
Introduction
Cancer is a leading cause of mortality worldwide. Global cancer burden increased to 14.1 million new cases and 8.2 million cancer deaths in 2012 from 12.7 million and 7.6 million respectively, in 2008. Cancer metastasis and reoccurrence are the major resources of cancer lethality. Cancer stem cells (CSCs) form a subpopulation of cells within tumors and are essential for tumor dissemination and relapse[1]. The acquired properties that make CSCs adaptive to micro-environmental stresses and enhanced resistance to chemo-/radiation-therapy, present a challenge for targeting CSCs within tumors[2-4]. Heterogenetic CSCs are believed to acquire diverse adaptive changes in genetic, epigenetic, signal transduction, metabolic, transcriptional and translational levels[5, 6]. Translation is essential for all aspects of tumor evolution, including tumorigenesis, tumorangiogenesis, tumor growth, metastasis, CSC heterogeneity, and drug resistance[7, 8]. Protein synthesis is central for CSCs self-renewal, maintenance, differentiation, growth and dissemination.
Translation generally initiates with the recruitment of eIF4F to the 5’- m7GpppN cap of messenger RNA (mRNA)[9, 10]. eIF4F is a complex composed of the cap-binding protein eIF4E1, scaffold protein eIF4G1 and RNA helicase eIF4A. The eIF4E1/eIF4G1 interaction is necessary for eIF4F assembly[10-12]. The mRNAs encoding proteins important for cell proliferation and growth frequently harbor secondary structure and/or long 5’-UTR, or other regulatory elements. Translation of mRNAs with structured and/or long 5’-UTR is highly eIF4E1 dependent[13, 14]. Consistently, enhanced eIF4E1 levels and/or activities have been demonstrated in a number of cancers in clinical observations[15-18]. The human eIF4E family has three members of eIF4E1, eIF4E2 and eIF4E3. The eIF4E2-RBM4-HIF-2α complex can initiate translation but eIF4E2 does not bind to eIF4G1[19, 20]. eIF4E3 competes with eIF4E1 in binding cap and acts as a tumor suppressor[21]. Multiple proteins have been demonstrated to be enhanced in breast CSCs, such as the CSC markers NANOG and OCT4[22], the metastasis regulator CXCR4[23, 24], the epithelial–mesenchymal transition (EMT)[25] regulator c-MYB[26], or the cell proliferation key factors c-MYC[27] and cyclin D1[28, 29].
Due to the central roles in cancer evolution, translation is targeted in anti-cancer drug development. Several agents targeting translation initiation have been developed[30, 31], including the small molecule 4EGI-1[32]. 4EGI-1 binds eIF4E1, prevents recruitment of eIF4E1 to eIF4G1. The compound mimics the function of 4E-binding protein 1 (4E-BP1), which regulates eIF4E1-eIF4G interaction in a phosphorylation–dependent manner; however, 4EGI-1 does not displace 4E-BP1 from eIF4E1[32]. 4EGI-1 exhibits inhibitory effects in several cancer cell lines[33-35].
Recently, we have isolated and identified a small fast adhesion subpopulation (CD44high/CD24low)FA from HMLER human mammary epithelial cancer cells [1, 2] that show the typical properties of cancer stem cells and are referred to as breast cancer stem cells (CSCs)[36]. These breast CSCs showed enhanced drug resistance to the anti-cancer drugs actinomycin D and camptothecin (about 10-fold) compared to non-CSCs [36]. However, it is unknown whether 4EGI-1 inhibits breast CSCs and suppresses breast CSC tumor growth, and whether 4EGI-1 interferes with translation selectively in breast CSCs is elusive.
We investigated the effects of 4EGI-1 on breast CSCs of HMLER (CD44high/CD24low)FA cells in vitro and breast CSC tumorangiogenesis and tumor growth in xenografted tumor model in vivo. We found that eIF4E1 is significantly increased in breast CSCs comparing to non-CSC breast cancer cells. 4EGI-1 selectively inhibits breast CSCs in comparison to non-CSC breast cancer cells. Both 4EGI-1 isomers repress breast CSC tumorangiogenesis and tumor growth in mouse model. They inhibit cell proliferation and increase cell apoptosis in breast CSC tumors. 4EGI-1 selectively inhibits translation of mRNAs of NANOG, OCT4, CXCR4, c-MYB, c-MYC and cyclin D1 in breast CSC tumors. These results suggest that 4EGI-1 targets breast CSCs through selectively inhibits translation of mRNAs essential for breast CSCs.
Results
Translation initiation factor eIF4E1 is enhanced in breast CSCs
To investigate the underlying translational mechanisms that are specifically required for CSC character maintenance of breast CSCs (BCSCs), we compared the cellular protein abundances of multiple translation initiation factors by Western blot assays in breast CSCs HMLER (CD44high/CD24low)FA cells and multiple non-CSC breast cancer cells (non-BCSCs). We found that eIF4E1 (also named eIF4E), but not eIF4E2, eIF4G1, eIF1A, eIF2α, or eIF5, is significantly increased in BCSCs, in comparison to SKBR-3, MCF-7 and MDA-MB-231 breast cancer cells and non-BCSCs of HMLER (CD44high/CD24low)SA cells (Fig.1A). eIF4E3 is hardly detectable in all these cancer cells. These results suggest that the activation of eIF4E-dependent translation is enhanced in these BCSCs.
