. In fact, Bcl-2 is over-expressed in 60%-90% of SCLC [7], while MYC family genes are amplified in 20%-30% of cases [6, 8]. Though these two obvious targets are common in SCLC, co-targeting of Bcl-2 and MYC pathway has not been explored previously.
Bcl-2 homology domains 3 (BH3) mimetics mimick the BH3 death domains and trigger apoptosis. They are an effective class of Bcl-2 inhibitor. A new generation of BH3 mimetic, ABT-263 directly binds Bcl-2 to block its interaction with Bim, thereby enabling Bim-mediated induction of apoptosis [9]. In a pan-cancer cell line study, ABT-263 has been shown to be more effective in inhibiting SCLC and hematologic malignancies, compared to other tumor types [7]. Further, a large scale drug screening study of Cancer Cell Line Encyclopedia (CCLE) also revealed a relatively lower IC50 values of ABT-263 in SCLC and hematologic cancer among other cancers [10].However, early phases of clinical trials showed that ABT-263 demonstrated unimpressive clinical activities in 26 SCLC patients, with only one patient having a partial response, while 9 patients had stable disease and 16 patients had disease progression [11]. These clinical findings revealed the limitation of ABT-263 monotherapy for SCLC.
Previous studies showed that cancer cells with high Bim/Mcl-1 ratios were sensitive to ABT-263 treatment [10]. Theoretically, the anti-tumor activity of ABT-263 can be enhanced by increasing Bim protein expression or reducing Mcl-1 expression. Recent study has shown that the target of rapamycin complex 1/2 (TORC1/2) inhibitor AZD8055, which reduces Mcl-1 protein level, is able to remarkably enhance antitumor activity of ABT-263 in SCLC [10]. But patients treated with TORC1/2 inhibitors were usually rash, mucositis, and fatigue. And based on previous experience, hyperglycemia was associated with mTOR inhibitors [12]. Therefore, we put forward a hypothesis that Bim induction by pharmacological approaches can potentially enhance ABT-263 activity in SCLC.
MYC family genes (MYC, MYCN and MYCL) are frequently amplified in SCLC [8]. JQ1, a Bromodomain and Extra-Terminal domain (BET) inhibitor can inhibit MYC transcription by disrupting the interaction of BET proteins and MYC promoter [13]. Furthermore, previous studies have demonstrated that MYC inactivation is able to increase Bim expression in lymphoma system [14]. Here, we hypothesized that targeting of MYC family oncogenes by JQ1 would induce Bim up-regulation in SCLC and results in sensitization to ABT-263. Our findings showed that MYCN-amplified SCLC cell lines were sensitive to JQ1. JQ1 induced Bim protein up-regulation, which sensitized MYCN-amplified SCLC cells to ABT-263. Co-treatment with ABT-263 and JQ1 effectively disrupted the interaction of Bim with Bcl-2 and prevented Bim’s interaction with Mcl-1. Further, MYCN-amplified SCLC xenografts are exquisitely sensitive to this JQ-1/ABT-263 combination. Our findings reveal a novel co-targeting strategy specifically for MYCN-amplified SCLC.
RESULTS
MYCN-amplified SCLC cell lines are sensitive to JQ1
Nearly 20%-30% of SCLC tumors harbor amplification of MYC family genes [6]. We firstly detected the expression of c-Myc and N-Myc proteins in seven SCLC cell lines. As shown in Figure 1A, N-Myc was highly expressed in H526 and H69 cell lines, which have MYCN gene amplification [15] and c-Myc was highly expressed in H82 SCLC cell line that harbors MYC gene amplification [15] (Figure 1A). All 7 SCLC cell lines were then subjected to JQ1 treatment for 72 hours. As shown in Figure 1B and Table 1, we found that the two cell lines that have N-Myc protein overexpression, H526 and H69, were the most JQ1-sensitive lines with the lowest IC50 (236.1 nM and 667.1 nM). The c-Myc overexpressed H82 line was moderate sensitive to JQ1 (IC50: 1.01 μM), while the remaining 4 SCLC cell lines were the least sensitive ones with higher IC50 values of >8 μM). Our results indicate that MYCN-amplified SCLC cell lines are highly sensitive to JQ1.
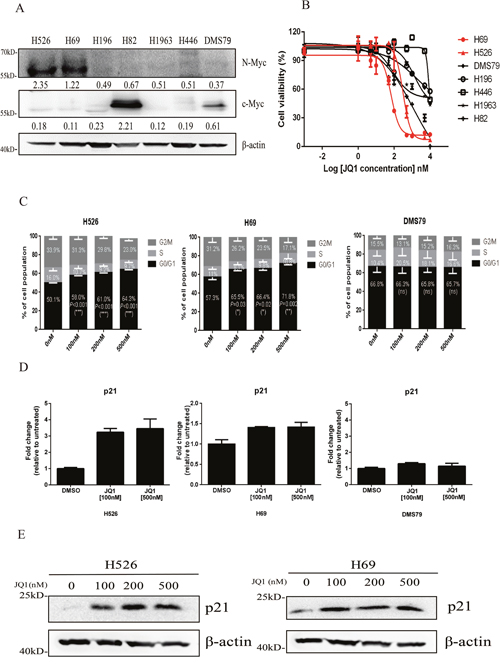
Figure 1: (A) Protein expression of c-Myc and N-Myc in a panel of SCLC cell lines by western blotting analysis. β-Actin was used as a loading control. Protein band intensities were quantified by ImageJ and normalized to Actin. (B) Growth inhibition curves of JQ1 in a panel of SCLC cell lines. SCLC cells were treated with different concentrations of JQ1 for 72 hours. CellTiter-Glo Luminescent assay was performed to evaluate the cell proliferation. Red curves represent MYCN-amplified cell lines, which are sensitive to JQ1. (C) Cell cycle distributions of H526, H69 and DMS79 cells treated with DMSO control or different concentrations of JQ1. Average percentage of cell population in different phases of cell cycle from 3 independent experiments was shown. Statistical significance of JQ1 treatment vs. DMSO control was indicated. (D) JQ1 induces p21 mRNA expression in MYCN-amplified SCLC cells. H526, H69 and DMS79 cells were treated with JQ1 or DMSO for 24 hours. Relative mRNA expression level of p21 was detected by quantitative RT-PCR. (E) JQ1 induces p21 protein expression in MYCN-amplified SCLC cells. H526 and H69 cells were treated with JQ1 or DMSO for 24 hours. Protein expression of p21 was detected by western blotting analysis. β-Actin was used as a loading control.
