
INTRODUCTION
Recent developments in genomic techniques have tremendously improved our knowledge about the complex genomic architecture of acute myeloid leukemia (AML) and have shed further light on the genetic heterogeneity of the disease. Despite the increasing number of genetic abnormalities only a minority of these findings has entered clinical practice by displaying prognostic and predictive relevance used for risk-adapted therapeutic strategies [1–6]. Most genetic studies have been descriptive and do not clarify functional relevance of specific candidate genes located in recurrently altered genomic regions.
Using high-resolution single-nucleotide polymorphism (SNP) analyses we have previously identified a recurrent microdeletion at chromosomal band 3p14.1-p13 in cytogenetically normal (CN-) AML [7, 8]. In addition, we found this commonly deleted region (CDR) also in complex karyotype (CK-) AML [9]. The CDR spans a genomic region of 2 Mbp including eight protein-coding genes [forkhead box P1 (FOXP1), eukaryotic translation initiation factor 4E family member 3 (EIF4E3), G protein-coupled receptor 27 (GPR27), prokineticin 2 (PROK2), RING1 and YY1 binding protein (RYBP), H/ACA ribonucleoprotein assembly factor (SHQ1), glucoside xylosyltransferase 2 (GXYLT2), and protein phosphatase 4 regulatory subunit 2 (PPP4R2)] and one microRNA (miR-1284). Recurrent copy number loss with focus on the region of chromosome 3p was recently reported for patients with solid cancers like prostate and cervical cancer [10–14]. Several genes affected by the CDR, e.g. FOXP1, RYBP, and SHQ1, have been implicated to cooperate tumor suppressive functions that are related to apoptosis, proliferation, metastasis, and chemoradioresistance [10, 12, 14]. Furthermore, loss and downregulation of candidate genes within the CDR (e.g. RYBP, SHQ1, FOXP1) have been associated with poor clinical outcome in different types of solid tumors like cervical cancer, non-small cell lung cancer, hepatocellular carcinoma, or breast cancer [11, 12, 15, 16]. The human myeloid leukemic cell line MEG-01 carries a homozygous deletion of PPP4R2 in addition to the heterozygous deletion of the other 3p candidate genes, which suggests a potential role of PPP4R2 in hematopoiesis and leukemia development. Regulatory subunits like PPP4R2 govern the activity and substrate specificity of the catalytic subunit of the protein phosphatase 4 complex (PPP4) [17]. Previous studies highlighted a functional role of PPP4R2 in cell development and differentiation, apoptosis, tumor progression, and DNA repair [18–27], which prompted us to focus the present work on the investigation of the candidate gene PPP4R2. Genetic inactivation studies identified that PPP4R2 deficiency impedes efficient repair of DNA damage in human cells [23–25, 27] by regulating the de-phosphorylation of critical DNA damage response (DDR) proteins that are required for efficient DNA repair like the phosphorylated KRAB-domain associated protein 1 (pKAP1), the phosphorylated histone variant H2AX (γH2AX), or the phosphorylated replication protein A2 (pRPA2) [23–27]. Furthermore, depletion of PPP4R2 resulted in elevated levels of DDR proteins γH2AX, pKAP1, or pRPA2, and has been associated with impaired homologous recombination- (HR) and non-homologous end-joining (NHEJ)-mediated DNA repair [23–27] representing the two major pathways that counteract DNA double strand breaks (DSB). DDR is an important mechanism to maintain genomic integrity, and is mediated by sequential phosphorylation of DDR proteins to recruit DNA repair and signaling proteins to the site of DNA damage [28]. The balance and termination upon activation of DDR signaling is carried out by protein phosphatases like PPP4.
The role of PPP4R2 in governing PPP4 and its de-phosphorylation activity has not been investigated in myelopoiesis and associated neoplasia so far. Here, we identified PPP4R2 as a critical regulator of cell survival and DDR signaling in normal hematopoietic and leukemic cells. Enhanced DNA damage as result of reduced PPP4R2 expression might be one possible mechanism by which the 3p CDR contributes to the pathogenesis of AML.
RESULTS
Differential PPP4R2 expression levels point to functional relevance during hematopoiesis and neoplasia
By screening the AML Study Group (AMLSG) metadatabase we were able to enlarge the cohort of AML patients encompassing the CDR at chromosomal band 3p14.1-p13 [7–9]. In total, we identified 10 AML patients harboring the 3p microdeletion within a normal karyotype, and additional 29 CK-AML cases with genomic loss at chromosome 3p affecting the CDR at 3p14.1-p13 including PPP4R2 (Supplementary Table 1). Deletion of this region was validated by cytogenetic and/or fluorescence in situ hybridization (FISH) analysis as well as SNP array analyses [7, 9]. First, we assessed whether PPP4R2 is mutated in AML patients (Supplementary Table 2). We screened a cohort of primary AML patient samples (n = 89) including CN-AML patients with 3p CDR for mutations in PPP4R2. This cohort was selected based on gene expression profiling (GEP) data and included patients with high (n = 30), intermediate (n = 29), and low (n = 30) global PPP4R2 expression. Despite a polymorphism in exon 8 (HetC+Tpos1, rs61188513) in 12% of patients (n = 11; high expression, n = 5; intermediate expression, n = 1; low expression, n = 5) and several polymorphisms at intron/exon boundaries, no PPP4R2 mutations were detected in this patient cohort. To our knowledge, none of the polymorphisms have been linked to leukemia or cancer so far.
We next investigated PPP4R2 mRNA expression levels during maturation of murine hematopoiesis as well as in primary human AML samples by quantitative real-time PCR (qRT-PCR). Single gene expression was analyzed in the following murine hematopoietic subpopulations: Lin-Sca+cKit+ cells (LSK), common myeloid progenitors (CMP), common lymphoid progenitors (CLP), megakaryocytic erythroid progenitors (MEP), granulocyte monocyte progenitors (GMP), macrophages, and granulocytes. Compared to LSK cells that comprise hematopoietic stem and progenitor cells (HSPC), Ppp4r2 expression decreases significantly during myeloid differentiation (Figure 1A). We detected similar Ppp4r2 mRNA levels in LSK and CLP, but decreased Ppp4r2 expression in CMP and GMP and even lower expression in macrophages (p = 0.002), and granulocytes (p = 0.0003). Of note, publicly available microarray datasets [29] displayed comparable expression pattern of PPP4R2 in normal human myelopoiesis with higher PPP4R2 expression in HSPC, and lower expression in more mature myeloid cells (Supplementary Figure 1).
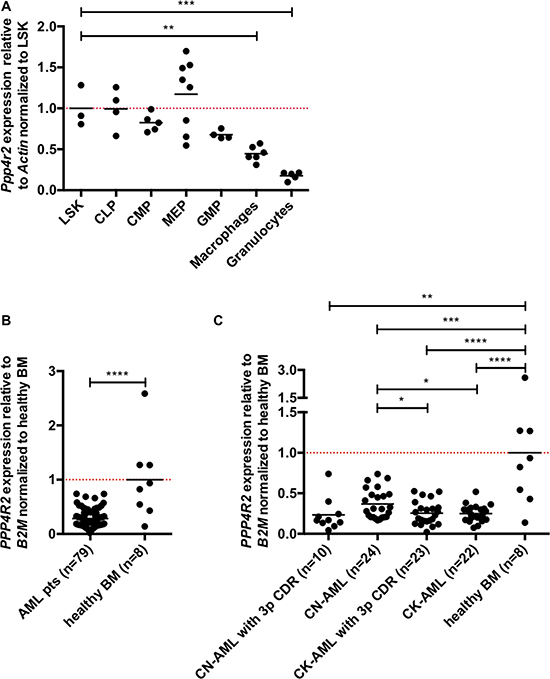
Figure 1: Ppp4r2 is differentially expressed in hematopoiesis and neoplasia. Expression of Ppp4r2 in murine hematopoietic subpopulations and PPP4R2 in AML patients was determined by qRT-PCR. (A) Ppp4r2 single gene expression among murine hematopoietic subpopulations relative to the housekeeping gene Actin and normalized to Lin-/Sca1+/ckit+ (LSK) cells [LSK (n = 3); common lymphoid progenitors (CLP, n = 4); common myeloid progenitors (CMP, n = 5); megakaryocyte/erythrocyte progenitors (MEP, n = 8); granulocyte/monocyte progenitors (GMP, n = 4); macrophages (n = 6); granulocytes (n = 5)]. (B) PPP4R2 mRNA expression relative to the housekeeping gene beta-2 microglobulin (B2M) in a selected cohort of AML patients (n = 79) normalized to healthy bone marrow (BM; n = 8). (C) PPP4R2 mRNA expression relative to B2M in distinct AML subgroups [CN-AML with 3p CDR (n = 10), CN-AML (n = 24), CK-AML with 3p CDR (n = 23), CK-AML (n = 22)] normalized to healthy BM (n = 8). Data are represented by each individual data point and the mean. Statistical analyses were carried out using unpaired two-tailed t-test. A p-value ≤0.05 was considered significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
To obtain first insights in the role of PPP4R2 in human leukemia, we investigated PPP4R2 mRNA expression levels in a selected cohort of AML patients with and without 3p CDR [n = 79; 3p CDR within CN-AML (n = 10), CN-AML no 3p deletion (n = 24), 3p CDR within CK-AML (n = 23), CK-AML no 3p deletion (n = 22)] in comparison to bone marrow (BM) of healthy controls (n = 8). In general, PPP4R2 was lower expressed in AML patients compared to healthy BM (p < 0.0001; Figure 1B). With regard to distinct subgroups compared to healthy BM, patients with 3p CDR within CN-AML (p = 0.007), 3p CDR within CK-AML (p < 0.0001) as well as CN-AML (p = 0.0005) and CK-AML (p < 0.0001) displayed significantly lower PPP4R2 mRNA levels (Figure 1C). Compared to CN-AML, PPP4R2 expression was significantly lower in the other subgroups 3p CDR within CN-AML (p = 0.07), 3p CDR within CK-AML (p = 0.02) as well as CK-AML (p = 0.01).
