
INTRODUCTION
Retinal degenerative diseases are characterized by progressive loss of retinal cells [1–3]. UV radiation (UVR) and subsequent oxidative stresses are known as the major cause of damages to the resident retinal cells, including the retinal pigment epithelium (RPE) cells (RPEs) and retinal ganglion cells (RGCs) [4, 5]. Intense recent studies have been focusing on exploring novel strategies to protect retinal cells from UVR/oxidative stresses [6–9].
NF-E2-related factor 2 (Nrf2) is one extensively studied anti-oxidant signaling [10–13]. As a transcription factor, activated Nrf2 binds to the antioxidant responsive elements (AREs). It thus dictates transcription and expression of multiple genes, including those of endogenous antioxidants, phase II detoxification enzymes, along with other cellular defensive proteins [10–13]. Several key Nrf2-dependent genes have been identified, including NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), γ-glutamylcysteine synthetase modifier subunit (GCLM) and catalytic subunit (GCLC), among others [10–13]. Studies have demonstrated that Nrf2-dependent antioxidant response is a pivotal protection system against oxidative insults in mammalian cells [7, 14–16].
Nrf2 activation is tightly controlled by its inhibitor protein, Keap1 (Kelch-like ECH-associated protein 1) [13, 17, 18]. Nrf2’s binding to Keap1 in the cytoplasm will dictate it to Cullin-3-dependent ubiquitination and proteasomal degradation. Following activation, Nrf2 will disassociate with Keap1 ubiquitin E3 ligase complex, causing it stabilization and accumulation [13, 17, 18]. Activated/stabilized Nrf2 translocates to cell nuclei and then binds to ARE, leading to multiple gene transcription.
Recent studies have developed a Nrf2-targeting thiazole-containing compound, namely MIND4-17 [19, 20]. This small molecule compound covalently modified a critical stress-sensor cysteine (C151) of Keap1, leading to Nrf2’s departure from the ubiquitin E3 ligase complex. This would lead to Nrf2 stabilization, accumulation and nuclear translocation [19, 20]. Thus, treatment of this compound would induce profound Nrf2 activation. In the current study, we demonstrate that MIND4-17 efficiently protects RPEs and RGCs from UVR.
RESULTS
MIND4-17 protects retinal pigment epithelium (RPE) cells (RPEs) and retinal ganglion cells (RGCs) from UVR
The molecule structure of MIND4-17 [19, 20] was presented in Figure 1A. We aim to study the potential effect of this Nrf2-inducing compound on UV radiation (UVR)-induced retinal cell damages. ARPE-19 cells are well-established human retinal pigmentation epithelial (RPE) cells [7]. Cultured ARPE-19 cells were treated with UVR (UVA2 + B, 30 mJ/cm2) [7, 21]. After 48 hours of culture, the Cell Counting Kit-8 (CCK-8) assay results in Figure 1B demonstrated that the viability of ARPE-19 cells was dramatically reduced following UVR, and the CCK-8 optic density (OD) decreased over 60% (Figure 1B). Significantly, pretreatment with MIND4-17 (at 1–10 μM) largely inhibited UVR-induced viability reduction in ARPE-19 cells (Figure 1B). MIND4-17-indued cytoprotection was concentration-dependent (Figure 1B). MIND4-17 at a low concentration (0.1 μM) was in-effective against UVR (Figure 1B).
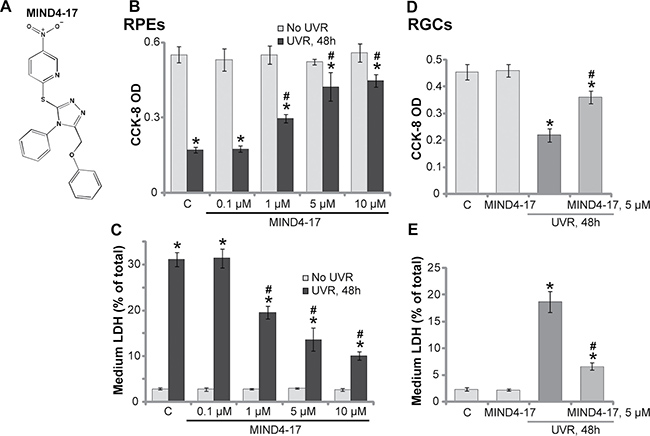
Figure 1: MIND4-17 protects retinal pigment epithelium (RPE) cells (RPEs) and retinal ganglion cells (RGCs) from UVR. The molecule structure of MIND4-17 was presented (A). ARPE-19 cells (B–C) or primary cultured human RGCs (D and E) were pretreated for 30 min with applied concentration of MIND4-17, cells were then subjected to UV radiation (UVR, UVA2 + B, 30 mJ/cm2) and were further cultured for 48 hours; Cell viability was tested by the CCK-8 assay (B and D); Cell death was examined by LDH release in the conditional medium (C and E). For each assay, n = 5. “C” stands for untreated control cells. *p < 0.05 vs. “C” cells. #p < 0.05 vs. “UVR” only cells. Experiments in this figure were repeated four times to insure consistency of results.