Table 1: Cells sensitivities to JQ1 as well as basal levels of c-Myc and N-Myc expression in SCLC cell lines
SCLC cell lines |
|||||||
|---|---|---|---|---|---|---|---|
High N-Myc |
High c-Myc |
Low c-Myc and N-Myc |
|||||
H526 |
H69 |
H82 |
DMS79 |
H196 |
H1963 |
H446 |
|
JQ1 IC50 (nM) |
236.1 |
667.1 |
1012 |
30383 |
26000 |
103755 |
8649 |
N-MYC expression |
2.35 |
1.22 |
0.67 |
0.37 |
0.49 |
0.51 |
0.51 |
C-MYC expression |
0.18 |
0.11 |
2.21 |
0.61 |
0.23 |
0.12 |
0.19 |
Previous studies have reported that JQ1 inhibits cancer cell growth through induction of cell cycle arrest in multiple cancer types, including leukemia, ovarian cancer and oral squamous cell carcinoma [16–19]. Therefore, we examined the effects of JQ1 on cell cycle progression in SCLC. As shown in Figure 1C and Supplementary Figure 1, in JQ1 sensitive SCLC cell line H526 and H69, JQ1 treatment significantly resulted in G1 arrest, with concomitant reduction in S phase population, when compared to DMSO control treatment. However, in JQ1 insensitive SCLC cell line DMS79, JQ1 did not induce any G1 changes. Since p21 protein is a key regulator of G1 checkpoint [20], we next examined if JQ1-induced G1 arrest was associated with p21 induction. As shown in Figure 1D, JQ1 treatment remarkably induced p21 mRNA expression in JQ1 sensitive cell line H526 and H69, but not in JQ1 insensitive cell line DMS79. Western blotting confirmed that p21 protein was induced by JQ1 treatment in H526 and H69 cells (Figure 1E). These results indicate that JQ1 induces growth inhibition of MYCN-amplified SCLC cells via G1 arrest.
JQ1 increases Bim expression through suppressing N-Myc
Previous study has shown that JQ1 is able to reduce the expression of c-Myc in multiple myeloma models [17] and inhibition of c-Myc can induce Bim expression in lymphoma models [14]. Next, we determined if JQ1 was able to increase Bim expression through suppressing N-Myc and c-Myc in SCLC. As shown in Figure 2A, JQ1 decreased N-Myc and c-Myc expression in MYCN-amplified H526 and H69 cells as well as MYC-amplified H82 cells. Further, using quantitative RT-PCR, we confirmed that JQ1 treatment was able to induce Bim mRNA levels in a dose dependent manner in H526 and H69 cells (Figure 2B). This is further confirmed at the protein levels with western blotting that Bim protein expression was induced by JQ1 in both H526 and H69 cells (Figure 2C). On the contrary, in H82 cells, JQ1 treatment did not increase Bim expression in mRNA level, with only a very slight increase in Bim protein level (Figure 2B and 2C). In order to investigate whether Bim up-regulation was mediated via N-Myc down-regulation, we knocked down N-Myc expression with two N-Myc specific siRNAs #1 and #2. As shown in Figure 2D, knockdown of N-Myc increased Bim expression in H69 cells. These data demonstrate that JQ1 specifically increases Bim expression through suppression of N-Myc in MYCN-amplified SCLC.
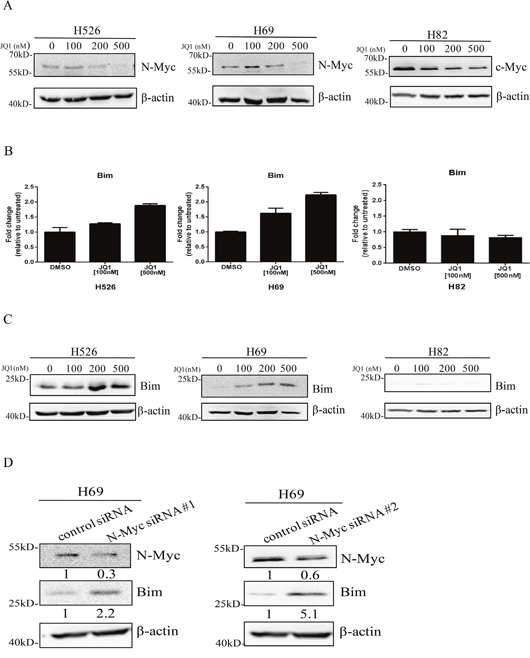
Figure 2: (A) JQ1 inhibits N-Myc and c-Myc expression in MYCN and MYC-amplified SCLC cells. H526, H69 and H82 cells were treated with JQ1 or DMSO for 24 hours. Protein expression of N-Myc and c-Myc was detected by western blotting. β-Actin was used as a loading control. (B) JQ1 induces mRNA expression of Bim in MYCN-amplified SCLC cells. H526, H69 and H82 cells were treated with JQ1 or DMSO for 24 hours. Relative mRNA level of Bim was detected by quantitative RT-PCR. (C) JQ1 induces protein expression of Bim in MYCN-amplified SCLC cells. H526, H69 and H82 cells were treated with JQ1 or DMSO for 24 hours. Protein expression of Bim was detected by western blotting. β-Actin was used as a loading control. (D) Knock-down of N-Myc increases Bim expression. H69 cells were treated with N-Myc siRNA #1, N-Myc siRNA #2 or their control siRNA for 48 hours, and then western blotting was performed to detect the expression levels of N-Myc and Bim. β-Actin was used as a loading control. The density of the bands was quantified by ImageJ and normalized to Actin.
