
Melanoma of the skin
In the United States, more than 68,000 new cases of melanoma were estimated to be diagnosed in 2010, continuing the trend of increased melanoma incidence seen over the past 40 years [1]. Melanoma is the fifth most commonly diagnosed type of cancer in men and seventh most common in women [1]. The lifetime risk of an American developing invasive melanoma is 1 in 55 as compared to 1 in 1,500 in the 1930s [2, 3]. More than 80% of melanomas are diagnosed at an early clinical stage (before regional metastasis) with more than 95% 5-year survival rates for patient’s with localized melanoma [2, 4]. However, when melanoma has spread to regional lymph nodes or metastasized to other organs, there is a significant decrease in survival. The 5-year survival rate for melanoma patients with distant metastasis is significantly decreased to 15% [2]. Therefore, it is imperative to determine the molecular events that lead to melanoma metastasis in search for possible therapeutic molecular targets to curtail the disease.
Early stage melanoma is easily treatable and highly curable with surgical resection [5]. However, once melanoma has metastasized, there are limited and ineffective treatment options available for patients [5, 6]. Decarbazine (DTIC) is the only FDA-approved chemotherapeutic agent for the treatment of metastatic melanoma despite response rates of only 15-20% [5, 6]. Furthermore, the duration of the response to DTIC is not sustained, often lasting as brief as 5 months, with only 5% of patients showing a complete response [7, 8]. Clinical studies utilizing adjuvant immunotherapies with interferon α for high-risk stages II and III melanoma, as well as treatment with high-dose interleukin (IL)-2 for stage IV melanoma, showed limited response rates of less than 15% [9-12]. Identification of melanoma oncogenes, tumor suppressor genes and information gathered from microarrays have advanced our understanding of the molecular mechanisms of melanoma [6]. Further studies in these areas will allow for a clearer understanding of the process of melanoma progression that might lead to the development of urgently needed clinical therapies for metastatic melanoma.
Our laboratory has been studying novel targets involved in the progression of melanoma to develop better therapeutic targets for metastatic melanoma. One of these genes that plays a key role in the progression from non-metastatic to metastatic melanoma is the thrombin receptor, Protease Activated Receptor-1 (PAR-1).
PAR-1
PAR-1 has been found to be involved in the progression of several cancers including breast [13, 14], colon [15, 16] prostate [17, 18] and melanoma [19-22]. As such, PAR-1 has significant roles not only in coagulation, wound healing and inflammation, but also in the progression of several cancer types. PAR-1 was first identified by Vu et al. in 1991 [23] and remains the most studied and best characterized protease-activated receptor.
The thrombin receptor belongs to a family of seven transmembrane G-protein-coupled receptors with a unique method of activation. Unlike typical ligand-binding interactions that occur with other types of G-protein receptors, PAR-1 activation occurs through proteolytic cleavage of the N-terminal domain of the receptor by serine proteases. Although thrombin is the most effective and potent activator of PAR-1, activation can occur through other ligands, such as coagulation factor Xa, trypsin, granzyme A and plasmin [24, 25]. PAR-1 can also be proteolytically cleaved and activated by matrix metalloprotease-1 (MMP-1) in breast cancer cells [26]. PAR-1 activation by thrombin involves the anion-binding exosite of thrombin binding to the PAR-1 amino terminus in an acidic region spanning amino acids 51 to 63 [23]. This highly acidic region, termed the hirudin-like binding site, is similar to the amino acid sequence of the leech anticoagulant peptide, hirudin [27]. This region increases the affinity and potency of thrombin for PAR-1 [27]. PAR-1 also contains a thrombin cleavage site between Arginine at position 41 and Serine at position 42 in the PAR-1 sequence. Thrombin proteolytically cleaves the PAR-1 N-terminus in an irreversible manner, thereby forming a new amino terminus with Serine 42 acting as a tethered ligand to activate the receptor [23, 28] (Figure 1)
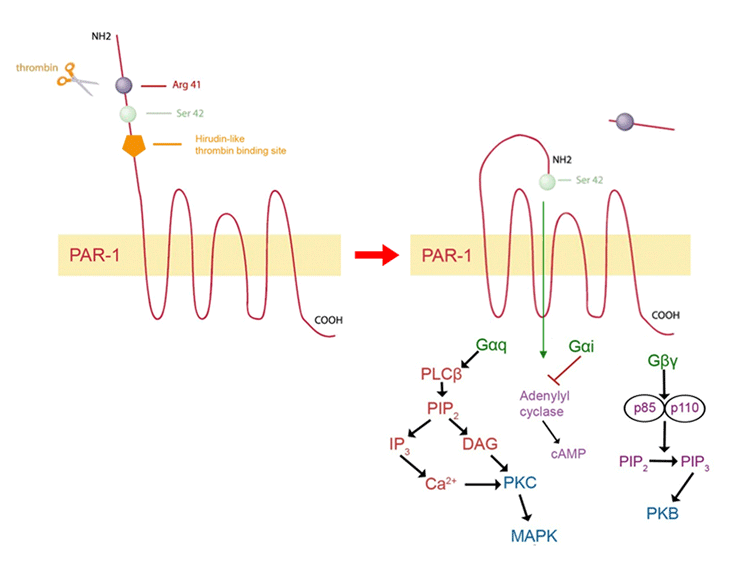
Figure 1: Activation and signaling of PAR-1. Thrombin, the most potent activator of PAR-1, binds to the hirudin-like binding domain on the N-terminus of the receptor and proteolytically cleaves between Arg 41 and Ser 42 in an irreversible manner. Ser 42 now acts as a tethered ligand to activate PAR-1. PAR-1 signals through G-protein subtypes, Gaq, Gai
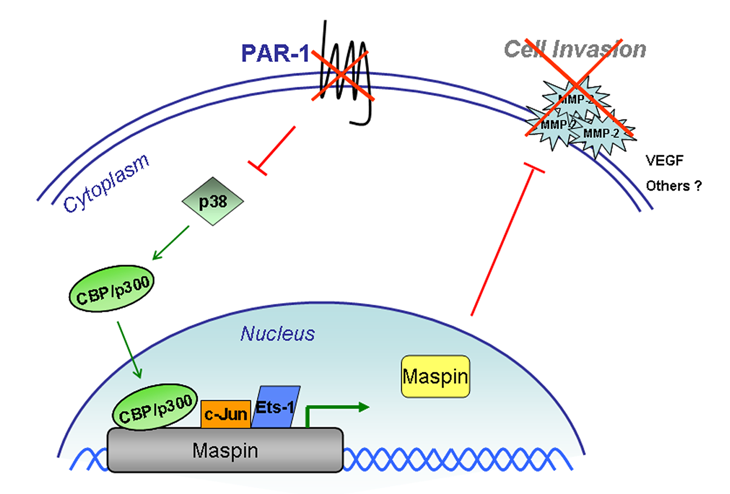
’
’
’