Ppp4r2 suppression regulates DNA damage response in normal murine BM cells
Differential PPP4R2 expression in hematopoietic cells and primary AML samples prompted us to hypothesize that PPP4R2 might play a functional role in leukemia. We genetically inactivated Ppp4r2 by RNAi in murine hematopoietic progenitor cells using two different shRNAs (Ppp4r2-sh3, Ppp4r2-sh4) and a non-targeting control (Supplementary Figure 2A). First, we assessed whether Ppp4r2 impacts on the ability of HSPC to divide and proliferate to form colonies and cell clusters. There was no apparent effect of Ppp4r2 knockdown on colony formation of murine hematopoietic progenitor cells (Figure 2A). PPP4R2 has been shown to be involved in DNA repair by governing the de-phosphorylation activity and substrate specificity of the PPP4 complex [23, 24, 26, 27]. To investigate the contribution of PPP4R2 to DNA repair of normal hematopoietic cells, we assessed for DDR signaling proteins pKAP1 (S824) and γH2AX (S139) by flow cytometry. Determination of DDR induced by IR with 2 Gy [directly after irradiation (further referred to as 0 h post IR), 0.5 h, 2 h, and 6 h post IR] showed that Ppp4r2 knockdown resulted in elevated levels of pKAP1 (Ppp4r2-sh4: 0 h post IR, p = 0.003; 0.5 h post IR, p = 0.008), most prominent at 0.5 h post IR as depicted in Figure 2B. DNA damage measured by γH2AX was increased upon Ppp4r2 knockdown followed by IR with 2 Gy (Figure 2C). Western Blot analysis confirmed elevated pKAP1 protein levels upon knockdown of Ppp4r2 as well as activated P53 [phosphorylated P53 (S15), pP53] after IR with 2 Gy (Figure 2D). Further investigation of apoptosis induction in response to DNA damage determined as percentage of AnnexinV+/7-AAD- cells displayed an increase in early apoptotic cells upon Ppp4r2 knockdown (Ppp4r2-sh3: 0 h post IR, p = 0.004; 24 h post IR, p = 0.007; Ppp4r2-sh4: 48 h post IR, p = 0.02; Figure 2E).
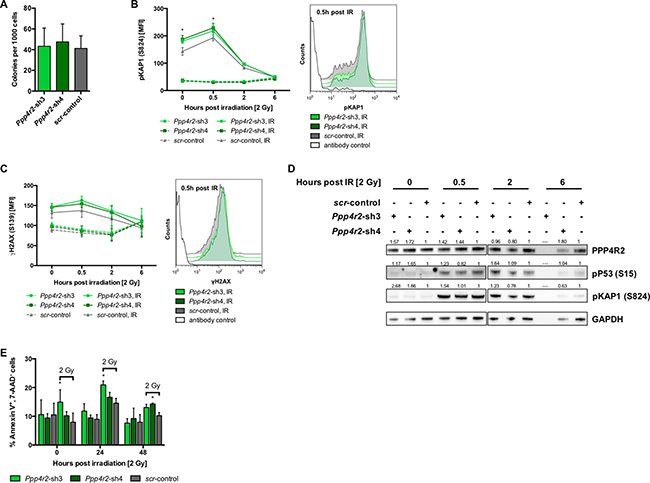
Figure 2: Ppp4r2 suppression regulates DNA damage response in normal murine hematopoietic cells. (A) Impact of Ppp4r2 knockdown in murine Lin- bone marrow (BM) cells on clonogenic growth determined by colony forming cells in methylcellulose (CFC-Assay; n = 4). (B) Impact of Ppp4r2 knockdown in murine Lin- BM cells on DNA damage response upon ionizing radiation (IR) with 2 Gy determined by the mean fluorescence intensity (MFI) of phosphorylated KRAB-domain associated protein 1 [pKAP1(S824)] at the indicated time post IR (n = 2). Representative histogram depicts the MFI shift of pKAP1 (S824) at 0.5 h post IR in either murine Lin- BM cells with Ppp4r2 knockdown or control. (C) DNA damage at indicated time post IR with 2 Gy in murine Lin- BM cells upon Ppp4r2 knockdown determined by the MFI of phosphorylated histone variant H2AX [γH2AX (S139); n = 3]. Representative histogram depicts the MFI shift of γH2AX (S139) at 0.5 h post IR in murine Lin- BM cells with either Ppp4r2 knockdown or control. (D) Representative Western Blot displaying the effect of IR on phosphorylation of KAP1 (S824) and P53 (S15) in Lin- BM cells with either Ppp4r2 knockdown or control. Vertical lines have been inserted to indicate a repositioned gel lane. (E) Apoptosis induction upon IR with 2 Gy displayed as the percentage of AnnexinV+/7AAD- Lin- BM cells with either Ppp4r2 knockdown or control at the indicated time post IR (n = 3). Data are represented by the mean ± SD. Statistical analyses were carried out using unpaired two-tailed t-test or multiple t-tests corrected for multiple comparisons using the Holm-Sidak method. A p-value ≤ 0.05 was considered significant, *p ≤ 0.05.
Ppp4r2 loss-of-function enhances DNA damage in murine leukemic BM cells
To determine effects of Ppp4r2 loss-of-function in a leukemic state, Ppp4r2 knockdown experiments were carried out in murine myeloid/lymphoid or mixed-lineage leukemia translocated to 3 - lysine methyltransferase 2A (MLLT3-KMT2A, formerly known as MLL-AF9) transformed Lin- BM cells, which fully recapitulate human myeloid KMT2A leukemia [30–32]. We genetically inactivated Ppp4r2 in murine MLLT3-KMT2A leukemic cells by lentiviral transduction with two shRNAs (Ppp4r2-sh3, Ppp4r2-sh4) targeting Ppp4r2 (Supplementary Figure 2B). Additionally, qRT-PCR analyses of the other PPP4 regulatory subunits Ppp4r1, Ppp4r3a, and Ppp4r3b were performed to validate specific knockdown of Ppp4r2 in the MLLT3-KMT2A leukemia model (Supplementary Figure 2C). To evaluate DNA damage and response to DSB, cells with either Ppp4r2 knockdown or control were exposed to IR with 2 Gy and DDR signaling proteins pKAP1 (S824) and γH2AX (S139) were determined by flow cytometry at 0 h, 0.5 h, 2 h, and 6 h post IR. DDR upon Ppp4r2 knockdown led to the accumulation of pKAP1 (Ppp4r2-sh3: 2 h post IR, p = 0.002; Figure 3A). DNA damage assessed by γH2AX showed elevated levels upon Ppp4r2 knockdown followed by IR (Ppp4r2-sh3: 2 h post IR, p = 0.002; Ppp4r2-sh4: 2 h post IR, p = 0.0009; 6 h post IR, p = 0.01; Figure 3B). Western Blot analyses displaying the phosphodynamics of key DDR proteins after IR with 2 Gy confirmed elevated pKAP1 and γH2AX protein levels in murine MLLT3-KMT2A leukemic cells with Ppp4r2 knockdown. In addition, they further revealed activation of P53 by phosphorylation as well as RPA2 hyperphosphorylation upon knockdown of Ppp4r2 in murine MLLT3-KMT2A leukemic cells (Figure 3C). Determination of early apoptotic cells in response to IR after 0 h, 24 h, and 48 h revealed increased sensitivity to IR upon Ppp4r2 knockdown displaying higher numbers of early apoptotic cells represented by the percentage of AnnexinV+/7-AAD- cells (Ppp4r2-sh3: 24 h post IR, p = 0.007; Ppp4r2-sh4: 0 h no IR, p = 0.007; 24 h no IR, p = 0.008; 0 h post IR, p = 0.002; 24 h post IR, p = 0.0002; 48 h post IR, p = 0.03; Figure 3D). Colony formation ability of MLLT3-KMT2A transformed leukemic cells was not affected upon Ppp4r2 suppression (Figure 3E). Proliferative capacity was further investigated using MTS-Assay, which revealed reduced metabolic activity upon Ppp4r2 knockdown in murine MLLT3-KMT2A transformed leukemic cells (Ppp4r2-sh3: day 7, p = 0.001; Ppp4r2-sh4: day 7, p < 0.0001; Figure 3F).