Bim up-regulation by JQ1 sensitizes MYCN-amplified SCLC cells to ABT-263
As we hypothesized that Bim up-regulation by JQ1 sensitizes SCLC cells to ABT-263, we thereby examined whether combination treatment of JQ1 and ABT-263 exerted synergistic activity in inhibiting SCLC cell growth. We firstly tested the antitumor ability of ABT-263 in 7 SCLC cell lines. As shown in Supplementary Figure 2, SCLC cells exhibited different responses to ABT-263. DMS79, H69 and H1963 were sensitive to ABT-263 (IC50<1μM), while H526, H82, H196 and H446 were insensitive to ABT-263 (IC50>1μM). MYCN- or MYC-amplified H526, H69 and H82 cell lines were then subjected to JQ1/ABT-263 combination treatment. Our findings indicated that co-treatment of JQ1 and ABT-263 dramatically inhibited proliferation of MYCN-amplified H526 and H69 cell lines (Figure 3A), but not that of the MYC-amplified H82 cell line (Supplementary Figure 3). Furthermore, by calculating the combination index (CI) for synergism, we concluded that JQ1 and ABT-263 co-treatment exerted very strong synergistic anti-proliferative effects (CI<0.1) at all concentrations tested in H526 cells (Table 2). Next, we examined if this drug combination also enhanced apoptosis in MYCN-amplified H526 and H69 cells. We detected PARP cleavage (apoptotic marker) when H526 and H69 cells were treated with JQ1, ABT-263 or the combination. As shown in Figure 3B, the combination induced higher levels of cleaved PARP in H526 and H69 cells, when compared with either JQ1 or ABT-263 treatment alone.
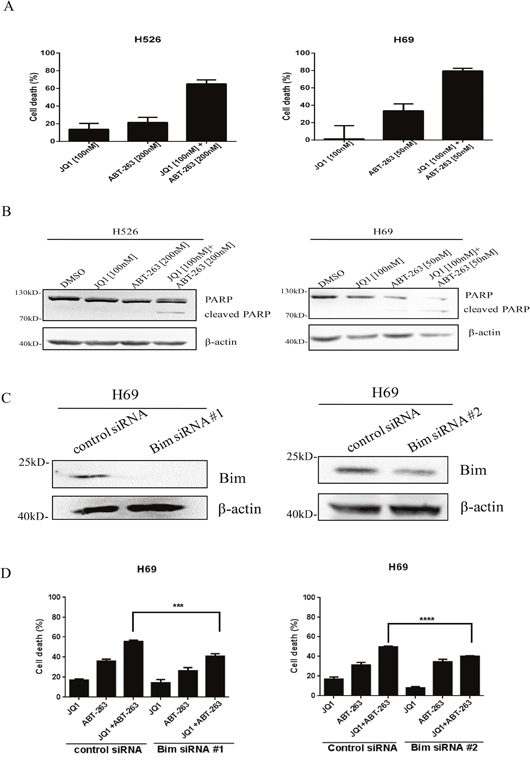
Figure 3: (A) Dramatical growth inhibition of MYCN-amplified SCLC cells induced by co-treatment of JQ1 and ABT-263. H526 and H69 cells were treated with DMSO control, JQ1, ABT-263 or the combination of JQ1 and ABT-263 for 72 hours. After treatment, growth inhibition was determined by CellTiter-Glo Luminescent assay. (B) JQ1 and ABT-263 co-treatment dramatically induces apoptosis in MYCN-amplified SCLC cells. H526 and H69 cells were treated with DMSO control, JQ1, ABT-263, or the combination of JQ1 and ABT-263 for 24 hours. PARP cleavage was detected by western blotting. β-Actin was used as a loading control. (C) Knock-down of Bim is confirmed by western blotting. After H69 cells were transfected by Bim siRNA #1, Bim siRNA #2 and control siRNA for 48 hours, cellular proteins were collected, and then Bim was detected by western blotting. (D) Knockdown of Bim significantly decreases the growth inhibition of H69 cells treated with combination of JQ1 and ABT-263. H69 cells were transiently transfected with Bim siRNA #1, Bim siRNA #2 and control siRNA for 24 hours. Then, transfected cells were plated in 96-well plate, and incubated with DMSO, JQ1 (100 nM), ABT-263 (50 nM) or the combination of JQ1 (100 nM) and ABT-263 (50 nM) for 72 hours. Cell proliferation was evaluated by CellTiter-Glo Luminescent assay.
Table 2: Combination index (CI) of JQ1 and ABT-263 in H526 cells
JQ1 dose (nM) |
ABT-263 dose (nM) |
Mean growth inhibition (%) |
Dose of JQ1 alone with same inhibition (nM) |
Dose of ABT-263 alone with same inhibition (nM) |
CI |
|---|---|---|---|---|---|
50 |
10 |
63.9 |
575 |
2454 |
0.004 |
50 |
100 |
78.6 |
1621 |
5248 |
0.049 |
100 |
200 |
87.8 |
1698 |
6025 |
0.092 |
200 |
50 |
94.7 |
2238 |
8511 |
0.094 |
200 |
200 |
99.8 |
10964 |
11220 |
0.035 |
In order to determine whether JQ1 sensitize SCLC cells to ABT-263 was through Bim up-regulation, we knocked down Bim expression by two Bim siRNA #1 and #2. As shown in Figure 3C, Bim was effectively knocked down by both Bim siRNA treatment in H69 cells. Upon Bim knockdown, the inhibition of cell growth by JQ1 and ABT-263 co-treatment was significantly reduced in H69 cells (vs. control siRNA transfection) (Figure 3D). These results demonstrate that JQ1 sensitizes MYCN-amplified SCLC cells to ABT-263 via Bim up-regulation.