Lactate dehydrogenase (LDH) release is often tested as a marker of cell death. Following UVR, the LDH level in the conditional medium of ARPE-19 cells was significantly increased (Figure 1C), indicating cell death. Pretreatment with MIND4-17 at 1–10 μM largely attenuated UVR-induced ARPE-19 cell death (LDH release, Figure 1C). Retinal ganglion cells (RGCs) are also main UVR-targeting cells in the retina [21, 22]. Here, we demonstrated that UVR similarly induced viability loss (CCK-8 OD reduction, Figure 1D) and cell death (LDH release, Figure 1E) in primary human RGCs [21, 22]. Importantly, such effects by UVR were largely attenuated with pretreatment of MIND4-17 (5 μM) (Figure 1D and 1E). It should be noted that treatment with MIND4-17 alone at tested concentration failed to change viability and death of the retinal cells (Figure 1B–1E). Together, these results demonstrate that MIND4-17 protects human RPEs and RGCs from UVR.
MIND4-17 inhibits UVR-induced apoptosis in RPEs and RGCs
The potential effect of MIND4-17 on UVR-induced retinal cell apoptosis was also tested. As shown in Figure 2A, in the ARPE-19 cells, 16 hours after UVR (UVA2 + B, 30 mJ/cm2), expressions of both cleaved-caspase-3 and cleaved-PARP [poly (ADP-ribosyl) transferase] were both increased (Figure 2A). Meanwhile, UVR-induced significant production of single strand DNA (ssDNA), which is the characteristic marker of cell apoptosis (Figure 2B). Such effects by UVR were largely inhibited with pretreatment of MIND4-17 (5 μM) in ARPE-19 cells (Figure 2A and 2B). These results suggest that MIND4-17 possibly inhibits UVR-induced RPE cell apoptosis. Indeed, further studies displayed that MIND4-17 (5 μM) pretreatment efficiently inhibited UVR-induced increase of Annexin V-positive (Figure 2C) and TUNEL-positive (Figure 2D) ARPE-19 cells. The similar results were also obtained in the primary human RGCs, where MIND4-17 (5 μM, 30 min pretreatment) inhibited UVR-induced apoptosis induction (TUNEL cell increase, Figure 2E). MIND4-17 alone was in-effective to cell apoptosis in the tested retinal cells (Figure 2A–2E). Together, we demonstrate that MIND4-17 inhibits UVR-induced apoptosis in human RPEs and RGCs.
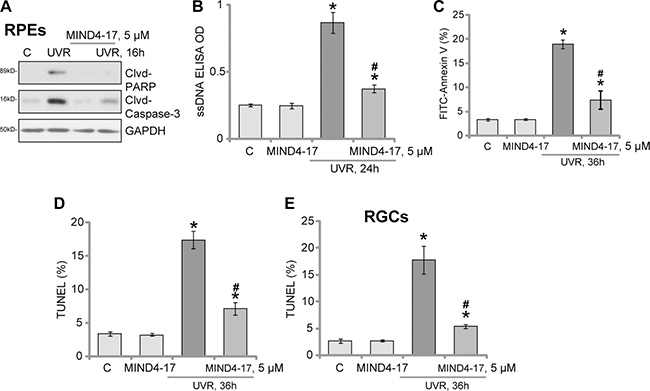
Figure 2: MIND4-17 inhibits UVR-induced apoptosis in RPEs and RGCs. ARPE-19 cells (A–D) or primary cultured human RGCs (E) were pretreated for 30 min with MIND4-17 (5 μM), cells were then subjected to UV radiation (UVR, UVA2 + B, 30 mJ/cm2) and were further cultured for applied time; Expressions of cleaved-PARP (“Clvd-PARP”) and cleaved-caspase-3 (“Clvd-Caspase-3”) were tested (A, GAPDH was shown as the loading control); Cell apoptosis was tested by the assays mentioned in the text (B–E). Annexin V ratio included both early (PI negative) and late (PI positive) apoptotic cells (C). For TUNEL assay, at least 200 cells in five random views (1×100 magnification) of each condition were analyzed to calculate TUNEL ratio (D and E). “C” stands for untreated control cells. *p < 0.05 vs. “C” cells. #p < 0.05 vs. “UVR” only cells. Experiments in this figure were repeated three times to insure consistency of results.