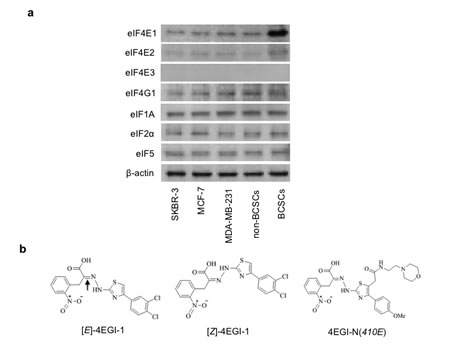
Figure 1: Translation initiation factor eIF4E1 is enhanced in breast CSCs and structures of compounds. (a) Western blot analyses of translation initiation factor abundance in SKBR-3, MCF-7, MDA-MB-231 breast cancer cells, breast CSCs (BCSCs) of HMLER (CD44high/CD24low)FA cells and non-BCSCs of HMLER (CD44high/CD24low)SA breast cancer cells. β-actin was used as a loading control. (b) Structures of 4EGI-1 isomers ([E]- and [Z]-isomer) and inactive analogue 4EGI-N(410E). Arrow shows the double bonds.
Selective inhibition of breast CSCs by 4EGI-1 comparing to non-CSCs
4EGI-1 consists of two isomers: [E]- and [Z]-4EGI-1. To evaluate the effects of 4EGI-1, we synthesized and identified an inactive analogue 4EGI-N(410E) (Fig.1B). We compared the inhibitory effects of 4EGI-1 isomers on breast CSCs and multiple non-CSC breast cancer cells by ATP concentration based cell viability assays. We observed that the IC50 of 4EGI-1 is about 30µM on SKBR-3, MCF-7 and MDA-MB-231 breast cancer cells, about 22µM on non-CSCs, and about 11µM ([E]-)/10µM ([Z]-) on breast CSCs (Table 1). The above data indicate that 4EGI-1 presents increased cytotoxicity (>2-fold) to breast CSCs comparing to non-CSC breast cancer cells, which is in agreement with the significantly enhanced eIF4E1 in breast CSCs (Fig. 1A).
Table 1: Cytotoxicity of 4EGI-1 isomers on breast CSCs and breast cancer cells. |
||
BCSCs and breast cancer cells |
IC50 of [E]-4EGI-1(µM) |
IC50 of [Z]-4EGI-1(µM) |
SKBR-3 |
30±1.5 |
29±1.3 |
MCF-7 |
30±0.9 |
30±1.1 |
MDA-MB-231 |
32±1.2 |
30±1.4 |
Non-BCSCs of HMLER(CD44high/CD24low)SA |
24±1.1 |
22±0.7 |
BCSCs of HMLER(CD44high/CD24low)FA |
11±0.8 |
10±0.6 |
Note: IC50 means inhibitory concentration that leads to 50% cell viability decrease.
4EGI-1 promotes breast CSC differentiation
CSC maintenance and differentiation are important for CSC tumorigenesis and tumor evolution[37]. CD44high/CD24low is one of the defining characteristics of these breast CSCs, whereas CD44low/CD24high represents the CSC-depleted population of these breast cancer cells. We treated the breast CSCs with 4EGI-1 isomers, an inactivate analogue 4EGI-N(410E) and vehicle with series of concentrations for three days followed by flow cytometry analyses with CD44 and CD24 antibodies. 4EGI-1 isomers significantly decreased the percentage of CD44high/CD24low population cells and increased the population of CD44low/CD24high cells (Fig. 2A-2B). 4µM [E]- or [Z]-4EGI-1 depleted most of breast CSCs. Importantly, inactive analogue 4EGI-N(410E) did not affect breast CSCs under the same conditions (Fig. 2A-2B). These results suggested that 4EGI-1 potently depletes breast CSCs and promotes breast CSC differentiation.
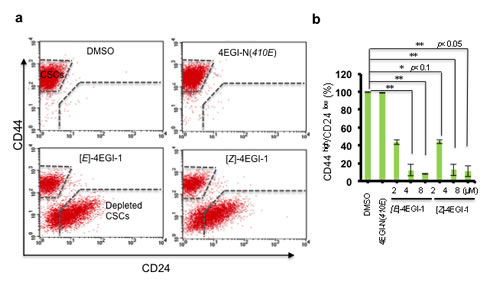
Figure 2: 4EGI-1 promotes breast CSC differentiation. (a) Representative fluorescence-activated cell sorting (FACS) images. CD44high/CD24low population cells and CD44low/CD24high population cells with DMSO, 4µM 4EGI-N(410E) , 4µM[E]-4EGI-1, or 4µM[Z]-4EGI-1 treatment for three days were shown. Both 4EGI-1 isomers promote breast CSC differentiation. (b) Statistical analyses of 4EGI-1 effects on CD44high/CD24low population cell percentage of breast CSCs. Average CD44high/CD24low percentages were shown (mean ± SD, t-test, two-tailed). *: p< 0.1; **: p< 0.05; ***: p< 0.01.