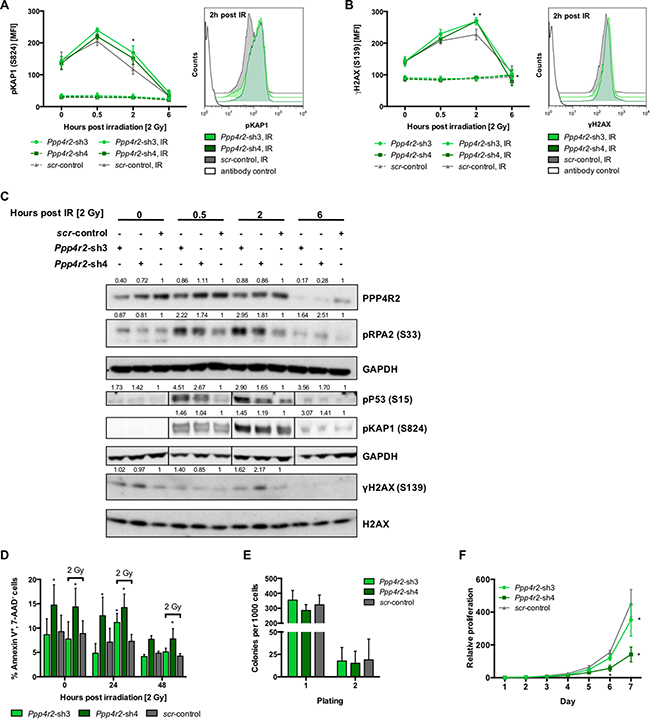
Figure 3: Ppp4r2 loss-of-function enhances DNA damage in murine leukemic bone marrow cells. (A) Impact of Ppp4r2 knockdown in murine MLLT3-KMT2A transformed Lin- bone marrow (BM) cells on DNA damage response upon ionizing radiation (IR) with 2 Gy measured by the mean fluorescence intensity (MFI) of phosphorylated KRAB-domain associated protein 1 [pKAP1 (S824)] at the indicated time post IR (n = 3). Representative histogram depicts the MFI shift of pKAP1 (S824) at 2 h post IR in either murine MLLT3-KMT2A transformed Lin- BM cells with Ppp4r2 knockdown or control. (B) DNA damage at indicated time post IR with 2 Gy in murine MLLT3-KMT2A Lin- BM cells upon Ppp4r2 knockdown determined by the MFI of phosphorylated histone variant H2AX [γH2AX (S139); n = 3]. Representative histogram depicts the MFI shift of γH2AX at 2 h post IR in murine MLLT3-KMT2A transformed Lin- BM cells with either Ppp4r2 knockdown or control. (C) Representative Western Blot displaying the effect of IR on phosphorylation of the key DDR protein RPA2 (S33), P53 (S15), KAP1 (S824), and H2AX (S139) in murine MLLT3-KMT2A Lin- BM cells with either Ppp4r2 knockdown or control. Vertical lines have been inserted to indicate a repositioned gel lane. (D) Apoptosis induction upon IR with 2 Gy displayed as the percentage of AnnexinV+/7AAD- murine MLLT3-KMT2A Lin- BM cells with either Ppp4r2 knockdown or control at the indicated time post IR (n = 5). Impact of Ppp4r2 knockdown in murine MLLT3-KMT2A transformed Lin- BM cells on (E) clonogenic growth and replating capacity determined by colony forming cells in methylcellulose (CFC-Assay; n = 5), and (F) proliferation potential measured by MTS-Assay (n = 3). Data are represented by the mean ± SD. Statistical analyses were carried out using unpaired two-tailed t-test or multiple t-tests corrected for multiple comparisons using the Holm-Sidak method. A p-value ≤ 0.05 was considered significant, *p ≤ 0.05.
Re-expression of PPP4R2 in 3p deleted human leukemic cells restores DNA repair upon irradiation
Next, we performed reconstitution experiments in the PPP4R2-deficient human leukemic cell line MEG-01 by lentiviral transduction of the PPP4R2 open reading frame (PPP4R2-ORF; Supplementary Figure 2D) to evaluate the role of PPP4R2 in hematologic neoplasia associated with 3p loss. DNA damage was induced by IR followed by investigation of DDR proteins pKAP1 (S824) and γH2AX (S139) by flow cytometry at 0 h, 0.5 h, 2 h, and 6 h post IR. In comparison to irradiated MEG-01 cells used as control, re-expression of PPP4R2 resulted in significantly lower pKAP1 levels in response to induction of DSB by IR (0.5 h post IR, p = 0.01; 2 h post IR, p = 0.0001), which is represented by the shift to the left of pKAP1 MFI at 2 h post IR (Figure 4A). DNA damage determined by γH2AX after irradiation showed less phosphorylation in PPP4R2 expressing MEG-01 cell line, more clearly represented by the respective histogram showing a MFI shift of γH2AX to the left (Figure 4B). Western Blot analysis confirmed reduced pKAP1 protein in MEG-01 cells upon re-expression of PPP4R2 and exposure to IR (Figure 4C). Additionally, we observed reduced phosphorylation of the DDR proteins pP53, γH2AX, and pRPA2 in comparison to MEG-01 control (Figure 4C). Although we did not observe induction of apoptosis upon DNA damage, independent of PPP4R2 expression (Figure 4D), re-expression of PPP4R2 impaired colony forming potential compared to control (p = 0.07; Figure 4E). Determination of cell proliferation and viability by metabolic activity with MTS-Assay showed reduced proliferation of the human myeloid leukemic cell line MEG-01 after re-expression of PPP4R2 (day 7, p < 0.0001; Figure 4F), which was also confirmed by counting viable cells by trypan blue exclusion method (data not shown).
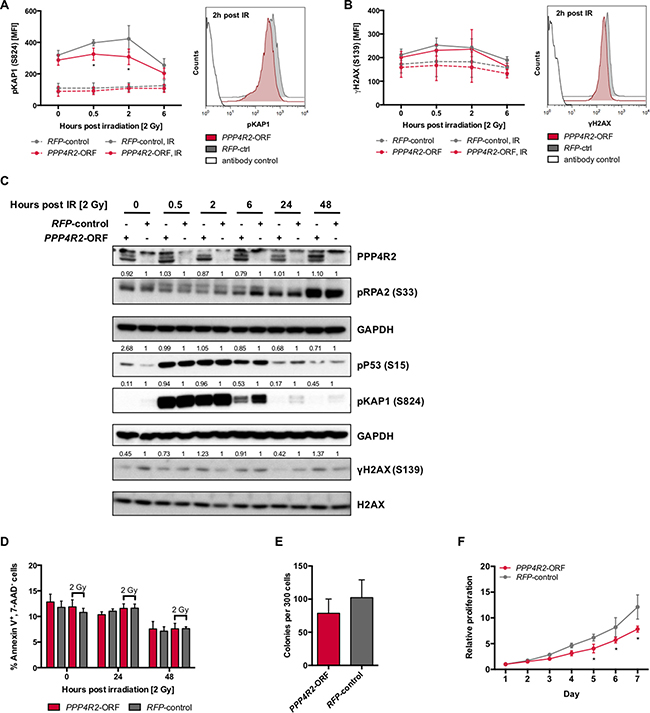
Figure 4: Re-expression of PPP4R2 restores DNA repair in leukemic cells with 3p microdeletion. (A) Impact of PPP4R2 restoration in human leukemic MEG-01 cells on DNA damage response upon ionizing radiation (IR) with 2 Gy determined by the mean fluorescence intensity (MFI) of phosphorylated KRAB-domain associated protein 1 [pKAP1 (S824)] at the indicated time post IR (n = 5). Representative histogram depicts the MFI shift of pKAP1 (S824) at 2 h post IR in either MEG-01 cells with PPP4R2 restoration or control. (B) DNA damage at indicated time post IR with 2 Gy in MEG-01 cells upon PPP4R2 re-expression determined by the MFI of phosphorylated histone variant H2AX [γH2AX (S139); n = 4]. Representative histogram depicts the MFI shift of γH2AX (S139) at 2 h post IR in MEG-01 cells with either PPP4R2 restoration or control. (C) Representative Western Blot displaying the effect of IR with 2 Gy on phosphorylation of the DDR protein RPA2 (S33), P53 (S15), KAP1 (S824), and H2AX (S139) in MEG-01 cells with either PPP4R2 restoration or control. (D) Effect of PPP4R2 re-expression and exposure to IR with 2 Gy on apoptosis induction displayed as the percentage of AnnexinV+/7AAD- cells in comparison to control (n = 4). Impact of PPP4R2 restoration on (E) clonogenic growth determined by colony forming cells in methylcellulose (CFC-Assay; n = 4), and (F) proliferation potential measured by MTS-Assay (n = 3). Data are represented by the mean ± SD. Statistical analyses were carried out using unpaired two-tailed t-test or multiple t-tests corrected for multiple comparisons using the Holm-Sidak method. A p-value ≤ 0.05 was considered significant, *p ≤ 0.05.
Gene expression profiling (GEP) and whole exome sequencing (WES) of primary AML samples identified enriched gene sets associated with DNA repair
Based on our findings that PPP4R2 is differentially expressed in hematopoiesis and leukemia as well as its involvement in DDR (Figure 5), we aimed to investigate whether our hypothesis that deregulated DNA repair promoted by PPP4R2 depletion may contribute to leukemia development is also reflected by global gene expression data of primary AML samples.