MIND4-17 activates Nrf2 signaling in retinal cells
Activation of Nrf2 signaling pathway can inhibit UVR-induced damages in retinal cells [6, 7, 23, 24]. MIND4-17 is a Nrf2-inducing compound [19, 20]. We therefore tested Nrf2 signaling in MIND4-17-treated retinal cells. The real-time quantitative PCR (“RT-qPCR”) assay results displayed that treatment with MIND4-17 at 1–10 μM significantly increased mRNA expressions of several Nrf2-dependent genes [14, 15, 25], including heme oxygenase-1 (HO-1, Figure 3A), NAD(P)H quinone oxidoreductase 1 (NQO1, Figure 3B) and γ-glutamyl cystine ligase modulatory subunit (GCLM, Figure 3C). The effect by MIND4-17 was dose-dependent (Figure 3A–3C). On the other hand, Nrf2 mRNA level was unchanged before and after t MIND4-17 treatment (Figure 3D). Nrf2 protein level was yet significantly increased in MIND4-17-treated RPE cells, suggesting Nrf2 stabilization (Figure 3E). Protein expressions of HO1, NQO1 and GCLM were also boosted following MIND4-17 (1–10 μM) treatment (Figure 3E). Importantly, we found that stabilized Nrf2 translocated to cell nuclei after treatment with MIND4-17, and the nuclear Nrf2 protein level was significantly increased (Figure 3F). Based on these results, we propose that MIND4-17 treatment possibly separates Nrf2 from Keap1, thus allowing stabilization and accumulation of Nrf2, which then translocates to cell nuclei, causing transcription of ARE-dependent genes, i.e. HO1, NQO1 and GCLM. In the primary human RGCs, MIND4-17 (5 μM, 3 hours) treatment also increased mRNA expressions of HO1, NQO1 and GCLM, but not Nrf2 (Figure 3G). Therefore, MIND4-17 apparently activates Nrf2 signaling in retinal cells.
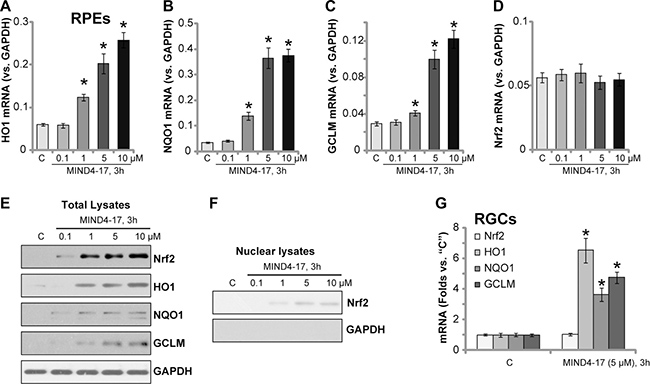
Figure 3: MIND4-17 activates Nrf2 signaling in retinal cells. ARPE-19 cells (A–F) or primary cultured human RGCs (G) were treated with applied concentration of MIND4-17, cells were further cultured for 3 hours; The real-time quantitative PCR (RT-qPCR) assay was employed to test mRNA expressions of listed genes (A–D and (G), GAPDH mRNA was tested as the internal control); Listed proteins in total cell lysates (E) and nuclear fraction lysates (F) were also tested by Western blotting assay (GAPDH was tested as the loading control, which was absent in the nuclear fractions). “C” stands for untreated control cells. *p < 0.05 vs. “C” cells. Experiments in this figure were repeated three times to insure consistency of results.
Nrf2 is required for MIND4-17-mediated retinal cytoprotection against UVR
In order to test that Nrf2 signaling activation is required for MIND4-17-mediated cytoprotection, short hairpin RNA (shRNA) method was employed to knockdown Nrf2 in retinal cells. As described [7, 24], the Nrf2 shRNA-containing lentiviral particles (purchased from Santa Cruz Biotech) were added to ARPE-19 cells. Stable cells were then selected by puromycin. The applied Nrf2 shRNA dramatically downregulated Nrf2 mRNA expression in the stable cells (Figure 4A). Since basal Nrf2 protein level was low in the ARPE-19 cells (see Figure 3), we stimulated cells with MIND4-17 (5 μM, 3 hours). Western blotting assay results confirmed that the applied Nrf2 shRNA also dramatically downregulated Nrf2 protein in MIND4-17-treated ARPE-19 cells (Figure 4B). Downstream HO1 expression was also largely inhibited (Figure 4B). Remarkably, UVR-induced cell death (CCK-8 OD reduction, Figure 4C) and apoptosis (ssDNA ELISA OD increase, Figure 4D) were intensified in Nrf2-shRNA-expressing ARPE-19 cells. These results suggest that Nrf2 activation might also be important for the protection against UVR, which is consistent with previously findings [6, 7]. Significantly, MIND4-17-mediated cytoprotection against UVR was almost completely abolished in Nrf2-silenced cells (Figure 4C and 4D). MIND4-17 was also most ineffective against UVR in Nrf2-shRNA expressing cells (Figure 4C and 4D). The similar results were also obtained in the primary human RGCs (Data not shown). These results suggest that Nrf2 is required for MIND4-17-mediated retinal cytoprotection against UVR.