4EGI-1 inhibits breast CSC induced HUVEC tube-like structure formation
Angiogenesis is the process of new vessel generation and growth, which is important for tumor growth and metastasis[38]. CSCs produce multiple proangiogenic factors to stimulate tumor angiogenesis[39]. To examine whether 4EGI-1 inhibits proangiogenic factor protein synthesis in breast CSCs, we employed human umbilical vein endothelial cells (HUVECs). We pretreated breast CSCs with compounds at the indicated concentrations for 3 hours. Then, we embedded these breast CSCs in 50% growth factor reduced Matrigel (mixed with 50% breast CSC medium MEGM) and planted HUVECs on the surface of these Matrigel mixtures with endothelial cell growth medium (EGM) (Fig. 3A). After incubation at 37oC for two days, breast CSCs pretreated with DMSO and inactivate analogue 4EGI-N(410E) grew into tumorspheres, while those treated with 4EGI-1 isomers did not (Fig. 3B). HUVECs cultured on the surface of Matrigel mixture with DMSO pretreated breast CSCs formed tube-like structures (33±2 tubes/HPF, high performance field, 20×), and 32.6±2.58 tubes/HPF with 4EGI-N(410E) pretreated breast CSCs. In contrast, HUVECs only formed 4.5±1 tubes/HPF and 4.1±0.7 tubes/HPF with 8µM [E]- and [Z]-isomer pretreated breast CSCs, respectively (Fig. 3C-D). Importantly, 4EGI-N(410E) did not affect HUVEC tube formation and HUVECs hardly form tubes on growth factor reduced Matrigel (Fig. 3D). These results demonstrated that 4EGI-1 inhibits proangiogenic factor production in breast CSCs and significantly represses breast CSC promoted HUVEC tube formation.
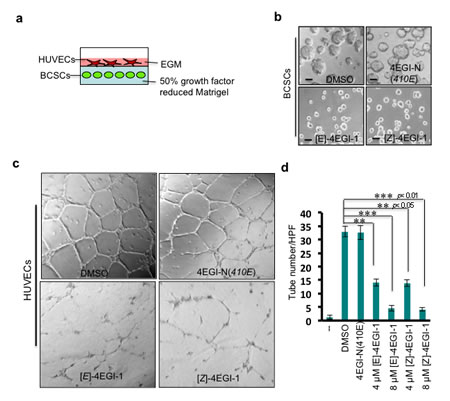
Figure 3: 4EGI-1inhibits breast CSC induced HUVEC tube-like structure formation. (a) Diagram of HUVEC-breast CSC sandwich. Breast CSCs were embedded in 50% growth factor reduced Matrigel (mixed with 50% MEGM medium). Human umbilical vein endothelial cells (HUVECs) were planted on Matrigel mixture in endothelial growth medium (EGM). (b) Representative images of breast CSC mammospheres (top panels) or breast CSCs (bottom panels) in Matrigel mixture. Breast CSCs were pretreated with DMSO, 8µM 4EGI-N(410E), 8µM[E]-4EGI-1, or 8µM[Z]-4EGI-1 for 1 day, followed by culturing in Matrigel mixture for three days. Images were taken at the third day in Matrigel mixture. Bar=10µm. (c) Representative images of HUVEC tube-like structures. HUVECs were cultured on Matrigel mixture containing 8µM compound pretreated breast CSCs for 24hr. Images were taken at 24hr. Breast CSCs (mammospheres) promote HUVEC tube formation, and 4EGI-1 inhibits this capacity of breast CSCs. (d) Statistical analyses of 4EGI-1 effects on 4EGI-1 effects on breast CSC stimulation of HUVEC tube formation. “ˍ ˍ” means Matrigel mixture contains no breast CSCs. 50% growth factor reduced Matrigel does not lead to HUVEC tube formation. Average tube numbers per high performance field (20×) were shown (mean ± SD, t-test, two-tailed). **: p< 0.05; ***: p< 0.01.
4EGI-1 suppresses breast CSC tumor growth and tumorangiogenesis
To investigate the effects of 4EGI-1 on breast CSC tumor growth in vivo, we employed xenografted tumor model in mouse. After the breast CSC tumor formation (tumor volume is about 75 mm3), we injected both 4EGI-1 isomers 75 mg/kg/day (5 mice for each group) by intraperitoneal injection (I.P.) for 30 days. Tumor volumes were measured every three days. At the 30th day, the average breast CSC tumor volume of vehicle treatment was 237.4±18.6 mm3, whereas the average breast CSC tumor volumes of [E]- and [Z]- isomer treated were 113.2±11.4 mm3 and 123.9±10.8 mm3, respectively (Fig. 4A). The average breast CSC tumor weights of vehicle, [E]-, [Z]- isomer treated were 0.0825±0.01145g, 0.036±0.00618g, 0.038±0.0077g, respectively (Fig. 4B). These data demonstrated that 4EGI-1 effectively suppressed breast CSC tumor growth in vivo. Then, we performed hematoxylin and eosin (H&E) staining and immunohistostaining with anti-mouse CD31 (vascular endothelial cell marker) antibodies with breast CSC tumor sections. 4EGI-1 significantly decreased vessel numbers in breast CSC tumors (Fig. 4C), suggesting that 4EGI-1 inhibits angiogenesis in breast CSC tumors in vivo. Next, we performed immunohistostaining to detect cell proliferation marker Ki-67 and cell apoptosis marker cleaved CASP3 (cCASP3) in these breast CSC tumor sections. 4EGI-1 decreased Ki-67 in most breast CSC tumor cells and increased cCASP3 in a few breast CSC tumor cells (Fig. 4D), suggesting that 4EGI-1 significantly inhibits breast CSC proliferation and can induce apoptosis in breast CSCs in vivo. Furthermore, we found 4EGI-1 strikingly decreased cell proliferation key regulators cyclin D1 and c-MYC in breast CSC tumor cells (Fig. 4E), indicating that 4EGI-1 inhibits translation essential for cell proliferation in breast CSCs.