Combination of JQ1 and ABT-263 disrupts the interaction of Bim/Bcl-2 and prevents Bim’s interaction with Mcl-1 in MYCN-amplifed SCLC cells
Anti-apoptotic proteins Bcl-2 and Mcl-1 can block apoptosis by interacting with the pro-apoptotic protein Bim. Previously, ABT-263 has been shown to block the binding of Bcl-2 to Bim, but does not block the binding of Mcl-1 to Bim [10]. To indicate whether combination treatment of JQ1 and ABT-263 is able to disrupt Bcl-2/Bim or Mcl-1/Bim interaction, we performed immunoprecipitation assay. As shown in Figure 4, ABT-263 treatment alone disrupted the interaction of Bim and Bcl-2. Surprisingly, we found that an even more substantial disruption of the Bcl-2/Bim complex was observed with the JQ1/ABT-263 combination treatment. Consistent with previous study [10], we found that ABT-263 treatment disrupted the interaction of Bim with Bcl-2, but the released Bim from Bcl-2/Bim complex was captured by Mcl-1 (Figure 4). Interestingly, combination treatment of JQ1 and ABT-263 prevented the released Bim from binding to Mcl-1. Our data suggest that the interaction of Bim with Bcl-2 or Mcl-1 is strongly disturbed by JQ1 and ABT-263 co-treatment, resulting in more Bim to induce apoptosis.

Figure 4: Combination treatment of JQ1 and ABT-263 strongly disrupts the interaction of Bim with Bcl-2 and Mcl-1. H526 cells were treated with DMSO control, JQ1 (100 nM), ABT-263 (200 nM), JQ1 (100 nM) plus ABT-263 (200 nM) for 24 hours. After treatment, Cells were lysed in immunoprecipitation lysis buffer. Cellular extracts were immunoprecipitated with 1 μg anti-Bim antibody. Precipitates were analyzed by western blotting to detect Bim, Mcl-1 and Bcl-2 proteins. The total lysates were also subjected to western blotting to detect the expression of Bim, Mcl-1 and Bcl-2 proteins. β-Actin was used as a loading control. Protein band intensities were quantified by ImageJ.
JQ1/ABT-263 combination substantially inhibits SCLC tumor growth in vivo
To evaluate the synergistic antitumor activity of JQ1 and ABT-263 in vivo, we established a human tumor xenograft model by subcutaneously injecting MYCN-amplified H526 SCLC cells into nude mice. Tumor-bearing mice were intraperitoneally treated with vehicle control, JQ1, ABT-263, or the combination of JQ1 and ABT-263 every two days for 14 days. As shown in Figure 5A, 5B and 5C, combination treatment with JQ1 and ABT-263 significantly inhibited tumor growth of H526 xenografts as compared with vehicle-treated control, but treatment with JQ1 or ABT-263 alone did not inhibit tumor growth. H&E staining of tumor sections revealed that combination treatment of JQ1 and ABT-263 decreased tumor cell density (Figure 5D). Ki67 expression was found to be strongly decreased in tumors treated by JQ1 plus ABT-263, whereas apoptosis (cleaved Caspase 3 staining) was increased in the tumors treated with the JQ1/ABT-263 combination (Figure 5D). No significant difference in body weight was observed among vehicle control, JQ1, ABT-263 or the combination group (Figure 5E). The results indicate that combination treatment of JQ1 and ABT-263 significantly inhibits tumor growth in MYCN-amplified SCLC xenografts.
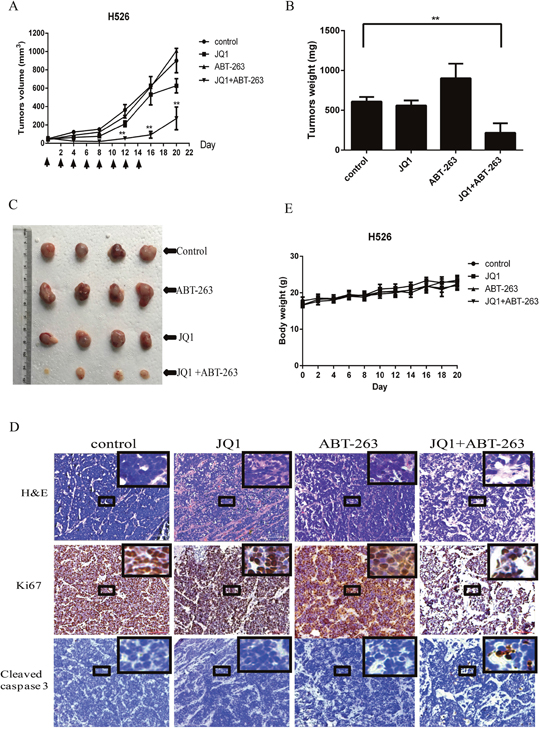
Figure 5: (A) Combination treatment with JQ1 and ABT-263 leads to robust antitumor activity in MYCN-amplified SCLC xenograft. H526 xenografts were treated by DMSO control, 25 mg/kg JQ1, 80 mg/kg ABT-263, or the combination of JQ1 (25 mg/kg) and ABT-263 (80 mg/kg) every two days for 2 weeks. The mean tumor size ± SEM is shown (**, P < 0.01 by an unpaired t test). (B) Tumor weights in JQ1+ABT-263 group were significantly decreased compared to control group (**, p < 0.01, by an unpaired t test). Data of tumor weights are expressed as means ±SEM. (C) Imaging of representative tumors from each group. The tumors were excised at the end of the experiment. (D) Histologic analysis of tumors, including H&E staining, immunohistochemical detection of Ki67 or cleaved Caspase 3. Scale bars represent 100 μm. Small boxed-areas were enlarged. (E). There are no obviously difference in mouse weights from H526 xenografts during the course of treatment, comparing to control group. Detection was conducted in every two days from day 0 to the end of experiment. The mean body weight ± SEM is shown.