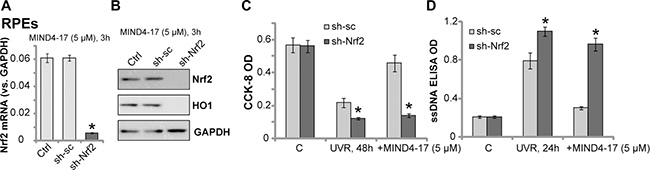
Figure 4: Nrf2 is required for MIND4-17-mediated retinal cytoprotection against UVR. Stable ARPE-19 cells, expressing scramble control shRNA (“sh-sc”) or Nrf2 shRNA (“sh-Nrf2”), were treated with MIND4-17 (5 μM) for 3 hours, Nrf2 mRNA (A) and listed proteins (B) were tested by RT-qPCR assay (GAPDH mRNA was tested as the internal control) and Western blotting assay (GAPDH was tested as the loading control), respectively; Cells were exposed UV radiation (UVR, UVA2 + B, 30 mJ/cm2) and cultured for applied time; Cell survival and apoptosis were tested by CCK-8 assay (C) and ssDNA ELISA assay (D), respectively. “Ctrl” stands for parental ARPE-19 cells (A and B). For each assay, n = 5. *p < 0.05 vs. “sh-sc” cells. Experiments in this figure were repeated three times to insure consistency of results.
MIND4-17 efficiently attenuates UVR-induced oxidative stress in retinal cells
Reactive oxygen species (ROS) production and following oxidative stress are major causes of cell death by UVR [26, 27]. Nrf2 signaling is a well-established anti-oxidant signaling [10, 12, 17, 28]. In line with previous findings [6, 7, 29, 30], UVR radiation to ARPE-19 cells induced significant ROS production, which was tested by increase of DCFH-DA intensity (Figure 5A). UVR-induced ROS production was followed by increase of DNA damages (γ-H2AX intensity increase, Figure 5B) and lipid peroxidation (TBAR intensity increase, Figure 5C). Remarkably, such effects by UVR were largely attenuated by pretreatment of MIND4-17 (5 μM) (Figure 5A–5C). In the primary human RGCs, MIND4-17 (5 μM) pretreatment also inhibited ROS production by UVR (Figure 5D). These results demonstrate that MIND4-17 efficiently attenuates UVR-induced oxidative stress in retinal cells.
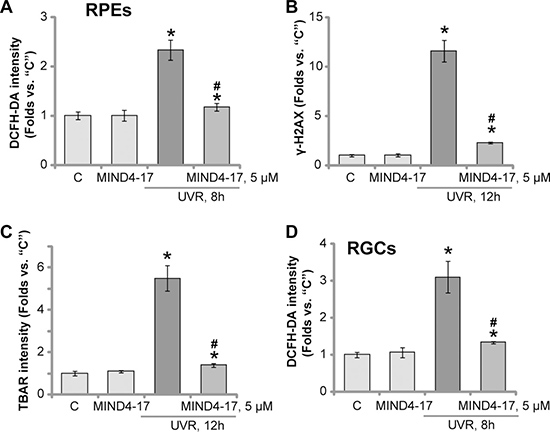
Figure 5: MIND4-17 efficiently attenuates UVR-induced oxidative stress in retinal cells. ARPE-19 cells (A–C) or primary cultured human RGCs (D) were pretreated for 30 min with MIND4-17 (5 μM), cells were then subjected to UV radiation (UVR, UVA2 + B, 30 mJ/cm2) and were further cultured for applied time; ROS production was tested by DCFH-DA intensity assay (A and D); DNA damages and lipid peroxidation were tested by γ-H2AX assay (B) and TBAR activity assay (C), respectively. “C” stands for untreated control cells. *p < 0.05 vs. “C” cells. #p < 0.05 vs. “UVR” only cells. Experiments in this figure were repeated three times to insure consistency of results.
DISCUSSION
In the pathology of AMD and other retinal degenerative diseases, UVR and subsequent oxidative stresses cause direct damages to resident retinal cells, i.e. RPEs and RGCs [6–8, 21, 31, 32]. Existing recent studies have demonstrated that activation of Nrf2 could be a fine strategy to protect retinal cells from UVR and/or oxidative stress [6–8, 21, 31, 32]. For instance, Zhang et al., showed that Salvianolic acid A protected RPEs from oxidative stress through activation of Nrf2-HO1 signaling [8]. Similarly, the other Traditional Chinese Medicine, 3H-1, 2-dithiole-3-thione (D3T), activated Nrf2 signaling to protect RPEs from UVR [6]. SC79, a novel AKT activator [7, 33, 34], protected RPEs from UVR through activating AKT downstream Nrf2 signaling [7]. Additionally, microRNA-mediated silence of Keap1 also activated Nrf2 signaling in RPEs and RGCs, therefore offering protection against UVR-induced oxidative stress [21].