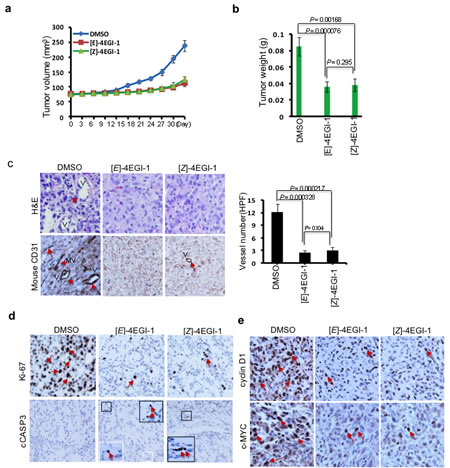
Figure 4: 4EGI-1 suppresses breast CSC tumor growth and tumorangiogenesis in vivo. (a) The graph represents average tumor volumes of each group over time. The average tumor volume from DMSO treated group increased from 74.30±2.43 to 237.4± 18.6mm3, whereas that from [E]-4EGI-1 treated group increased from 76.87±1.96 to 113.2±11.4 mm3, and that from [Z]-4EGI-1 increased from 75.01±1.75 to 123.9 ±10.8 mm3. Bar represents tumor volume mean ± SD (n=5 mice, t-test, p<0.001). (b) Average tumor weights of each group. The average tumor weight from DMSO treated group is 0.08524±0.01145g, whereas that from [E]-4EGI-1 treated group is 0.036± 0.00618g and that from [Z]-4EGI-1 treated group is 0.038±0.0077g (mean ± SD, n=5, t-test). (c) Representative H&E staining (top panels) and immunohistostaining (bottom panels) images of anti-mouse CD31 antibody in tumor cross-sections. Left panel: Statistical data from immunohistostaining analyses expressed as average tumor vessel number per high performance field (HPF, 200×) with 12.2±1.92 vessel/HPF in DMSO treated tumor group, 2.4±0.54 vessel/HPF in [E]-4EGI-1treated tumor group, and 3±0.7 vessel/HPF in [E]-4EGI-1treated tumor group (mean ± SD, n=6, t-test). Arrows indicate vessels and microvessels. V: vessels; MV: microvessels. (d) Representative immunohistostaining images of tumor cross-sections with anti-Ki-67 and anti-cleaved Caspase 3 (cCASP3) antibodies. Arrows indicate signals of Ki-67 or cCASP3. Framed areas show the enlarged images of cCASP3 signals. (e) Representative immunohistostaining images of cross-sections with anti-cyclin D1 and anti-c-MYC antibodies. Arrows indicate signals of cyclin D1 or c-MYC.
4EGI-1 selectively inhibits translation that persists in CSC maintenance and dissemination
To examine the 4EGI-1 effects on translation essential for CSC maintenance and dissemination, we performed Western blot with cellular proteins extracted from these breast CSC tumors. 4EGI-1 treated tumor cells presented decreased CSC pluripotency markers NANOG and OCT4, tumor metastasis regulator CXCR4, EMT mediator c-MYB and proangiogenic factor VEGF[40, 41], but not translation initiation factors eIF1A or eIF5, comparing to vehicle treated breast CSC tumor cells in vivo (Fig. 5A). Previously, we reported that 4EGI-1 inhibits translation of several oncogene mRNAs without affecting their transcription in multiple cancer cell lines at 24hr under normoxia and at 12hr under hypoxia conditions in vitro[16, 32]. These observations suggested that the indicated decreased protein levels in 4EGI-1 treated BCSC tumors were primarily, if not all, caused by selective inhibition of translation of mRNAs encoding these proteins. Importantly, we found that 4EGI-1 isomers inhibit the eIF4E1-eIF4G1 interaction and increase 4E-BP1 stability of binding to eIF4E1 in breast CSC tumor cells in vivo (Fig. 5B). These results demonstrated that 4EGI-1 selectively inhibits translation of mRNAs encoding proteins that persists in CSC maintenance, pro-angiogenesis and metastasis.