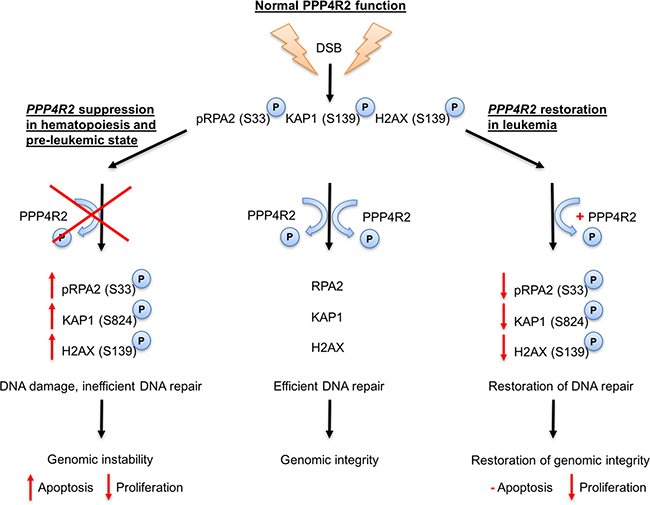
Figure 5: Proposed model for PPP4R2 and its involvement in DNA damage response in normal hematopoietic and leukemic cells.
Therefore, we performed a supervised analysis determining PPP4R2 co-expressed genes by comparing 10% of AML cases with the highest and lowest PPP4R2 expression. This analysis revealed 5463 significantly differentially expressed genes at a false discovery rate below 1%. Notably, the top 1000 most significant genes of this signature were significantly enriched for members of pathways associated with cancer [e.g. ABL proto-oncogene 1 (ABL1), mutS homolog 6 (MSH6), nucleoporin 98 (NUP98), tumor protein 53 binding protein 2 (TP53BP2)] and DNA repair [e.g. breast cancer 1 (BRCA1), MSH6, TP53BP2, small ubiquitin-like modifier 1 (SUMO1)] as well as apoptosis following DNA damage [e.g. protein tyrosine phosphatase non-receptor type 12 (PTPN12), nuclear factor kappa B subunit 1 (NFKB1)] (Supplementary Table 3). In accordance, an analysis comparing CN-AML cases with 3p CDR (n = 9) with CN-AML without 3p deletion (n = 161) revealed a significant enrichment of gene sets associated with cancer, DNA repair as well as cell death and survival, which further supports a strong impact of PPP4R2 in 3p deleted AML (Supplementary Table 4).
Next, we were interested whether primary AML patients with 3p microdeletion also harbor a distinct gene mutation pattern that might further support the deregulation of the respective pathways. Whole exome sequencing of five paired diagnosis/remission samples from CN-AML cases with 3p CDR showed on average 14 tumor-specific mutations per AML (Table 1). Beside significant enrichment for hits and pathways typically associated with AML [e.g. nucleophosmin (NPM1), fms related tyrosine kinase 3 (FLT3), NRAS proto-oncogene GTPase (NRAS)], we also found a notable number of hits in pathways associated with response to DNA damage as well as apoptosis and cell survival [e.g. mutS homolog 2 (MSH2), forkhead box O3 (FOXO3), cell division cycle 27 (CDC27)] (Supplementary Table 5).
Table 1: Mutations detected by whole exome sequencing of AML cases with 3p microdeletion (n = 5)
Patient |
Tumor-specific mutations (somatic) |
Pre-leukemic mutations (germline/remission) |
|---|---|---|
1 |
TEX11, GATA2a, ZNF687, CCT7, CATSPER4, NRASa, NPM1a, TLR4a, SYNE2, POSTNa, CTDSP2, GXYLT1a, GXYLT1a, CTBP2a |
DNMT3Aa |
2 |
CACNA1A, BMP5a, TRPC6, CYFIP2a, RBX1, PRLR, IRF2BP2, UNC13Ca, ARMC9, SENP3, NPM1a |
RUNX1a, DNMT3Aa |
3 |
ADAMTS12, DISP1, KIAA1551, OR5H1, DNAH9, TJP1, PLPPR1, RIT1a, NPM1a, HSPBAP1, CCDC178 |
NPAT, DNMT3Aa |
4 |
DNMT3Aa, RBM46, PHGR1, APOA5, HAUS1, ZNF469, CHFR, MSH2, SLC12A7, KAT6B, FAM184A, PCDH15, AGAP1, PFN3, NPM1a, ADGRA3a, FOXO3, FLT3 |
|
5 |
HIPK3, PKP1, CDC42EP1, CMTM3a, SPDYC, WT1a, OSMR, MYT1La, FCN1, ACLY, COL19A1, DMBT1, CDC27, NPM1a, CTNNA3 |
amutated in cancer according to the Catalogue of Somatic Mutations in Cancer (COSMIC).
Genes in bold are commonly mutated.
TEX11, testis expressed gene 11; GATA2, GATA binding protein 2; ZNF687, zinc finger protein 687; CCT7, chaperon containing TCP1 subunit 7; CATSPER4, cation channel sperm associate 4; NRAS, NRAS proto-oncogene GTPase; NPM1, nucleophosmin; TLR4, toll like receptor 4; SYNE2, spectrin repeat containing nuclear envelope protein 2; POSTN, periostin; CTDSP2, CTD small phosphatase 2; GXYLT1, glucoside xylosyltransferase 1; CTBP2, C-terminal binding protein 2; CACNA1A, calcium voltage-gated channel subunit alpha1 A; BMP5, bone morphogenetic protein 5; TRPC6, transient receptor potential cation channel subfamily C member 6; CYFIP2, cytoplasmic FMR1 interacting protein 2; RBX1, ring-box 1; PRLR, prolactin receptor; IRF2BP2, interferon regulatory factor 2 binding protein 2; UNC13C, unc-13 homolog C; ARMC9, armadillo repeat containing 9; SENP3, SUMO1/sentrin/SMT3 specific peptidase 3; ADAMTS12, ADAM metallopeptidase with thrombospondin type 1 motif 12; DISP1, dispatched RND transporter family member 1; OR5H1, olfactory receptor family 5 subfamily H member 1; DNAH9, dynein axonemal heavy chain 9; TJP1, tight junction protein 1; PLPPR1, phospholipid phosphatase related 1; RIT1, Ras like without CAAX1; HSPBAP1, HSPB1 associated protein 1; CCDC178, coiled-coil domain containing 178; DNMT3A, DNA methyltransferase 3 alpha; RBM46, RNA binding motif protein 46; PHGR1, proline, histidine and glycine rich 1; APOA5, apolipoprotein A5; HAUS1, HAUS augmin like complex subunit 1; ZNF469, zinc finger protein 469; CHFR, checkpoint with forkhead and ring finger domains; MSH2, mutS homolog 2; SLC12A7, solute carrier family 12 member 7; KAT6B, lysine acetyltransferase 6B; FAM184A, family with sequence similarity 184 member A; PCDH15, protocadherin related 15; AGAP1, ArfGAP with GTPase domain, ankyrin repeat and PH domain 1; PFN3, profilin 3; ADGRA3, adhesion G protein-coupled receptor A3; FOXO3, forkhead box O3; FLT3, fms related tyrosine kinase 3; HIPK3, homeodomain interacting protein kinase 3; PKP1, plakophilin 1; CDC42EP1, CDC42 effector protein 1; CMTM3, CKLF like MARVEL transmembrane domain containing 3; SPDYC, speedy/RINGO cell cycle regulator family member C; WT1, Wilms tumor 1; OSMR, oncostatin M receptor; MYT1L, myelin transcription factor 1 like; FCN1, ficolin 1; ACLY, ATP citrate lyase; COL19A1, collagen type XIX alpha 1 chain; DMBT1, deleted in malignant brain tumors 1; CDC27, cell division cycle 27; CTNNA3, catenin alpha 3; RUNX1, runt related transcription factor 1; NPAT, nuclear protein, coactivator of histone transcription.
Finally, we performed a comprehensive analysis of the gene expression profiles and mutational patterns of AML patients obtained by GEP and WES analyses to identify common deregulated genes and functional-related pathways. To this end, we determined overlapping differentially expressed and mutated genes or enriched gene sets obtained by comparison of analyses of 1) AML with high versus low global PPP4R2 expression, 2) CN-AML with 3p microdeletion versus CN-AML without 3p microdeletion, and 3) mutated genes of AML patients with 3p microdeletion (Supplementary Figure 3). Mutated or differentially expressed DNA methyltransferase 3 alpha (DNMT3A) was shared among all three types of analyses. Common leukemia-associated genes have been identified by the investigation of gene sets obtained by comparison of analysis 1 with analysis 3 [FLT3 and C-terminal binding protein 2 (CTBP2)] and analysis 2 with analysis 3 [NRAS and GATA binding protein 2 (GATA2)]. The comparison of analysis 1 to analysis 2 identified 98 common deregulated genes. Of note, the comparison of enriched gene sets among all three analyses identified one common deregulated gene set (DACOSTA_UV_RESPONSE_VIA_ERCC3_DN). This gene set comprises genes that were downregulated in fibroblasts expressing mutant forms of ERCC excision repair 3 (ERCC3) after ultraviolet irradiation, representing a transcriptional profile that was specifically regulated upon ultraviolet irradiation depending on the DNA repair capacity of the cell [33].