Recent studies have characterized MIND4-17 as a unique and highly selective small molecule activator of Nrf2 [19, 20]. MIND4-17-induced Nrf2 activation is initially through covalent modification of the Keap1 sensor-cysteine C151 [19, 20]. This modification mimics the effects of oxidative stress, which causes Keap1 conformational change and arrests of the Keap1-Nrf2 complex [19, 20]. This would release Nrf2 from the from the ubiquitin E3 ligase complex, causing its stabilization and accumulation in the cytoplasm. It is followed by nuclear translocation of Nrf2 and de novo synthesis of ARE-response genes [19, 20]. Unlike other known Nrf2 activators, this small molecule compound directly and uniquely disassociates the Nrf2-Keap1 complex. It therefore activates Nrf2 signaling at an extremely high efficiency [19, 20].
In the present study, we show that MIND4-17 treatment presumably separated Nrf2 from Keap1, thus allowing its stabilization and accumulation in retinal cells. Stabilized Nrf2 by MIND4-17 translocated to cell nuclei, causing transcription and expression of multiple ARE-dependent anti-oxidant genes, including HO1, NQO1 and GCLM. Subsequently, activation of Nrf2 by MIND4-17 largely ameliorated UVR-induced ROS production, lipid peroxidation and DNA damages in RPEs and RGCs. Retinal cell death and apoptosis by UVR were also dramatically alleviated by MIND4-17. Importantly, we demonstrate that Nrf2 is required for MIND4-17’s actions in retinal cells. shRNA-mediated knockdown of Nrf2 almost completely reversed MIND4-17-induced retinal cytoprotection against UVR.
MATERIALS AND METHODS
Reagents, chemicals and antibodies
Based on the structure in previous studies [19, 20] (also see Figure 1A), MIND4-17 was synthesized and verified by Minde Biotech (Suzhou, China). Puromycin was obtained from Sigma-Aldrich (Nanjing, China). The reagents for cell culture were provided by Gibco (Guangzhou, China). Antibodies against Nrf2, HO1, NQO1, GCLM and GAPDH were purchased from Santa Cruz Biotech (Beijing, China). Antibodies for cleaved-caspase-3 and cleaved-PARP were provided by Cell Signaling Tech (Nanjing, China).
Culture of RPEs
The established ARPE-19 cells were provided as a gift from Dr. Jiang [6, 35, 36]. The DMEM plus fetal bovine serum (FBS) medium was employed for ARPE-19 cell culture.
Culture of primary human RGCs
The primary-cultured human RGCs were also provided by Dr. Jiang [21]. The primary human cells were maintained as described previously [21]. Primary human RGCs at passage 3–8 were utilized for further experiments.
UV radiation
UV radiation (UVR, UVA2 + B, 30 mJ/cm2) to the cultured RPEs and RGCs was described previously [6, 7, 29, 30]. After UVR, cells were maintained in the conditional medium, and cultured for described time.
Cell viability and cell death assays
Testing of cell viability by the Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) [37] as well as cell death assay by lactate dehydrogenase (LDH) release assay (Takara, Tokyo, Japan) were extensively described in early studies [38]. CCK-8 optic density at 450 nm was recorded. LDH content in the conditional medium was normalized to total LDH [38, 39].
ssDNA ELISA assay of cell apoptosis
DNA formamide denaturation and subsequent the production of single strand DNA (ssDNA) is characteristic marker of cell apoptosis, which was tested by the ApoStrandTM ELISA apoptosis detection kit (BIOMOL International, Plymouth Meeting, PA). ELISA OD at 450 nm was recorded.
Assessment of apoptosis by Annexin V FACS assay
Following the applied treatment, retinal cells were harvested, washed and resuspended in binding buffer with 1 μL of Annexin V-FITC and 1 μL of propidium iodide (PI) (Biyuntian, Wuxi, China). After incubation for 20 min at room temperature, cells were then tested by flow cytometry via CellQuest software (BD Biosciences, Shanghai, China). Annexin V ratio was recorded [8, 39].
TUNEL assay
After the described treatment, retinal cells were further stained with TUNEL and DAPI florescence dyes (Sigma, Shanghai, China). TUNEL ratio (vs. DAPI staining) was calculated [40].
Western blotting assay
Cells were lysed via the described lysis buffer [41]. Determination of the protein concentration was though the Bio-Rad Protein Assay (Bio-Rad, Shanghai, China). Protein concentration (μg/mL) was measured at 690 nm. The detailed protocol for Western blotting assay and data quantification were described previously [42, 43]. Cell nuclei were isolated by the “cell nuclei isolation kit” (Sigma) [8].