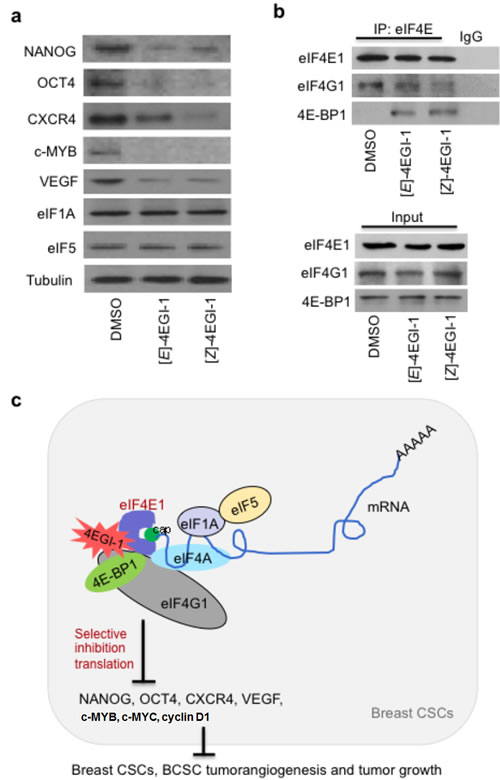
Figure 5: 4EGI-1 selectively inhibits translation in breast CSC tumors. (a) 4EGI-1 decreases NANOG, OCT4, CXCR4, c-MYB and VEGF, but not eIF1A or eIF5, in breast CSC tumors. Western blot assays of 4EGI-1 effects on translation in breast CSC tumors. Total proteins were extracted from tumor tissues. Western blot were performed three independent experiments with indicated antibodies. Tubulin was used as a loading control. (b) 4EGI-1 disrupts eIF4E1-eIF4G1 interaction and increases stability of 4E-BP1 binding to eIF4E1 in breast CSC tumors. Co-immunoprecipitation assays were performed using anti-eIF4E antibody or control IgG. “Input” represents 10% of total protein used in immunoprecipitation. (c) Schematic diagram for enhanced eIF4E1 in breast CSCs, selective translation initiation interference by 4EGI-1 in breast CSCs, and inhibitory effects of 4EGI-1 on breast CSC tumorangiogenesis and tumor growth. 4EGI-1 binds to eIF4E1, inhibits eIF4E1-eIF4G1 interaction, and increase 4E-BP1 binding to eIF4E1. 4EGI-1 selectively inhibits translation essential for breast CSC maintenance, proliferation and metastasis, and suppresses breast CSC tumorangiogenesis and tumor growth.
Discussion
Cancer stem cells are critical for tumor metastasis and reoccurrence, which are the major resources of cancer-related lethality. CSCs have acquired enhanced drug resistance. Here we found that small molecule 4EGI-1 preferentially inhibits breast CSCs compared to non-CSC breast cancer cells in vitro and effectively suppresses breast CSC tumor growth and tumorangiogenesis in vivo. 4EGI-1 promotes breast CSC differentiation, inhibits breast CSC tumorsphere growth and suppresses HUVEC tube formation. 4EGI-1 significantly inhibits breast CSC proliferation and can induce apoptosis in breast CSC tumor cells. Furthermore, we found that 4EGI-1 selectively inhibits translation of mRNAs encoding proteins that are enhanced in CSC maintenance, proliferation and metastasis, such as NANOG, OCT4, c-MYC, cyclin D1, CXCR4, VEGF and c-MYB, in breast CSC tumor cells. Our study suggests that selective translation initiation interference by 4EGI-1 inhibits breast CSC tumor growth and tumorangiogenesis (Fig. 5C).
Malignant cancer cells typically undergo extensive adaptive genetic, epigenetic and metabolic changes to support the heterogeneous malignant phenotypes, yet cell survival and proliferation may dependent on one or a subset of driver mutations[42, 43]. This continued dependence on the driver mutation is deferred to oncogene addiction[42]. Inhibition of the addictive oncoproteins can lead to therapeutic intervention, which is the basis for a considerable promise of current chemotherapy. However, the followed tumor reoccurrence, tumor metastasis and drug resistance suggest limits of using inhibitors targeting one or a few addictive oncoproteins. Although it is known that: 1) CSC quiescence decreases drug sensitivity; and 2) CSCs may never acquire addiction to the oncogene that drives disease development, the mechanisms of enhanced drug resistance of CSCs are elusive[4]. Despite of the extreme complexity of increased drug resistance of CSCs[45], it is possible to target the CSC-specific adaptive translational mechanisms that constantly produce proteins to support CSC maintenance, self-renewal and metastasis. Here, we found that translation initiation factor eIF4E1, but not eIF4E2, eIF4G1, eIF1A, eIF2α, or eIF5, is significantly enhanced in breast CSCs in comparison to non-CSC breast cancer cells. We show that the eIF4E1-eIF4G1 interaction inhibitor 4EGI-1 preferentially inhibits breast CSCs compared to non-CSC breast cancer cells, effectively suppresses breast CSC tumor growth, selectively inhibiting translation of mRNAs encoding NANOG, OCT4, CXCR4, c-MYC, cyclin D1, c-MYB and VEGF, but not eIF1A or eIF5. CXCR4 and c-MYC are addictive oncoproteins of many malignant tumors. Therefore, 4EGI-1 both inhibits addictive oncoproteins and interferes with the CSC adaptive protein synthesis machinery in breast CSCs. These results not only demonstrate that 4EGI-1 is a potential candidate for CSC-targeted cancer therapy, but more importantly, suggest that the eIF4E1-eIF4G1 interaction is an available target for the selective inhibition of CSCs.