DISCUSSION
We previously identified a recurrent microdeletion at chromosome 3p14.1-p13 in AML [7–9] that was also described in prostate and cervical cancer, and was supposed to harbor several potential cooperative tumor suppressor genes [10, 11]. In the present work we report on the functional characterization of PPP4R2, one candidate gene located in the CDR, in the context of hematopoiesis and leukemia.
Mutation analyses of primary AML patient samples revealed that the function of PPP4R2 is not affected by mutations in AML. This is in line with public sequencing data from The Cancer Genome Atlas (TCGA), which showed neither mutation nor amplification of PPP4R2 but deletion in 1.6% of AML cases [3]. This finding indicates that PPP4R2 function might rather be affected by gene deletions or epigenetic deregulation than gene mutations.
Our data concerning PPP4R2 expression levels suggest a functional relevance of PPP4R2 in HSPC and might indicate its requirement in normal hematopoietic cells during early myelopoiesis. Publicly available microarray data provide evidence for such an expression pattern also in human myelopoiesis [29]. The functional properties of PPP4R2 are not thoroughly investigated so far, thus we also interpreted the results of our study with respect to the catalytic subunit of PPP4, whose activity is controlled by regulatory subunits like PPP4R2. Besides embryonic lethality of Ppp4-null mice, PPP4 has been shown to be essential for development of hematopoietic cells. T-/B-cell specific Ppp4 deletion led to an early block in T-/B-cell development and abnormal thymocyte maturation [18–20]. Furthermore, PPP4R2 expression has been associated with differentiation in neuronal NSC-34 cells, in which PPP4R2 depletion resulted in abnormal and strongly inhibited differentiation towards differentiation block [21]. Based on our data decreased expression in primary AML cases may contribute to leukemia initiation or development. Loss of PPP4R2 expression in CK-AML, an AML subgroup associated with substantial genomic instability and unfavorable prognosis, might point to AML cells that select for PPP4R2 depletion along with perturbed maintenance of genomic integrity. Recent expression analyses of primary lung cancer cases revealed lower PPP4R2 levels in late stages of tumor progression characterized by lymph node metastases than in matched primary lung cancer tissue [22]. Genetic inactivation of PPP4R2 in the lung cancer cell line A549 displayed significant more metastatic progression in mice, indicating that lower levels of PPP4R2 support metastatic processes e.g. migration and invasion to promote tumor progression.
Our functional studies implicate that Ppp4r2 contributes to DNA repair of normal murine HSPC by positive regulation of the PPP4 de-phosphorylation activity of DDR signaling proteins as well as induction of apoptosis. Clonogenicity was not apparently affected upon suppression of Ppp4r2 in normal murine Lin- BM cells. Comparable results have been described for PPP4 in the context of lymphopoiesis. Loss of Ppp4 in B-cells of conditional knockout mice was associated with impaired cell development, increased apoptosis, and persistent DNA damage upon immunoglobulin recombination that is typically repaired by NHEJ [19, 20]. Interestingly, downregulation of PPP4 and its regulatory subunits in non-malignant human embryonic kidney cell line reduced HR- and NHEJ-mediated DNA repair and resulted in increased DNA damage [23, 27].
Our report further provides evidence for Ppp4r2 affecting DNA repair of leukemic hematopoietic cells. Response to DNA damage upon Ppp4r2 knockdown in the murine MLLT3-KMT2A transformed leukemia model was accompanied by enhanced DNA damage and/or impaired DNA repair. Previous reports have shown that PPP4R2 depletion in MCF-7 breast cancer cells caused increased KAP1 phosphorylation in response to camptothecin- or etoposide-mediated DNA damage, and was associated with a significant reduction of NHEJ-mediated DNA repair [27]. Phosphorylation of KAP1 caused global decondensation of chromatin that might increase susceptibility for DNA damage [34], and transcriptional de-repression of e.g. p21 and P53 target genes [35, 36]. Moreover, γH2AX as a readout for DNA damage has also been shown to be controlled by PPP4R2 in osteosarcoma U2OS or cervix carcinoma HeLa cell line, even independent of exogenous DNA damage [23, 24]. Depletion of PPP4R2 or the catalytic subunit of PPP4 delayed γH2AX de-phosphorylation during recovery from irradiation [24]. PPP4 deficiency particularly impeded HR-mediated repair of DSB by unresolved γH2AX during replication [23]. Although Nakada et al. proposed that de-phosphorylation of γH2AX upon DNA damage by irradiation is rather a function attributed to PPP4, other PP2A phosphatases also target γH2AX to facilitate DSB repair and might compensate the effect of Ppp4r2 knockdown [24, 37]. Furthermore, the main PPP4 complex that de-phosphorylates pKAP1 or γH2AX includes the PPP4R3β subunit, which could explain mitigated effects in response to DNA damage seen in our study upon genetic inactivation of Ppp4r2 [23, 24, 26]. Knockdown of Ppp4r2 in murine MLLT3-KMT2A leukemia followed by induction of DSB was further accompanied by elevated levels of pRPA2, which is a specific and relevant substrate of the PPP4R2-containing PPP4 complex that impaired HR-mediated DSB repair by inefficient DDR [25]. Hence, suppression of Ppp4r2 in the murine MLLT3-KMT2A leukemia model reduced the de-phosphorylation of key DDR proteins, which resulted in elevated DDR presenting inefficient DNA repair followed by accumulation of DNA damage. Intriguingly, downregulation of the catalytic subunit of the PPP4 complex in human T-cell leukemic cell lines Jurkat and CEM-C7 by RNAi displayed substantially increased mutation frequency upon ultraviolet irradiation, supporting an important role of the PPP4 complex in DNA repair of leukemic cells [38]. Impaired DNA repair by loss of Ppp4r2 function potentially promotes genomic instability, leading to apoptosis induction and compromised proliferative capacity represented by lower metabolic activity. Colony formation capability of MLLT3-KMT2A transformed hematopoietic progenitors was not apparently affected by Ppp4r2 knockdown. A more detailed examination of colonies could associate smaller colonies upon Ppp4r2 knockdown with reduced growth rate. However, the impact of downregulated PPP4 or its regulatory subunit PPP4R2 on proliferation and apoptosis is controversially discussed. Comparable results were described upon PPP4R2 knockdown in neuronal NSC-34 cells, which revealed increased apoptosis rate upon DNA damage by etoposide treatment [21]. PPP4 depletion in murine thymocytes or HEK293T cells resulted in decreased proliferation and enhanced apoptosis [18, 39]. In contrast, downregulation of the catalytic subunit in T-leukemic cell lines, human peripheral blood T-lymphocytes, or HEK293T cells increased proliferation and inhibited apoptosis in response to different apoptotic stimuli [38, 40, 41]. Such cell type-dependent differences imply that the PPP4 complex needs to be tightly regulated. Altered PPP4R2 expression might impact on multiple cellular functions such as cell survival, not at least due to its involvement in centrosome maturation and microtubule organization [39, 42].
Our reconstitution experiments indicate that re-expression of PPP4R2 reduces DNA damage probably through improved DNA repair in leukemic cells. Restoration of DNA repair by PPP4R2 expression in human myeloid leukemic cell line MEG-01 and additional deregulation of apoptosis pathways may account for lack of DNA damage triggered apoptosis. It is feasible that the involvement of the PPP4R2-containing PPP4 complex in centrosome maturation or spliceosome assembly is responsible for lower proliferation rate and colony formation upon ectopic expression of PPP4R2 [39, 43]. This observation suggests that a certain level of PPP4R2 is relevant while decrease as well as increase of PPP4R2 perturbs cell homeostasis and viability. In osteosarcoma cell line U2OS it has been demonstrated that PPP4R2 in complex with the catalytic subunit was able to de-phosphorylate γH2AX in a dose-dependent manner [24]. In further accordance with our data, pKAP1 was described to be a substrate of the PPP4 complex, and overexpression of the catalytic subunit in breast cancer cell line MCF-7 resulted in decreased levels of camptothecin-induced pKAP1 [27]. The more specific de-phosphorylation activity of the PPP4R3β-containing PPP4 complex on pKAP1 and γH2AX could explain moderate effects upon ectopic PPP4R2 expression in our study [23, 24, 26]. Moreover, highly proliferative and aggressive leukemic cells have to cope with DNA damage and activated DNA repair signaling. Additional genetic alterations in the PPP4R2-deficient human myeloid leukemic cell line MEG-01 permit survival of cells that acquired genomic instability providing an advantage to select for subclones, and indicating a cooperative role for PPP4R2 deficiency in leukemia progression. In line with this, neuronal NSC-34 cells overexpressing PPP4R2 were protected from apoptosis under basal conditions as well as in response to the DNA damaging agent etoposide [21].