DISCUSSION
Currently, no targeted therapy is available for SCLC due to the lack of therapeutically relevant genome alterations. MYC family oncogene (MYC, MYCN or MYCL) is amplified in 20%-30% of the SCLC patients, thereby representing a potential therapeutic target in SCLC [6]. However, MYC family oncogene, as a non-kinase oncogene, is a challenging target for drug development. BET domain proteins act as epigenetic factors associated with acetylated histones and facilitate transcription of target genes, including MYC, MYCN and MYCL[17, 21, 22]. Recently, several groups reported that indirectly targeting MYC by the BET bromodomain inhibitor JQ1 exhibited anti-tumor activity in childhood sarcoma, thyroid tumor and endometrial cancer [23–25]. In SCLC, previous studies indicated that JQ1 targeted MYCL and ASCL1 to inhibit cancer cell growth [26, 27]. In this study, we demonstrated that JQ1 was able to down-regulate MYCN gene encoding protein N-Myc, leading to growth inhibition of MYCN-amplified SCLC cells. Interestingly, our study found that MYCN-amplified SCLC cells were the most sensitive to JQ1, which agree with a previous report by Baratta et al. indicating that JQ1 sensitivity strongly correlated with MYCN expression levels [28]. Thus, our study demonstrates that JQ1 is more effective in MYCN-amplified SCLC cells.
In this study, we found that JQ1 induced Bim in SCLC, which is consistent with recently several reports indicating that JQ1 is able to up-regulate Bim in hepatocellular carcinoma and leukemia [29, 30]. Furthermore, our study found that Bim induction by JQ1 is through N-Myc inhibition, as N-Myc inhibition by siRNA treatment up-regulated Bim. To our knowledge, our study is the first report to indicate that N-Myc inhibition is able to up-regulate Bim expression. Previous study has demonstrated that c-MYC inactivation up-regulated Bim through suppressing miR-17-92, a key post-transcriptional repressor of Bim in MYC-induced lymphomas model [14]. However, in SCLC, our study found that c-Myc inhibition did not up-regulate Bim expression by unknown reason. Further study needs to investigate how N-Myc inhibition up-regulates Bim, and why c-Myc inhibition does not up-regulate Bim in SCLC.
Previous study has indicated that cancer cells with higher Bim and lower Mcl-1 expression is more sensitivity to ABT-263. The down-regulation of Mcl-1 by mTOR inhibitor AZD8055 sensitizes SCLC cells to ABT-263 [10]. Ham et al. also reported that Arora Kinase inhibitor MLN8237 sensitized MYCN-amplified neuroblestoma to BH3 mimics ABT-199 partially via Mcl-1 reduction [31]. In our study, we found that JQ1 up-regulated Bim, but had no impact on Mcl-1 expression (Supplementary Figure 4). Furthermore, we indicated that Bim up-regulation by JQ1 is able to sensitize MYCN-amplified SCLC cells to ABT-263. In contrast, JQ1 did not induce Bim in MYC-amplified H82 cells, and thereby JQ1 did not sensitize H82 cells to ABT-263. In addition, we found that Bcl-2 expression was lower in H82 cells than that in H526 and H69 cells (Supplementary Figure 5), which may also be a reason why no synergistic effect of JQ1 combined with ABT-263 was observed in H82 cells. Our study suggests that the elevation of Bim level by pharmacological treatment may be an effective strategy to sensitize cancer cells to ABT-263.
The proapoptotic activity of Bim is regulated by the interaction of Bim with anti-apoptotic protein Bcl-2 and Mcl-1. Our immunoprecipitation data of Bim showed that ABT-263 treatment disrupted the interaction of Bim with Bcl-2, whereas the released Bim bound to Mcl-1. These results are consistent with previous study by Faber et al.[10], indicating that Mcl-1 binds to the Bim that is released from Bim/Bcl-2 complex upon ABT-263 treatment. Therefore, the inability of disrupting the interaction of Bim with Mcl-1 limits the activity of ABT-263. Notably, our study found that combination treatment of ABT-263 and JQ1 not only led to more release of Bim from the Bim/Bcl-2 complex, but also prevented the released Bim binding to Mcl-1. Taken together, our study suggests that combination of JQ1 and ABT-263disrupts the interaction of Bim/Bcl-2 and prevents the interaction of the released Bim with Mcl-1, which may promote strong apoptotic response.
In summary, our study demonstrates that targeting N-Myc by JQ1 sensitizes MYCN-amplified SCLC cells to ABT-263. The synergistic anti-tumor activity of JQ1 and ABT-263 is through up-regulation of Bim, and disturbing the interaction of Bim with Bcl-2 and Mcl-1 (Figure 6). Furthermore, our in vivo experiment demonstrated that the combination of JQ1 and ABT-263 leads to marked tumor regression in MYCN-amplfied SCLC xenografts. In clinical medicine, patients with MYCN-amplified SCLC have been associated with tumor aggressiveness and shorter survival [32]. Altogether, our study reveals for the first time that co-targeting of N-Myc and Bcl-2 by JQ1/ABT-263 combination is a novel and effective strategy specifically for MYCN-amplified SCLC.