It has been demonstrated that translation of mRNAs encoding proteins important for cell proliferation and survival is highly eIF4E1 dependent [15, 44]. Here we show that 4EGI-1 selectively inhibits translation essential for breast CSCs, including NANOG, OCT4, CXCR4, c-MYC, c-MYB and VEGF. Data shown here and elsewhere[32] have consistently evidenced that 4EGI-1 performs selective translation interference in breast CSCs and multiple cancer cell lines. Here we show that 4EGI-1 increases cytotoxicity to breast CSCs that exhibit enhanced drug resistance to anti-cancer drugs of Actinomycin D and Camptothecin (about 10-fold). This suggests that 4EGI-1 can selectively target breast CSCs. The intraperitoneal injection and suppression of breast CSC tumor growth, and the low side effects on mice (data not shown) in this study reflect the selective breast CSC inhibition by 4EGI-1. In short, 4EGI-1 exhibits capacities of both selective translation initiation interference and selective breast CSC inhibition.
CSC maintenance facilitates tumorigenesis and tumor reoccurrence[22], and CSC quiescence decreases sensitivity to inhibitors[45, 46]. Furthermore, CSC depletion reduces CSC tumorigenesis capacity. We found that 4EGI-1 inhibits breast CSC maintenance and promotes breast CSC differentiation with decreased CD44high/CD24low population and increased CD44low/CD24high population cells. Furthermore, 4EGI-1 decreased CSC markers of NANOG and OCT4 in breast CSC tumors, which associate with hypoxia (low oxygen) within solid tumors. These consistent results demonstrate that 4EGI-1 effectively promotes breast CSC differentiation by selectively impairing translation required for CSC maintenance.
Cancer cells have acquired capacities of uncontrolled proliferation and apoptosis evasion[47]. We found that 4EGI-1 significantly decreases cell proliferation, and c-MYC and cyclin D1 protein levels in breast CSC tumors. 4EGI-1 can induce apoptosis in CSCs within breast CSC tumors. These observations evidenced that selective translation initiation interference may be an avenue to effectively suppress proliferation in and induce apoptosis into CSCs. On the other hand, targeting CSC adaptive translation machinery may overcome: 1) inherent resistance to drugs targeting one or a few oncoproteins, which is caused by CSC heterogeneity and/or compensation of diverse/redundant oncoproteins in a type of CSCs[41]; and 2) acquired resistance due to mutations after drug treatment[48, 49].
We present here the first small molecule activities against breast cancer stem cells through selective translation initiation interference. Selective inhibition of translation of mRNAs encoding proteins essential for CSC maintenance, self-renewal, proliferation, differentiation, and dissemination could be an avenue to restrict and inhibit CSC activities, including CSC tumorangiogenesis and tumor growth. Targeting selective translation initiation is expected to overcome resistance due to mutations acquired during drug treatment and the related inherent CSC heterogeneity.
Material and Methods
Cells, antibodies, reagents and mice
HMLER cell line was kindly provided by Robert Weinberg (Whitehead Institute for Biomedical Research of Massachusetts Institute of Technology). HMLER(CD44high/ CD24low)FA cells were prepared as previously described[36]. MEGM and MEBM media, EGM media, and Human Umbilical Vein Endothelial Cells (HUVECs) were purchased from Lonza. The [E]- and [Z]-4EGI-1were either self-synthesized or ordered from Speed Chemical. 4EGI-N (410E) was self-synthesized. All compounds were dissolved in DMSO. NOD-SCID (strain name: NOD.CB17-Prkdcscid/J) mice were ordered from The Jackson Laboratory. Anti-Ki-67(SP6) antibody was ordered from Vector (#VP-RM04). Anti-cleaved CASP3 (5A1E) antibody for immunostaining was ordered from Cell Signaling (#9664s). Anti-CD31 (mouse) antibody was ordered from Dako (#N1596). Anti-c-Myc (N-term) antibody used for immunostaining was ordered from Epitomics (#1472-1). Anti-cyclin D1 antibody used for immunostaining was ordered from Neomarkers (#RM-9104-S). Anti-eIF4E1, anti-eIF4G1, anti-4E-BP1, and anti-β-actin antibodies were ordered from Cell Signaling. Cancer cell lines of SKBR-3, MCF-7, MDA-MB-231 were ordered from ATCC. Anti-c-MYC and anti-cyclin D1 antibodies were ordered from Cell Signaling.
Western blot assay
Cellular protein extraction and Western blot assays were performed as previously described with RIPA buffer (50mM Tris-HCl pH 7.4, 150mM NaCl, 1% Triton-X100, 0.1% SDS, 0.25% Na-deoxycholate, 1mM PMSF, 1×Roche complete mini protease inhibitor cocktail). Immuno blots were performed with indicated antibodies. All Western blot experiments were performed three times.