GEP and WES data obtained from primary AML samples further support our model that PPP4R2 is needed in immature cells to ensure genomic stability during the proliferative processes prior to cellular differentiation. Alterations in AML by either loss of PPP4R2 (3p deletion) and/or downregulation of PPP4R2 (possibly by epigenetic silencing) might enable the malignant cells to promote and rather tolerate genomic instability allowing faster cell turnover. Co-expressed genes and pathway enrichment analysis strongly highlight the involvement of PPP4R2 in DNA repair mechanisms also in primary AML. In further accordance, GEP signatures and exome sequencing of leukemias with 3p microdeletion support the disturbance of the respective pathways by low PPP4R2 expression, which might provide an advantage for subclones with 3p microdeletion. Despite co-occurrence of leukemia-associated differentially expressed and mutated genes, gene set enrichment analyses identified one common gene set comprising downregulated genes expressed in fibroblasts with mutant forms of ERCC3 upon ultraviolet irradiation [33]. Intriguingly, mutations in ERCC3, a known cancer gene, can result in xeroderma pigmentosum with Cockayne syndrome or trichothiodystrophy, which are prominent diseases associated with defective DNA repair and predisposition to skin cancer [44–47].
In conclusion, we identified involvement of PPP4R2 in DDR of both normal hematopoietic and leukemic cells via deregulation of the PPP4 complex. Impaired DNA repair and enhanced DNA damage by PPP4R2 suppression might promote genomic instability, one possible mechanism by which 3p microdeletions could potentially contribute to the pathogenesis of AML. Intriguingly, tumors that are enriched for defects in DNA repair process were impressively susceptible to poly(ADP-ribose) polymerase (PARP) inhibitors, most prominent examples represent BRCA1- or BRCA2-deficient cells [48, 49]. Nevertheless, also BRCA-independent defects in HR-mediated DNA repair e.g. by downregulation of protein phosphatase 2 regulatory subunit B alpha (PPP2R2A) in non-small cell lung cancer cells promoted increased sensitivity to PARP inhibitors in vitro and in vivo [50]. Whether compromised DNA repair caused by PPP4R2 inactivation may contribute to enhanced sensitivity to targeted therapy with e.g. PARP inhibitors remains to be elucidated. Further studies will determine the potential of PPP4R2 deficiency as possible new target for AML therapy.
MATERIALS AND METHODS
Primary patient samples
Diagnostic BM and/or peripheral blood (PB) samples from AML patients (n = 85) and healthy BM controls (n = 8) were analyzed. Patients were enrolled into different consecutive AMLSG multicenter treatment trials [AML HD98A, NCT00146120 (n = 5); AML HD98B [51] (n = 4); AMLSG 06-04, NCT001512255 (n = 4); AMLSG 07-04, NCT00151242 (n = 57); AMLSG BiO, NCT01252485 (n = 4); AML 43, NCT00121303 (n = 2); RATIFY, NCT00651261 (n = 1); AMLSG-R1, NCT00744081 (n = 1); no study (n = 1); unknown (n = 6)]. All patients gave informed consent for treatment and genetic analysis according to the Declaration of Helsinki.
Cell line models
BM from C57BL/6J wildtype mice was harvested and enriched for lineage-negative cells [Lin-, cells negative for lineage markers CD3, CD4, CD11b, CD19, TCRγ/δ, (BD Biosciences, Franklin Lakes, NJ, USA), CD8a, NK1.1, Gr-1, TCRα/β, Ter119 (eBioscience, San Diego, CA, USA)] using Dynabeads Sheep anti-rat IgG (Thermo Fisher Scientific, Waltham, MA, USA). Lin- BM cells were cultured in StemSpan SFEM (StemCell Technologies, Vancouver, Canada) supplemented with 10 ng/ml thrombopoietin, 6 ng/ml interleukin (IL-)3, 10 ng/ml IL-6, 100 ng/ml stem cell factor, and 60 ng/ml granulocyte-colony stimulating factor (PeproTech, Rocky Hill, NJ, USA).
BM cells from C57BL/6J wildtype mice transformed by MLLT3-KMT2A transduction were cultured in IMDM+GlutaMAX (Thermo Fisher Scientific, Waltham, MA; USA) supplemented with 10% FBS (Hyclone, Thermo Fisher Scientific, Waltham, MA, USA), 1 mM sodium pyruvate (Thermo Fisher Scientific, Waltham, MA; USA), 0.002% 1thioglycerol (Sigma Aldrich, St. Louis, MO, USA), 1% IL-3 supernatant, and 1% penicillin/streptomycin (PAN-Biotech GmbH, Aidenbach, Germany). Hematopoietic subpopulations from C57BL/6J mice were sorted using fluorescent activated cell sorting as previously published [52].
Human leukemic cell line MEG-01 (DSMZ, Braunschweig, Germany) was cultured in RPMI1640 (biochrom, Berlin, Germany) supplemented with 10% FBS (Sigma Aldrich, St. Louis, MO, USA), 1% penicillin/streptomycin (PAN-Biotech GmbH, Aidenbach, Germany), and 2 mM L-glutamine (PAN-Biotech GmbH, Aidenbach, Germany).
Phosphoprotein flow cytometry
Cells with either Ppp4r2 knockdown/PPP4R2 re-expression or control were exposed to 2 Gy IR or not followed by fixation and permeabilization at indicated time post IR with BD Cytofix/Cytoperm Buffer and BD Permeabilization Buffer Plus, respectively (Fixation/Permeabilization Solution Kit, BD Biosciences, Franklin Lakes, NJ, USA).
For measurement of pKAP1, cells were incubated with anti-pKAP1 antibody (S824; Bethyl Laboratories, Montgomery, TX, USA) and subsequently stained with anti-Dylight 649 donkey anti-rabbit IgG (BioLegend, San Diego, CA, USA).
Determination of γH2AX was performed by incubation with anti-phospho-Histone H2A.X antibody (Ser139; Clone JBW 301; Merck Millipore, Darmstadt, Germany) followed by staining with anti-Mouse IgG1 eFluor® 660 (eBioscience, San Diego, CA, USA).
Cells were acquired by flow cytometry (FACSCalibur, BD Biosciences, Franklin Lakes, NJ, USA) and analyzed by Flow Jo Software (FlowJo, Ashland, OR, USA).
Apoptosis analysis
Cells with either Ppp4r2 knockdown/PPP4R2 re-expression or control were exposed to IR with 2 Gy or not. At indicated time post IR, apoptotic cells were determined using the AnnexinV Apoptosis Detection Kit I (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. Briefly, cells were washed twice with cold PBS and double-stained with APC AnnexinV (BD Biosciences, Franklin Lakes, NJ, USA) and 7-AAD. Data were acquired by flow cytometry (FACSCalibur, BD Biosciences, Franklin Lakes, NJ, USA) and analyzed with Flow Jo Software (FlowJo, Ashland, OR, USA).
Colony formation assay (CFC-assay)
Murine BM cells (Lin-, MLLT3-KMT2A) with either Ppp4r2 knockdown or control were plated in duplicates with 1 × 103 cells per plate in methylcellulose MethoCult M3434 (StemCell Technologies, Vancouver, Canada) and colony forming cells were counted after 7 days.
Human leukemic MEG-01 cells with ectopic expression of PPP4R2 or control were plated in duplicates with 300 cells per plate in methylcellulose MethoCult H4330 (StemCell Technologies, Vancouver, Canada) and colonies were counted after 10–14 days.
Cell proliferation analysis (MTS-assay)
Cell viability and proliferation was assessed by determination of metabolic activity using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). 2 × 104 cells/ml with either Ppp4r2 knockdown/PPP4R2 re-expression or control were cultured in triplicates and examined daily according to the manufacturer’s instructions.
Analysis of gene expression profiling (GEP) data
A previously published GEP data set of our group comprising 436 AML cases accessible at gene expression omnibus (GEO accession GSE22778) was available for further studies [53]. Analyses were performed as previously reported by ClassComparison analysis using BRB Array Tools Version 3.8.0 – Beta_2 Release of BRB-Array Tools and using R, version 2.9.0 [54]. Pathway enrichment analysis was performed using the Molecular Signatures Database (MSigDB, http://broadinstitute.org/gsea/index.jsp).
Whole exome sequencing (WES)
Whole exome sequencing was performed on five diagnosis and remission samples from cases with 3p CDR. DNA libraries were generated from 50 ng DNA using the Nextera Rapid Capture Expanded Exome kit (Illumina, San Diego, CA, USA) and the Nextera Rapid Capture Exome kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Pooled DNA libraries were sequenced on an Illumina HiSeq2000 with the 200-cycle TruSeq SBS v3 kit (Illumina, San Diego, CA, USA). Following de-multiplexing the paired-end sequences were aligned to the human reference genome hg19 with BWA-MEM23. BAM files were sorted and indexed using SortSam and BuildBamIndex (both Picard 1.138, http://picard.sourceforge.net). Variants were called using VarScan2 (http://varscan.sourceforge.net) by comparing tumor samples to the matching remission sample and annotated using ANNOVAR. Intronic variants, synonymous variants, variants with less than 2 tumor variant supporting reads in forward and reverse direction, variants with an entry in dbSNP13827 (http://www.ncbi.nlm.nih.gov/SNP), but not in COSMIC28 (http://cancer.sanger.ac.uk/cosmic), variants located in segmental duplication areas, variants which are located in polymer-repeat regions, and variants with a variant allele frequency <10% were filtered out. Pathway enrichment analysis was performed using the Molecular Signatures Database (MSigDB, http://broadinstitute.org/gsea/index.jsp). Statistical significance was assessed using a nonparametric Mann-Whitney test.