Real-time quantitative PCR
Total RNA was extracted by TRIzol reagents (Invitrogen, Shanghai, China) and then reversely transcribed using a PrimeScript RTreagent kit (Takara Biotechnology, Japan). Real-time quantitative PCR (“RT-qPCR”) assay was performed by a SYBR Premix Ex TaqTM kit on the ABI-7700 fast PCR system (Takara Biotechnology, Japan). mRNA primers for human HO-1, NQO1 and GLCM and GAPDH were described previously [44]. GAPDH mRNA was tested as the reference gene. The 2–ΔΔCt method was employed to calculate relative expression of targeted mRNAs.
Nrf2 shRNA
The Nrf2 shRNA lentiviral particles (sc-37030-V, Santa Cruz Biotech, Shanghai, China) and the scramble control shRNA lentiviral particles (sc-108080, Santa Cruz Biotech) were both commercial available. The shRNA lentivirus was added to ARPE-19 cells for 12 hours. Afterwards, puromycin (2.5 μg/mL) was added to select stable cells for 5–6 passages. Nrf2 knockdown was verified by both Western blotting assay and RT-qPCR assay.
Reactive oxygen species (ROS) assay
ROS intensity in the control and UVR-treated retinal cells was measured by the carboxy-H2DCF-DA dye (Invitrogen, Shanghai, China) assay [45]. Following the treatment, cells were incubated with 1 μM of carboxy-H2-DCFDA for 120 min under the dark, which were then washed with warm PBS and immediately tested by a spectrofluorometer at excitation and emission wavelengths of 485 and 535 nm, respectively. The results were expressed as increase in fluorescence with respect to control cells.
Lipid peroxidation assay
Thiobarbituric acid reactive substances (TBAR) level was examined to reflect the production of toxic aldehyde resulting from oxidative fatty acyl degradation, the malondialdehyde (MDA). The detailed protocol was described previously [45, 46].
γ-H2AX assay of DNA damages. ARPE-19 cells were fixed in ice-cold ethanol, which were then incubated with a mouse monoclonal anti-γ-H2AX antibody (Santa Cruz Biotech). Afterwards, the FITC-conjugated anti-mouse secondary antibody (Santa Cruz) was then added. Cells were then subjected to FACS assay to determine the γ-H2AX percentage, reflecting DNA damage intensity [47].
Statistics
The results were presented as the mean ± standard deviation (SD). One-way ANOVA was employed to examine the significant differences between groups using SPSS 17.0 (SPSS, Chicago, CA). Values of p < 0.05 were considered statistically significant.
CONCLUSIONS
Together, we conclude that targeting Nrf2 by MIND4-17 efficiently protects RPEs and RGCs from UVR.
Author contributions
All the listed authors carried out the experiments, participated in the design of the study and performed the statistical analysis, participated in its design and coordination and helped to write the manuscript and proof readings.
ACKNOWLEDGMENTS
This study is partly supported by the Science Foundation of Huai’an First People’s Hospital, Nanjing Medical University.
CONFLICTS OF INTEREST
The listed authors have no conflicts of interests.
REFERENCES
1. van Lookeren Campagne M, LeCouter J, Yaspan BL, Ye W. Mechanisms of age-related macular degeneration and therapeutic opportunities. J Pathol. 2014; 232:151–164.
2. Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000; 45:115–134.
3. Young RW. Solar radiation and age-related macular degeneration. Surv Ophthalmol. 1988; 32:252–269.
4. Jin J, Ying H, Huang M, Du Q. Bioactive compounds in green tea leaves attenuate the injury of retinal ganglion RGC-5 cells induced by H2O2 and ultraviolet radiation. Pak J Pharm Sci. 2015; 28:2267–2272.
5. Guo D, Bi H, Liu B, Wu Q, Wang D, Cui Y. Reactive oxygen species-induced cytotoxic effects of zinc oxide nanoparticles in rat retinal ganglion cells. Toxicol In Vitro. 2013; 27:731–738.
6. Li KR, Yang SQ, Gong YQ, Yang H, Li XM, Zhao YX, Yao J, Jiang Q, Cao C. 3H-1,2-dithiole-3-thione protects retinal pigment epithelium cells against Ultra-violet radiation via activation of Akt-mTORC1-dependent Nrf2-HO-1 signaling. Sci Rep. 2016; 6:25525.
7. Gong YQ, Huang W, Li KR, Liu YY, Cao GF, Cao C, Jiang Q. SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget. 2016; 7:60123–60132. http://doi.org/10.18632/oncotarget.11164.
8. Zhang H, Liu YY, Jiang Q, Li KR, Zhao YX, Cao C, Yao J. Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic Biol Med. 2014; 69:219–228.