Cell viability assay
1 × 104 breast CSCs HMLER (CD44high/CD24low)FA cells and other indicated breast cancer cells were treated with DMSO, or [E]-4EGI-1 or [Z]-4EGI-1 at series of concentrations for 24 hours. The cells were performed cell viability assays with CellTiter-Glo® luminescent cell viability assay kit (Promega) according to the manual description. Three independent experiments were performed. Average IC50 results were shown (mean ± SD, t-test, two-tailed).
Fluorescence-activated cell sorting (FACS)
Breast CSCs were treated with the indicated compounds at indicated concentrations for three days, followed by fluorescence-activated cell sorting (FACS) assay with FITC-conjugated anti-CD44 (Biosciences, G44-26) antibodies and PE-conjugated anti-CD24 (Biosciences, ML15) antibodies.
HUVEC tube-like structure formation
A 200μl 50% growth factor reduced Matrigel mixture (BD Medical, mixed with MEGM at ratio of 1:1) was added to each well of 24-well plates followed by incubation at 37oC for 30 minutes. Breast CSCs were treated with DMSO, 4EGI-N(410E), [E] and [Z] at indicated concentrations for one day, and cultured on Matrigel mixture, and covered by another 150 μl Matrigel mixture followed by incubation at 37oC for 30 minutes. Then 0.3 ml MEGM was added to each well and the plates were incubated at 37oC, 5% CO2 for 3 days. Images of breast CSCs or tumorspheres were taken by Nikon camera. After aspiration of the MEGM medium, 4×104 HUVECs were cultured into each well together with 0.3 ml EGM medium followed by incubation at 37oC, 5% CO2 for 24 hours. Images of HUVEC tube-like structure were taken by Nikon camera. The HUVEC tube-like structures in high performance fields (20×) were counted. The experiments were performed three independent times.
Tumor xenografted assay
In the tumor xenografted assay, 1×105 breast CSCs were mixed with 100μl Matrigel/DMEM mixture (Matrigel: DMEM = 1:2) (BD Bioscience). Breast CSCs/Matrigel/DMEM mixtures were injected into NOD/SCID female mice (the Jackson Laboratory) mammary glands by subcutaneous injection. After the tumor formation (about 75 mm3 in volume, 5 mice/group), DMSO, or 75mg/kg [E]-4EGI-1, or 75mg/kg [Z]-4EGI-1 was injected into the mice by intraperitoneal injection daily for 30 days. Tumor volumes were measured every three days. At the 30th day, mice were sacrificed and tumors were excised. Tumors weights were measured. Tumor tissue samples were used for immunohistostaining, Western blot and immunoprecipitation analyses. The mouse experiments were performed according to the policies of Harvard Medical Animal Committee.
Statistical analysis
Quantitative data were statistically analyzed (mean ± SD, t-test, two-tailed). Statistical significance was determined by t-test. Significance was expressed as: *: p< 0.1; **: p< 0.05; ***: p< 0.01, or with the p-value. P<0.05 was considered significant[2].
Acknowledgements
We appreciate Dr. Robert Weinberg of Massachusetts Institute of Technology for the generous gift of HMLER cell line and NIH grants CA127990 (to GW).
REFERENCES
1. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ and Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100:3983-8.
2. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA and Lander ES (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138:645-59.
3. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756-60.
4. Dean M, Fojo T and Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5:275-84.
5. Hanahan D and Weinberg RA (2000) The hallmarks of cancer. Cell 100:57-70.
6. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL and Clarke MF (2009) Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458:780-3.
7. Avdulov S, Li S, Michalek V, Burrichter D, Peterson M, Perlman DM, Manivel JC, Sonenberg N, Yee D, Bitterman PB and Polunovsky VA (2004) Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 5:553-63.
8. Silvera D, Formenti SC and Schneider RJ Translational control in cancer. Nat Rev Cancer 10:254-66.
9. Marintchev A and Wagner G (2004) Translation initiation: structures, mechanisms and evolution. Q Rev Biophys 37:197-284.
10. Gingras AC, Raught B and Sonenberg N (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68:913-63.
11. Gross JD, Matsuo H, Fletcher M, Sachs AB and Wagner G (2001) Interactions of the eukaryotic translation initiation factor eIF4E. Cold Spring Harb Symp Quant Biol 66:397-402.
12. Gross JD, Moerke NJ, von der Haar T, Lugovskoy AA, Sachs AB, McCarthy JE and Wagner G (2003) Ribosome loading onto the mRNA cap is driven by conformational coupling between eIF4G and eIF4E. Cell 115:739-50.
13. Koromilas AE, Lazaris-Karatzas A and Sonenberg N (1992) mRNAs containing extensive secondary structure in their 5’ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. Embo J 11:4153-8.
14. Pickering BM and Willis AE (2005) The implications of structured 5’ untranslated regions on translation and disease. Semin Cell Dev Biol 16:39-47.
15. Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW and Sonenberg N (2004) eIF4E--from translation to transformation. Oncogene 23:3172-9.
16. Yi T, Papadopoulos E, Hagner PR and Wagner G (2013) Hypoxia-inducible factor-1alpha (HIF-1alpha) promotes cap-dependent translation of selective mRNAs through up-regulating initiation factor eIF4E1 in breast cancer cells under hypoxia conditions. J Biol Chem 288:18732-42.