Statistical analysis
Data are represented by each individual data point and the mean or mean ± SD, and number of replicates is indicated in the respective figure legend. Statistical analyses were carried out using unpaired two-tailed t-test or multiple t-tests corrected for multiple comparisons using the Holm-Sidak method (Prism 6, GraphPad Software, La Jolla, CA, USA). A p-value ≤ 0.05 was considered significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Abbreviations
ABL1: ABL proto-oncogene 1; AML: acute myeloid leukemia; AMLSG: AML Study Group; B2M: beta-2 microglobulin; BM: bone marrow; BRCA: breast cancer; CDC27: cell division cycle 27; CDR: commonly deleted region; CK: complex karyotype; CLP: common lymphoid progenitor(s); CMP: common myeloid progenitor(s); CN: cytogenetically normal; COSMIC: Catalogue of Somatic Mutations in Cancer; CTBP2: C-terminal binding protein 2; DDR: DNA damage response; DNMT3A: DNA methyltransferase 3 alpha; DSB: double strand break(s); EIF4E3: eukaryotic translation factor 4E family member 3; ERCC3: ERCC excision repair 3; FISH: fluorescence in situ hybridization; FLT3: fms related tyrosine kinase 3; FOXO3: forkhead box O3; FOXP1: forkhead box P1; GATA2: GATA binding protein 2; GEP: gene expression profiling; GMP: granulocyte monocyte progenitor(s); GPR27: G protein-coupled receptor 27; GXYLT2: glucoside xylosyltransferase 2; HR: homologous recombination; HSPC: hematopoietic stem and progenitor cell(s); IL: interleukin; KMT2A: lysine methyltransferase 2A; Lin-: lineage-negative; LSK: Lin-Sca+cKit+; MEP: megakaryocytic erythroid progenitor(s); MLLT3: myeloid/lymphoid or mixed-lineage leukemia translocated to 3; MSH: mutS homolog; MSH2: mutS homolog 2; NFKB1: nuclear factor kappa B subunit 1; NHEJ: non-homologous end-joining; NPM1: nucleophosmin; NRAS: NRAS proto-oncogene GTPase; NUP98: nucleoporin 98; PARP: poly(ADP-ribose) polymerase; PB: peripheral blood; pKAP1: phosphorylated KRAB-domain associated protein 1; pP53: phosphorylated P53; PPP2R2A: protein phosphatase 2 regulatory subunit B alpha; PPP4: protein phosphatase 4; PPP4R2: protein phosphatase 4 regulatory subunit 2; PROK2: prokineticin 2; PTPN12: protein tyrosine phosphatase non-receptor type 12; qRT-PCR: quantitative real-time PCR; RPA2: replication protein A2; RYBP: RING1 and YY1 binding protein; SHQ1: H/ACA ribonucleoprotein assembly factor; SNP: single-nucleotide polymorphism; SUMO1: small ubiquitin-like modifier 1; TCGA: The Cancer Genome Atlas; TP53BP2: tumor protein 53 binding protein 2; WES: whole exome sequencing; γH2AX: phosphorylated histone variant H2AX.
Author contributions
Conception and design of the study by JKH, LB, AT, HD, KD, VIG. Acquisition of data by JKH, LB, AT, PZ, MS, VT, DW, FGR, PP, AD, ES, FK, VIG. Analysis and interpretation of data by JKH, LB, AT, PZ, MS, VT, DW, FGR, PP, AD, ES, FK, FHH, CB, HD, KD, VIG. Drafting/revising of the article by JKH, LB, AT, FHH, HD, KD, VIG.
ACKNOWLEDGMENTS
We thank all members of the AML Study Group for providing patient samples. We gratefully thank Martina Enz and Gabriele Allies for technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts interests.
FUNDING
This study was supported by the Sonderforschungsbereich 1074 (SFB 1074, projects B3 and B4) funded by the Deutsche Forschungsgemeinschaft. LB is Heisenberg Professor of the Deutsche Forschungsgemeinschaft (DFG, BU 1339/3-1). AT was funded by the International Graduate School Ulm and thereafter with a stipend from the Else Kröner-Forschungskolleg. FK was supported by grants from the Deutsche Krebshilfe (Max-Eder program, grant 109420), the Sonderforschungsbereich 1074 (SFB 1074, project A5) funded by the Deutsche Forschungsgemeinschaft, and the Wilhelm Sander Stiftung (2015.153.1). FHH was funded by the Thuringian state program ProExzellenz (RegenerAging - FSU-I-03/14) of the Thuringian Ministry for Research (TMWWDG). VG is a grant recipient of the Medical Faculty of Ulm University and received the Translational Research Training in Hematology (TRTH) by the American Society of Hematology and the European Hematology Association.
REFERENCES
1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015; 373:1136–52. https://doi.org/10.1056/NEJMra1406184.
2. Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010; 453–74. https://doi.org/10.1182/blood-2009-07-235358.
3. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013; 368:2059–74. https://doi.org/10.1056/NEJMoa1301689.
4. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, Gundem G, Van Loo P, Martincorena I, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016; 374:2209–21. https://doi.org/10.1056/NEJMoa1516192.
5. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127:2391–405. https://doi.org/10.1182/blood-2016-03-643544.
6. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2016. https://doi.org/10.1182/blood-2016-08-733196.
7. Bullinger L, Krönke J, Schön C, Radtke I, Urlbauer K, Botzenhardt U, Gaidzik V, Carió A, Senger C, Schlenk RF, Downing JR, Holzmann K, Döhner K, et al. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia. 2010; 24:438–49. https://doi.org/10.1038/leu.2009.263.
8. Bullinger L, Krönke J, Gaidzik V, Döhner H, Döhner K. Comment on ‘Integrative genomic profiling of human prostate cancer’. Leukemia. 2010; 24:1970–2. https://doi.org/10.1038/leu.2010.194.
9. Rücker FG, Bullinger L, Schwaenen C, Lipka DB, Wessendorf S, Fröhling S, Bentz M, Miller S, Scholl C, Schlenk RF, Radlwimmer B, Kestler HA, Pollack JR, et al. Disclosure of candidate genes in acute myeloid leukemia with complex karyotypes using microarray-based molecular characterization. J Clin Oncol. 2006; 24:3887–94. https://doi.org/10.1200/JCO.2005.04.5450.
10. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11–22. https://doi.org/10.1016/j.ccr.2010.05.026.
11. Lando M, Holden M, Bergersen LC, Svendsrud DH, Stokke T, Sundfør K, Glad IK, Kristensen GB, Lyng H. Gene dosage, expression, and ontology analysis identifies driver genes in the carcinogenesis and chemoradioresistance of cervical cancer. PLoS Genet. 2009; 5:e1000719. https://doi.org/10.1371/journal.pgen.1000719.
12. Lando M, Wilting SM, Snipstad K, Clancy T, Bierkens M, Aarnes EK, Holden M, Stokke T, Sundfør K, Holm R. Identification of eight candidate target genes of the recurrent 3p12–p14 loss in cervical cancer by integrative genomic profiling. J Pathol. 2013. https://doi.org/10.1002/path.4168.
13. Krohn A, Seidel A, Burkhardt L, Bachmann F, Mader M, Grupp K, Eichenauer T, Becker A, Adam M, Graefen M, Huland H, Kurtz S, Steurer S, et al. Recurrent deletion of 3p13 targets multiple tumour suppressor genes and defines a distinct subgroup of aggressive ERGfusion-positive prostate cancers. J Pathol. 2013; 231:130–41. https://doi.org/10.1002/path.4223.
14. Cho H, Herzka T, Stahlhut C, Watrud K, Robinson BD, Trotman LC. Rapid in vivo validation of candidate drivers derived from the PTEN-mutant prostate metastasis genome. Methods. 2015; 77–78:197–204. https://doi.org/10.1016/j.ymeth.2014.12.022.
15. Shigekawa T, Ijichi N, Ikeda K, Horie-Inoue K, Shimizu C, Saji S, Aogi K, Tsuda H, Osaki A, Saeki T, Inoue S. FOXP1, an estrogen-inducible transcription factor, modulates cell proliferation in breast cancer cells and 5-year recurrence-free survival of patients with tamoxifen-treated breast cancer. Horm Cancer. 2011; 2:286–97. https://doi.org/10.1007/s12672-011-0082-6.
16. Wang W, Cheng J, Qin JJ, Voruganti S, Nag S, Fan J, Gao Q, Zhang R. RYBP expression is associated with better survival of patients with hepatocellular carcinoma (HCC) and responsiveness to chemotherapy of HCC cells in vitro and in vivo. Oncotarget. 2014; 5:11604–19. https://doi.org/10.18632/oncotarget.2598.
17. Cohen PTW, Philp A, Vázquez-Martin C. Protein phosphatase 4--from obscurity to vital functions. FEBS Lett. 2005; 579:3278–86. https://doi.org/10.1016/j.febslet.2005.04.070.
18. Shui JW, Hu MCT, Tan TH. Conditional knockout mice reveal an essential role of protein phosphatase 4 in thymocyte development and pre-T-cell receptor signaling. Mol Cell Biol. 2007; 27:79–91. https://doi.org/10.1128/MCB.00799-06.