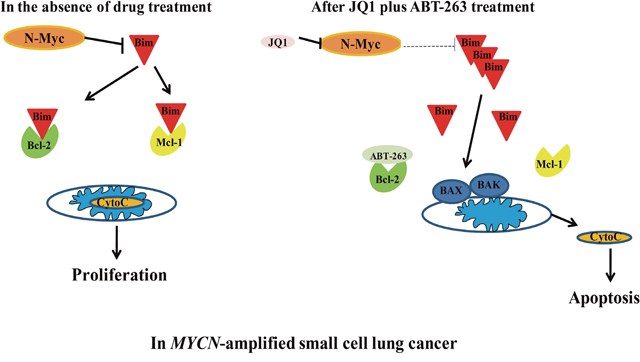
Figure 6: Schematic of MYCN-amplified SCLC before and after treatment with JQ1/ABT-263 combination. In MYCN-amplified SCLC, N-Myc inhibits Bim. JQ1 treatment decreases N-Myc to up-regulate Bim. Combination of JQ1 with ABT-263 disrupts Bim’s interaction with BCL-2 and Mcl-1, leading to the liberation of Bim and apoptosis.
MATERIALS AND METHODS
Materials
JQ1 and ABT-263 were purchased from Selleck chemical (Shanghai, China) and stock solutions were prepared in DMSO (Sigma–Aldrich, Saint Louis, MO, USA) at a concentration of 10 mM. Antibodies against p21, Bcl-2, N-Myc, c-Myc, Bim, PARP, cleaved-Caspase 3 and Mcl-1 were from Cell Signaling Technology, Danvers, MA, USA. Ki67 antibody was from ZSGB-BIO, Beijing, China. Actin antibody was from TransBionovo, Beijing, China. The siRNA #1 and #2 against human Bim as well as its negative control siRNA were purchased from Cell Signaling Technology (Danvers, MA, USA). And the siRNA #1 against human N-Myc as well as its negative control siRNA were purchased from Santa Cruz Biotechnology, Dallas, Texas, USA. The siRNA #2 against human N-Myc as well as its negative control siRNA were pruchased from OriGene Technologies, Rockville, MD, USA.
Cell lines
The human SCLC cell lines, H82, H526, DMS79, H69, H1963, H446 and H196 were maintained in RPMI-1640 media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a humidified incubator at 37ºC in 5% CO2. All of these SCLC cell lines were kindly provided by Dr. Matthew Meyerson at Dana-Farber Cancer Institute, USA. RPMI-1640 media, FBS and penicillin/streptomycin were purchased from Gibco, Life Technologies, Carlsbad, CA, USA.
Cell viability assay
SCLC cells were treated with DMSO control or drugs for 72 hours, and then cell viability was measured by using CellTiter-Glo Luminescent assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Luminescence was measured in a multi-label plate reader (Envision PerkinElmer, USA). Data were normalized to DMSO control and represented by the mean of three independent measurements with SEM. The IC50 values were determined from the sigmoidal dose–response curves using PRISM4 software (GraphPad Software, Inc., La Jolla, CA, USA).
Cell cycle analysis
After treatment with DMSO or JQ1 for 24 hours, cells were fixed by dropwise addition of ice-cold EtOH. Fixed cells were stained with PI/RNase staining buffer (BD Biosciences, Franklin, NJ, USA), and then analyzed by a FACS Calibur (BD Biosciences, Franklin, NJ, USA). The proportion of cells in each cell cycle phase was determined using ModFit software (Verity Software House, Topsham, ME, USA).
Western blotting
After drug treatment, cells were homogenized in lysis buffer (150mM NaCl, 50mM Tris-HCl pH 8.0, 1% Triton X-100, and 1mM EDTA). Protein extracts were quantified by BCA Protein Assay Kit (Beyotime, Shanghai, China), separated by SDS-PAGE, and then transferred onto nitrocellulose blotting membrane (Cell Signaling Technology, Danvers, MA, USA). The membranes were hybridized with a primary antibody at 4 °C overnight followed by incubation with a secondary antibody for 2 hours at room temperature. Signals were visualized using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA).
Quantitative RT-PCR
Total RNA was isolated using Trizol (Thermo Scientific, Rockford, IL, USA) together with Qiagen RNeasy Mini Kit (Qiagene, Hilden, Germany). cDNA was synthesized by Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. The quantitative real-time PCR was performed in triplicate using a FastStart Essential DNA Green Master (Roche, Mannheim, Germany) on a Roche LightCycler 96 Real Time PCR System. The expression levels of all tested genes were normalized to the expression level of β-actin. The cDNA was amplified with the following primers.
Bim
forward 5′-CCCCGCTTTTCATCTTTATG-3′,
reverse5′-GGGCTCCTGTCTGTGTCAA-3′
p21
forward 5′-GGCAGACCAGCATGACAGATT-3′,
reverse 5′-GCGGATTAGGGCTTCCTCT-3′
β-actin
forward 5′-CATGTACGTTGCTATCCAGGC-3′,
reverse 5′-CTCCTTAATGTCACGCACGAT-3′
Immunoprecipitation (IP)
After treatment, cells were lysed with immunoprecipitation lysis buffer (250mM NaCl, 50mM Tris-HCl pH7.4, 0.5% Triton X-100, 10% glycerol). Five hundred micrograms of whole-cell extracts were incubated with 1 μg of Bim antibody followed by rocking at 4°C for overnight. After incubation, 50 μL of protein A Megnetic beads (Thermo Scientific, Rockford, IL, USA) were added, and then the samples were rocked at 4°C for 4 hours. After washed three times, samples were resuspended in 20 μL of 2 X sample buffer, and boiled for 10 minutes, and followed by SDS-PAGE.
siRNA treatment
For Bim siRNA transfection, H69 cells (2 x105) were seeded onto 6-well plates and incubated for 24 hours. Cells were then transfected with Bim siRNA or control siRNA using Effectene Transfection Reagent (QIAGEN, Hilden, Germany). Two days after transfection, one part of transfectants was collected for western blot to check whether Bim was knocked down. Other part of transfectants was replated onto 96-well plate and was treated with DMSO or drugs for 72 hours, and then cell viability was measured by CellTiter-Glo Luminescent assay.