17. Lazaris-Karatzas A, Montine KS and Sonenberg N (1990) Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5’ cap. Nature 345:544-7.
18. Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C and Pandolfi PP (2004) The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med 10:484-6.
19. Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, Payette J, Holcik M, Pause A and Lee S (2012) An oxygen-regulated switch in the protein synthesis machinery. Nature 486:126-9.
20. Marcotrigiano J, Gingras AC, Sonenberg N and Burley SK (1999) Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol Cell 3:707-16.
21. Osborne MJ, Volpon L, Kornblatt JA, Culjkovic-Kraljacic B, Baguet A and Borden KL (2013) eIF4E3 acts as a tumor suppressor by utilizing an atypical mode of methyl-7-guanosine cap recognition. Proc Natl Acad Sci U S A 110:3877-82.
22. Mohyeldin A, Garzon-Muvdi T and Quinones-Hinojosa A (2010) Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7:150-61.
23. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E and Zlotnik A (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50-6.
24. Trautmann F, Cojoc M, Kurth I, Melin N, Bouchez LC, Dubrovska A and Peitzsch C (2014) CXCR4 as Biomarker for Radioresistant Cancer Stem Cells. Int J Radiat Biol.
25. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J and Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704-15.
26. Cesi V, Casciati A, Sesti F, Tanno B, Calabretta B and Raschella G (2011) TGFbeta-induced c-Myb affects the expression of EMT-associated genes and promotes invasion of ER+ breast cancer cells. Cell Cycle 10:4149-61.
27. Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, Rao PH and Ruggero D (2008) Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456:971-5.
28. Knudsen KE, Diehl JA, Haiman CA and Knudsen ES (2006) Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene 25:1620-8.
29. Liao DJ, Thakur A, Wu J, Biliran H and Sarkar FH (2007) Perspectives on c-Myc, Cyclin D1, and their interaction in cancer formation, progression, and response to chemotherapy. Crit Rev Oncog 13:93-158.
30. Bordeleau ME, Cencic R, Lindqvist L, Oberer M, Northcote P, Wagner G and Pelletier J (2006) RNA-mediated sequestration of the RNA helicase eIF4A by Pateamine A inhibits translation initiation. Chem Biol 13:1287-95.
31. Cencic R, Carrier M, Galicia-Vazquez G, Bordeleau ME, Sukarieh R, Bourdeau A, Brem B, Teodoro JG, Greger H, Tremblay ML, Porco JA, Jr. and Pelletier J (2009) Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS One 4:e5223.
32. Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA and Wagner G (2007) Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 128:257-67.
33. Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, Supko JG, Chorev M, Wagner G and Halperin JA (2012) Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 3:869-81.
34. Willimott S, Beck D, Ahearne MJ, Adams VC and Wagner SD (2013) Cap-translation inhibitor, 4EGI-1, restores sensitivity to ABT-737 apoptosis through cap-dependent and -independent mechanisms in chronic lymphocytic leukemia. Clin Cancer Res 19:3212-23.
35. Descamps G, Gomez-Bougie P, Tamburini J, Green A, Bouscary D, Maiga S, Moreau P, Le Gouill S, Pellat-Deceunynck C and Amiot M (2012) The cap-translation inhibitor 4EGI-1 induces apoptosis in multiple myeloma through Noxa induction. Br J Cancer 106:1660-7.
36. Yi T, Zhai B, Yu Y, Kiyotsugu Y, Raschle T, Etzkorn M, Seo HC, Nagiec M, Luna RE, Reinherz EL, Blenis J, Gygi SP and Wagner G (2014) Quantitative phosphoproteomic analysis reveals system-wide signaling pathways downstream of SDF-1/CXCR4 in breast cancer stem cells. Proc Natl Acad Sci U S A. 111:E2182-90.
37. Smalley M and Ashworth A (2003) Stem cells and breast cancer: A field in transit. Nat Rev Cancer 3:832-44.
38. Folkman J (2007) Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov 6:273-86.
39. Liotta LA, Steeg PS and Stetler-Stevenson WG (1991) Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell 64:327-36.
40. Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S, Chimenti S, Landsman L, Abramovitch R and Keshet E (2006) VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 124:175-89.
41. Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1:27-31.
42. Meacham CE and Morrison SJ (2013) Tumour heterogeneity and cancer cell plasticity. Nature 501:328-37.
43. Holzel M, Bovier A and Tuting T (2013) Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer 13:365-76.
44. Matsuo H, Li H, McGuire AM, Fletcher CM, Gingras AC, Sonenberg N and Wagner G (1997) Structure of translation factor eIF4E bound to m7GDP and interaction with 4E-binding protein. Nat Struct Biol 4:717-24.
45. Kusumbe AP and Bapat SA (2009) Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res 69:9245-53.
46. Li L and Bhatia R (2011) Stem cell quiescence. Clin Cancer Res 17:4936-41.
47. Igney FH and Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2:277-88.
48. Bouwman P and Jonkers J (2012) The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer 12:587-98.
49. Lito P, Rosen N and Solit DB (2013) Tumor adaptation and resistance to RAF inhibitors. Nat Med 19:1401-9.