19. Su YW, Chen YP, Chen MY, Reth M, Tan TH. The Serine/Threonine Phosphatase PP4 Is Required for Pro-B Cell Development through Its Promotion of Immunoglobulin VDJ Recombination. PLoS ONE. 2013; 8:e68804. https://doi.org/10.1371/journal.pone.0068804.
20. Chen MY, Chen YP, Wu MS, Yu GY, Lin WJ, Tan TH, Su YW. PP4 is essential for germinal center formation and class switch recombination in mice. PLoS ONE. 2014; 9:e107505. https://doi.org/10.1371/journal.pone.0107505.
21. Bosio Y, Berto G, Camera P, Bianchi F, Ambrogio C, Claus P, Di Cunto F. PPP4R2 regulates neuronal cell differentiation and survival, functionally cooperating with SMN. European Journal of Cell Biology. 2012; 91:662–74. https://doi.org/10.1016/j.ejcb.2012.03.002.
22. Ho MY, Liang CM, Liang SM. PATZ1 induces PP4R2 to form a negative feedback loop on IKK/NF-κB signaling in lung cancer. Oncotarget. 2016; 7:52255–69. https://doi.org/10.18632/oncotarget.10427.
23. Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP, Lieberman J. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008; 31:33–46. https://doi.org/10.1016/j.molcel.2008.05.016.
24. Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a γH2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008; 9:1019–26. https://doi.org/10.1038/embor.2008.162.
25. Lee DH, Pan Y, Kanner S, Sung P, Borowiec JA, Chowdhury D. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat Struct Mol Biol. 2010; 17:365–72. https://doi.org/10.1038/nsmb.1769.
26. Lee DH, Goodarzi AA, Adelmant GO, Pan Y, Jeggo PA, Marto JA, Chowdhury D. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J. 2012; 31:2403–15. https://doi.org/10.1038/emboj.2012.86.
27. Liu J, Xu L, Zhong J, Liao J, Li J, Xu X. Protein phosphatase PP4 is involved in NHEJ-mediated repair of DNA double-strand breaks. Cell Cycle. 2012; 11:2643–9. https://doi.org/10.4161/cc.20957.
28. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010; 40:179–204. https://doi.org/10.1016/j.molcel.2010.09.019.
29. Bagger FO, Sasivarevic D, Sohi SH, Laursen LG, Pundhir S, Sønderby CK, Winther O, Rapin N, Porse BT. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 2016; 44:D917–24. https://doi.org/10.1093/nar/gkv1101.
30. Dobson CL, Warren AJ, Pannell R, Forster A, Lavenir I, Corral J, Smith AJ, Rabbitts TH. The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 1999; 18:3564–74. https://doi.org/10.1093/emboj/18.13.3564.
31. Somervaille TCP, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006; 10:257–68. https://doi.org/10.1016/j.ccr.2006.08.020.
32. Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, Golub TR, Armstrong SA. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006; 442:818–22. https://doi.org/10.1038/nature04980.
33. da Costa RMA, Riou L, Paquola A, Menck CFM, Sarasin A. Transcriptional profiles of unirradiated or UV-irradiated human cells expressing either the cancer-prone XPB/CS allele or the noncancer-prone XPB/TTD allele. Oncogene. 2005; 24:1359–74. https://doi.org/10.1038/sj.onc.1208288.
34. Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006; 8:870–6. https://doi.org/10.1038/ncb1446.
35. Lee YK, Thomas SN, Yang AJ, Ann DK. Doxorubicin Down-regulates Krüppel-associated Box Domain-associated Protein 1 Sumoylation That Relieves Its Transcription Repression on p21WAF1/CIP1 in Breast Cancer MCF-7 Cells. J Biol Chem. 2007; 282:1595–606. https://doi.org/10.1074/jbc.M606306200.
36. Li X, Lee YK, Jeng JC, Yen Y, Schultz DC, Shih HM, Ann DK. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J Biol Chem. 2007; 282:36177–89. https://doi.org/10.1074/jbc.M706912200.
37. Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005; 20:801–9. https://doi.org/10.1016/j.molcel.2005.10.003.
38. Mourtada-Maarabouni M, Williams GT. Protein phosphatase 4 regulates apoptosis in leukemic and primary human T-cells. Leuk Res. 2009; 33:1539–51. https://doi.org/10.1016/j.leukres.2009.05.013.
39. Martin-Granados C, Philp A, Oxenham SK, Prescott AR, Cohen PTW. Depletion of protein phosphatase 4 in human cells reveals essential roles in centrosome maturation, cell migration and the regulation of Rho GTPases. Int J Biochem Cell Biol. 2008; 40:2315–32. https://doi.org/10.1016/j.biocel.2008.03.021.
40. Mourtada-Maarabouni M, Williams GT. Protein phosphatase 4 regulates apoptosis, proliferation and mutation rate of human cells. Biochim Biophys Acta. 2008; 1783:1490–502. https://doi.org/10.1016/j.bbamcr.2008.03.005.
41. Mourtada-Maarabouni M, Kirkham L, Jenkins B, Rayner J, Gonda TJ, Starr R, Trayner I, Farzaneh F, Williams GT. Functional expression cloning reveals proapoptotic role for protein phosphatase 4. Cell Death Differ. 2003; 10:1016–24. https://doi.org/10.1038/sj.cdd.4401274.
42. Toyo-oka K, Mori D, Yano Y, Shiota M, Iwao H, Goto H, Inagaki M, Hiraiwa N, Muramatsu M, Wynshaw-Boris A, Yoshiki A, Hirotsune S. Protein phosphatase 4 catalytic subunit regulates Cdk1 activity and microtubule organization via NDEL1 dephosphorylation. J Cell Biol. 2008; 180:1133–47. https://doi.org/10.1083/jcb.200705148.
43. Carnegie GK, Sleeman JE, Morrice N, Hastie CJ, Peggie MW, Philp A, Lamond AI, Cohen PTW. Protein phosphatase 4 interacts with the Survival of Motor Neurons complex and enhances the temporal localisation of snRNPs. J Cell Sci. 2003; 116:1905–13. https://doi.org/10.1242/jcs.00409.
44. Oh KS, Khan SG, Jaspers NGJ, Raams A, Ueda T, Lehmann A, Friedmann PS, Emmert S, Gratchev A, Lachlan K, Lucassan A, Baker CC, Kraemer KH. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat. 2006; 27:1092–103. https://doi.org/10.1002/humu.20392.
45. Oh KS, Imoto K, Boyle J, Khan SG, Kraemer KH. Influence of XPB helicase on recruitment and redistribution of nucleotide excision repair proteins at sites of UV-induced DNA damage. DNA Repair (Amst). 2007; 6:1359–70. https://doi.org/10.1016/j.dnarep.2007.03.025.
46. Oh KS, Bustin M, Mazur SJ, Appella E, Kraemer KH. UV-induced histone H2AX phosphorylation and DNA damage related proteins accumulate and persist in nucleotide excision repair-deficient XP-B cells. DNA Repair (Amst). 2011; 10:5–15. https://doi.org/10.1016/j.dnarep.2010.09.004.
47. Fassihi H, Sethi M, Fawcett H, Wing J, Chandler N, Mohammed S, Craythorne E, Morley AMS, Lim R, Turner S, Henshaw T, Garrood I, Giunti P, et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A. 2016; 113:E1236–45. https://doi.org/10.1073/pnas.1519444113.
48. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–7. https://doi.org/10.1038/nature03443.
49. Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NMB, Jackson SP, Smith GCM, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–21. https://doi.org/10.1038/nature03445.
50. Kalev P, Simicek M, Vazquez I, Munck S, Chen L, Soin T, Danda N, Chen W, Sablina A. Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition. Cancer Res. 2012; 72:6414–24. https://doi.org/10.1158/0008-5472.CAN-12-1667.
51. Schlenk RF, Fröhling S, Hartmann F, Fischer JT, Glasmacher A, del Valle F, Grimminger W, Götze K, Waterhouse C, Schoch R, Pralle H, Mergenthaler HG, Hensel M, et al. Phase III study of all-trans retinoic acid in previously untreated patients 61 years or older with acute myeloid leukemia. Leukemia. 2004; 18:1798–803. https://doi.org/10.1038/sj.leu.2403528.
52. Petriv OI, Kuchenbauer F, Delaney AD, Lecault V, White A, Kent D, Marmolejo L, Heuser M, Berg T, Copley M, Ruschmann J, Sekulovic S, Benz C, et al. Comprehensive microRNA expression profiling of the hematopoietic hierarchy. Proc Natl Acad Sci USA. 2010; 107:15443–8. https://doi.org/10.1073/pnas.1009320107.
53. Kharas MG, Lengner CJ, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, Morgan K, Tam W, Paktinat M, Okabe R, Gozo M, Einhorn W, Lane SW, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. 2010; 16:903–8. https://doi.org/10.1038/nm.2187.
54. Lück SC, Russ AC, Du J, Gaidzik V, Schlenk RF, Pollack JR, Döhner K, Döhner H, Bullinger L. KIT mutations confer a distinct gene expression signature in core binding factor leukaemia. Br J Haematol. 2010; 148:925–37. https://doi.org/10.1111/j.1365-2141.2009.08035.x.