For N-Myc siRNA transfection, H69 cells (2 x105) were seeded onto 6-well plates and incubated for 24 hours. Cells were then transfected with N-Myc siRNA or control siRNA using Effectene Transfection Reagent (QIAGEN, Hilden, Germany). Two days after transfection, cell lysis was collected for western blotting.
Xenograft experiments
Animal experiments were carried out according to a protocol approved by Institutional Animal Care and Use Committee of Hefei Institutes of Physical Science. Athymic nude mice were injected subcutaneously in dorsal flank, with a 100 μL suspension of 2 x 106 H526 cells in an equal volume of Matrigel (BD Biosciences, Franklin, NJ, USA). When tumors grew to 4 to 5 mm in diameter, the mice were treated by intraperitonel injection with DMSO control, JQ1 (25 mg/kg), ABT-263 (80 mg/kg) and the combination of JQ1 (25 mg/kg) with ABT-263 (80 mg/kg) every 2 days. The tumor size was monitored by caliper measurements and calculated by the formula: Volume=(length×width×width)/2.
H&E staining
Tumors were harvested from euthanized mice, fixed in 4% paraformaldehyde for 24 hours and embedded in paraffin wax. After sections were cut in 6 μm, sections were dewaxed, rehydrated and dipped into Mayer’s hematoxylin with agitation for 3-5 min. After rinsed by H2O, sections were stained with 1% eosin Y solution with agitation for 1-3 min. Subsequently, the sections were dehydrated with alcohol and xylene. Finally, mounting medium was added prior to covering with a cover slip.
Immunohistochemistry
Tissues were cut as 6 μm sections. After dewaxed and reydrated, sections were placed in citrate buffer (0.01 M, pH 6.0) antigen retrieval solution boiled for 3 min in a pressure cooker. After blocked with 5% BSA, sections were incubated with Ki-67 antibody or cleaved-Caspase 3 antibody at 4°C overnight. Then, sections were incubated with secondary antibody and stained with DAB (ZSGB-BIO, Beijing, China). Finally, sections were counterstained with hematoxylin, and dehydrated, and then mounted.
Assessment of drug synergy
Drug synergy was determined quantitatively using the combination index (CI) method of Chou and Talalay [33]. CI was calculated using the formula: CI=D1/DX1+D2/DX2, in which D1 and D2 are the doses used to achieve a specific growth inhibition when two drugs combined, and DX1 and DX2 are individual drug doses needed to achieve similar growth inhibition. CI<1 indicates synergism, whereas CI>1 indicates antagonism.
Statistical analysis
All data were analyzed using PRISM4 Software (GraphPad Software, Inc., La Jolla, CA, USA). Statistical analysis was performed using an unpaired t-test. Results were considered as statistically significant when P< 0.05.
Abbreviations
Small cell lung cancer (SCLC), Cancer Cell Line Encyclopedia (CCLE), Combination index (CI), Non-small cell lung cancer (NSCLC), Epidermal Growth Factor Receptor (EGFR), Anaplastic Lymphoma Kinase (ALK), Bcl-2 homology domains 3 (BH3), Bromodomain and Extra-Terminal (BET), Target of rapamycin complex 1/2 (TORC1/2).
ACKNOWLEDGMENTS
We thank members of the Lin laboratory for critical reading of the manuscript and helpful discussions. We also thank Dr. Matthew Meyerson (Dana-Farber Cancer Institute, USA) for kindly providing SCLC cell lines.
CONFLICTS OF INTEREST
Authors have no financial/commercial conflicts of interest regarding the study.
FUNDING
This study was supported by National Natural Science Foundation of China (Grant Numbers: 81372214 and 81502632), Natural Science Foundation of Anhui Province (Grant Number: 1608085MH179), Key Technologies R & D Programme of Anhui Province (Grant Number: 1604a0802103), The Major/Innovative Program of Development Foundation of Hefei Center for Physical Science and Technology (Grant Number: 2016FXCX006) and the 100-Talent Program of Chinese Academy of Sciences. VWYL is funded by the Research Grant Council, Hong Kong (#17114814, #17121616, General Research Fund), (Theme-based Research Scheme: T12-401/13-R), and the Start-up Fund from the School of Biomedical Sciences, Faculty of Medicine, the Chinese University of Hong Kong.
REFERENCES
1. Mamdani H, Induru R, Jalal SI. Novel therapies in small cell lung cancer. Trans Lung Cancer Res. 2015; 4:533-544.
2. Kahnert K, Kauffmann-Guerrero D, Huber RM. SCLC-state of the art and what does the future have in store? Clin Lung Cancer. 2016; 17:325-333.
3. Alvarado-Luna G, Morales-Espinosa D. Treatment for small cell lung cancer, where are we now?-a review. Trans Lung Cancer Res. 2016; 5:26-38.
4. Li J, Deng H, Hu M, Fang Y, Vaughn A, Cai X, Xu L, Wan W, Li Z, Chen S, Yang X, Wu S, Xiao J. Inhibition of non-small cell lung cancer (NSCLC) growth by a novel small molecular inhibitor of EGFR. Oncotarget. 2015; 6:6749-6761. https://doi.org/10.18632/oncotarget.3155.
5. Santarpia M, Altavilla G, Rosell R. Alectinib: a selective, next-generation ALK inhibitor for treatment of ALK-rearranged non-small-cell lung cancer. Expert Rev Respir Med. 2015; 9:255-268.
6. Semenova EA, Nagel R, Berns A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015; 29:1447-1462.
7. Gardner EE, Connis N, Poirier JT, Cope L, Dobromilskaya I, Gallia GL, Rudin CM, Hann CL. Rapamycin rescues ABT-737 efficacy in small cell lung cancer. Cancer Res. 2014; 74:2846-2856.
8. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, Muller C, Dahmen I, Jahchan NS, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015; 524:47-53.
9. S Soderquist R, Eastman A. BCL2 inhibitors as anticancer drugs: a plethora of misleading BH3 mimetics. Mol Cancer Ther. 2016; 15:2011-2017.
10. Faber AC, Farago AF, Costa C, Dastur A, Gomez-Caraballo M, Robbins R, Wagner BL, Rideout WM 3rd, Jakubik CT, Ham J, Edelman EJ, Ebi H, Yeo AT, et al. Assessment of ABT-263 activity across a cancer cell line collection leads to a potent combination therapy for small-cell lung cancer. Proc Natl Acad Sci U S A. 2015; 112:E1288-1296.
11. Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, Ranson MR, Dive C, McKeegan EM, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012; 18:3163-3169.
12. Basu B, Dean E, Puglisi M, Greystoke A, Ong M, Burke W, Cavallin M, Bigley G, Womack C, Harrington EA, Green S, Oelmann E, de Bono JS, et al. First-in-human pharmacokinetic and pharmacodynamic study of the dual m-TORC 1/2 inhibitor AZD2014. Clin Cancer Res. 2015; 21:3412-3419.
13. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, et al. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067-1073.
14. Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014; 26:262-272.
15. Kaur G, Reinhart RA, Monks A, Evans D, Morris J, Polley E, Teicher BA. Bromodomain and hedgehog pathway targets in small cell lung cancer. Cancer Lett. 2016; 371:225-239.
16. Wang L, Wu X, Huang P, Lv Z, Qi Y, Wei X, Yang P, Zhang F. JQ1, a small molecule inhibitor of BRD4, suppresses cell growth and invasion in oral squamous cell carcinoma. Oncol Rep. 2016; 36:1989-1996.
17. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146:904-917.
18. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011; 108:16669-16674.
19. Shao Q, Kannan A, Lin Z, Stack BC Jr, Suen JY, Gao L. BET protein inhibitor JQ1 attenuates Myc-amplified MCC tumor growth in vivo. Cancer Res. 2014; 74:7090-7102.
20. Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair. 2016; 42:63-71.
21. Suzuki K, Yamamoto K, Arakawa Y, Yamada H, Aiba K, Kitagawa M. Antimyeloma activity of bromodomain inhibitors on the human myeloma cell line U266 by downregulation of MYCL. Anticancer Drugs. 2016; 27:756-765.
22. Shahbazi J, Liu PY, Atmadibrata B, Bradner JE, Marshall GM, Lock RB, Liu T. The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce N-Myc expression and induce anticancer effects. Clin Cancer Res. 2016; 22:2534-2544.
23. Bid HK, Phelps DA, Xaio L, Guttridge DC, Lin J, London C, Baker LH, Mo X, Houghton PJ. The bromodomain BET inhibitor JQ1 suppresses tumor angiogenesis in models of childhood sarcoma. Mol Cancer Ther. 2016; 15:1018-1028.
24. Zhu X, Enomoto K, Zhao L, Zhu YJ, Willingham MC, Meltzer P, Qi J, Cheng SY. Bromodomain and extraterminal protein inhibitor JQ1 suppresses thyroid tumor growth in a mouse model. Clin Cancer Res. 2017; 23:430-440.
25. Qiu H, Li J, Clark LH, Jackson AL, Zhang L, Guo H, Kilgore JE, Gehrig PA, Zhou C, Bae-Jump VL. JQ1 suppresses tumor growth via PTEN/PI3K/AKT pathway in endometrial cancer. Oncotarget. 2016; 11:66809-66821. https://doi.org/10.18632/oncotarget.11631.
26. Kato F, Fiorentino FP, Alibes A, Perucho M, Sanchez-Cespedes M, Kohno T, Yokota J. MYCL is a target of a BET bromodomain inhibitor, JQ1, on growth suppression efficacy in small cell lung cancer cells. Oncotarget. 2016; 7:77378-77388. https://doi.org/10.18632/oncotarget.12671.
27. Lenhart R, Kirov S, Desilva H, Cao J, Lei M, Johnston K, Peterson R, Schweizer L, Purandare A, Ross-Macdonald P, Fairchild C, Wong T, Wee S. Sensitivity of small cell lung cancer to BET inhibition is mediated by regulation of ASCL1 gene expression. Mol Cancer Ther. 2015; 14:2167-2174.
28. Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, Curtis J, Piao H, Wong LC, Kung AL, Beroukhim R, Bradner JE, Drapkin R, et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci U S A. 2015; 112:232-237.
29. Li GQ, Guo WZ, Zhang Y, Seng JJ, Zhang HP, Ma XX, Zhang G, Li J, Yan B, Tang HW, Li SS, Wang LD, Zhang SJ. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget. 2016; 7:2462-2474. https://doi.org/10.18632/oncotarget.6275.
30. Saenz DT, Fiskus W, Manshouri T, Rajapakshe K, Krieger S, Sun B, Mill CP, DiNardo C, Pemmaraju N, Kadia T, Parmar S, Sharma S, Coarfa C, et al. BET protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary AML cells. Leukemia. 2017; 31:678-687.
31. Ham J, Costa C, Sano R, Lochmann TL, Sennott EM, Patel NU, Dastur A, Gomez-Caraballo M, Krytska K, Hata AN, Floros KV, Hughes MT, Jakubik CT, et al. Exploitation of the apoptosis-primed state of MYCN-amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell. 2016; 29:159-172.
32. Beltran H. The N-myc oncogene: maximizing its targets, regulation, and therapeutic potential. Mol Cancer Res. 2014; 12:815-822.
33. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010; 70:440-446.