9. Li KR, Zhang ZQ, Yao J, Zhao YX, Duan J, Cao C, Jiang Q. Ginsenoside Rg-1 protects retinal pigment epithelium (RPE) cells from cobalt chloride (CoCl2) and hypoxia assaults. PLoS One. 2013; 8:e84171.
10. Krajka-Kuzniak V, Paluszczak J, Baer-Dubowska W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol Rep. 2017; 69:393–402.
11. Done AJ, Traustadottir T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016; 10:191–199.
12. Zhang H, Davies KJ, Forman HJ. Oxidative stress response and Nrf2 signaling in aging. Free Radic Biol Med. 2015; 88:314–336.
13. Vriend J, Reiter RJ. The Keap1-Nrf2-antioxidant response element pathway: a review of its regulation by melatonin and the proteasome. Mol Cell Endocrinol. 2015; 401: 213–220.
14. Zhao H, Mitchell S, Ciechanowicz S, Savage S, Wang T, Ji X, Ma D. Argon protects against hypoxic-ischemic brain injury in neonatal rats through activation of nuclear factor (erythroid-derived 2)-like 2. Oncotarget. 2016; 7:25640–25651. http://doi.org/10.18632/oncotarget.8241.
15. Jang HJ, Hong EM, Kim M, Kim JH, Jang J, Park SW, Byun HW, Koh DH, Choi MH, Kae SH, Lee J. Simvastatin induces heme oxygenase-1 via NF-E2-related factor 2 (Nrf2) activation through ERK and PI3K/Akt pathway in colon cancer. Oncotarget. 2016; 7:46219–46229. http://doi.org/10.18632/oncotarget.10078.
16. Chen Z, Zhang Y, Ma L, Ni Y, Zhao H. Nrf2 plays a pivotal role in protection against burn trauma-induced intestinal injury and death. Oncotarget. 2016; 7:19272–19283. http://doi.org/10.18632/oncotarget.8189.
17. Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015; 88:93–100.
18. Chapple SJ, Puszyk WM, Mann GE. Keap1-Nrf2 regulated redox signaling in utero: Priming of disease susceptibility in offspring. Free Radic Biol Med. 2015; 88:212–220.
19. Quinti L, Casale M, Moniot S, Pais TF, Van Kanegan MJ, Kaltenbach LS, Pallos J, Lim RG, Naidu SD, Runne H, Meisel L, Rauf NA, Leyfer D, et al. SIRT2- and NRF2-Targeting Thiazole-Containing Compound with Therapeutic Activity in Huntington’s Disease Models. Cell Chem Biol. 2016; 23:849–861.
20. Quinti L, Dayalan Naidu S, Trager U, Chen X, Kegel-Gleason K, Lleres D, Connolly C, Chopra V, Low C, Moniot S, Sapp E, Tousley AR, Vodicka P, et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc Natl Acad Sci USA. 2017; 114:E4676–E4685.
21. Cheng LB, Li KR, Yi N, Li XM, Wang F, Xue B, Pan YS, Yao J, Jiang Q, Wu ZF. miRNA-141 attenuates UV-induced oxidative stress via activating Keap1-Nrf2 signaling in human retinal pigment epithelium cells and retinal ganglion cells. Oncotarget. 2017; 8:13186–13194. http://doi.org/10.18632/oncotarget.14489.
22. Xie L, Cheng L, Xu G, Zhang J, Ji X, Song E. The novel cyclophilin D inhibitor compound 19 protects retinal pigment epithelium cells and retinal ganglion cells from UV radiation. Biochem Biophys Res Commun. 2017; 487: 807–812.
23. Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005; 25:10321–10335.
24. Ji C, Huang JW, Xu QY, Zhang J, Lin MT, Tu Y, He L, Bi ZG, Cheng B. Gremlin inhibits UV-induced skin cell damages via activating VEGFR2-Nrf2 signaling. Oncotarget. 2016; 7:84748–84757. http://doi.org/10.18632/oncotarget.12454.
25. Ning S, Sekar TV, Scicinski J, Oronsky B, Peehl DM, Knox SJ, Paulmurugan R. Nrf2 activity as a potential biomarker for the pan-epigenetic anticancer agent, RRx-001. Oncotarget. 2015; 6:21547–21556. http://doi.org/10.18632/oncotarget.4249.
26. Muthusamy V, Piva TJ. The UV response of the skin: a review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch Dermatol Res. 2010; 302: 5–17.
27. de Gruijl FR, van Kranen HJ, Mullenders LH. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J Photochem Photobiol B. 2001; 63:19–27.
28. Schafer M, Werner S. Nrf2--A regulator of keratinocyte redox signaling. Free Radic Biol Med. 2015; 88:243–252.
29. Yao J, Bi HE, Sheng Y, Cheng LB, Wendu RL, Wang CH, Cao GF, Jiang Q. Ultraviolet (UV) and hydrogen peroxide activate ceramide-ER stress-AMPK signaling axis to promote retinal pigment epithelium (RPE) cell apoptosis. Int J Mol Sci. 2013; 14:10355–10368.
30. Cao G, Chen M, Song Q, Liu Y, Xie L, Han Y, Liu Z, Ji Y, Jiang Q. EGCG protects against UVB-induced apoptosis via oxidative stress and the JNK1/c-Jun pathway in ARPE19 cells. Mol Med Rep. 2012; 5:54–59.
31. Wang L, Ebrahimi KB, Chyn M, Cano M, Handa JT. Biology of p62/sequestosome-1 in Age-Related Macular Degeneration (AMD). Adv Exp Med Biol. 2016; 854: 17–22.
32. Lambros ML, Plafker SM. Oxidative Stress and the Nrf2 Anti-Oxidant Transcription Factor in Age-Related Macular Degeneration. Adv Exp Med Biol. 2016; 854:67–72.
33. Moreira JB, Wohlwend M, Alves MN, Wisloff U, Bye A. A small molecule activator of AKT does not reduce ischemic injury of the rat heart. J Transl Med. 2015; 13:76.
34. Hao S, Song C, Shang L, Yu J, Qiao T, Li K. Phosphorylation of Akt by SC79 Prevents Iron Accumulation and Ameliorates Early Brain Injury in a Model of Experimental Subarachnoid Hemorrhage. Molecules. 2016; 21:325.
35. Yao J, Liu Y, Wang X, Shen Y, Yuan S, Wan Y, Jiang Q. UVB radiation induces human lens epithelial cell migration via NADPH oxidase-mediated generation of reactive oxygen species and up-regulation of matrix metalloproteinases. Int J Mol Med. 2009; 24:153–159.
36. Jiang Q, Zhou C, Bi Z, Wan Y. EGF-induced cell migration is mediated by ERK and PI3K/AKT pathways in cultured human lens epithelial cells. J Ocul Pharmacol Ther. 2006; 22:93–102.
37. Ren K, Lu X, Yao N, Chen Y, Yang A, Chen H, Zhang J, Wu S, Shi X, Wang C, Sun X. Focal adhesion kinase overexpression and its impact on human osteosarcoma. Oncotarget. 2015; 6:31085–31103. http://doi.org/10.18632/oncotarget.5044.
38. Guo S, Chen C, Ji F, Mao L, Xie Y. PP2A catalytic subunit silence by microRNA-429 activates AMPK and protects osteoblastic cells from dexamethasone. Biochem Biophys Res Commun. 2017; 487:660–665.
39. Yang L, Zheng LY, Tian Y, Zhang ZQ, Dong WL, Wang XF, Zhang XY, Cao C. C6 ceramide dramatically enhances docetaxel-induced growth inhibition and apoptosis in cultured breast cancer cells: a mechanism study. Exp Cell Res. 2015; 332:47–59.
40. Zhang G, Wang C, Sun M, Li J, Wang B, Jin C, Hua P, Song G, Zhang Y, Nguyen LL, Cui R, Liu R, Wang L, et al. Cinobufagin inhibits tumor growth by inducing intrinsic apoptosis through AKT signaling pathway in human nonsmall cell lung cancer cells. Oncotarget. 2016; 7:28935–28946. http://doi.org/10.18632/oncotarget.7898.
41. Qin LS, Jia PF, Zhang ZQ, Zhang SM. ROS-p53-cyclophilin-D signaling mediates salinomycin-induced glioma cell necrosis. J Exp Clin Cancer Res. 2015; 34:57.
42. Tung MC, Lin PL, Wang YC, He TY, Lee MC, Yeh SD, Chen CY, Lee H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget. 2015; 6:41692–41705. http://doi.org/10.18632/oncotarget.6150.
43. Ryoo IG, Choi BH, Kwak MK. Activation of NRF2 by p62 and proteasome reduction in sphere-forming breast carcinoma cells. Oncotarget. 2015; 6:8167–8184. http://doi.org/10.18632/oncotarget.3047.
44. Yu M, Li H, Liu Q, Liu F, Tang L, Li C, Yuan Y, Zhan Y, Xu W, Li W, Chen H, Ge C, Wang J, et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell Signal. 2011; 23:883–892.
45. Fernandez-Blanco C, Font G, Ruiz MJ. Oxidative stress of alternariol in Caco-2 cells. Toxicol Lett. 2014; 229:458–464.
46. Cortizo AM, Bruzzone L, Molinuevo S, Etcheverry SB. A possible role of oxidative stress in the vanadium-induced cytotoxicity in the MC3T3E1 osteoblast and UMR106 osteosarcoma cell lines. Toxicology. 2000; 147:89–99.
47. Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007; 6:1239–1248.