
INTRODUCTION
Domestic pigs (Sus scrofa domesticus) are the major species for meat consumption worldwide. Fat deposition in pigs varies with breeds. Backfat thickness has a strong influence on fattening efficiency, reproductive performance, growth rate and intramuscular fat content of pig. However, breeding pig with high proportion of lean meat in the long term have led to lower intramuscular fat content and finally decreased pork quality [1]. Therefore, how to breed pig species with low backfat thickness that can produce high quality meat remains a challenge. Meanwhile, excessive fat deposition in human body can result in obesity and disorders of energy metabolism, which further leads to obesity-associated diseases, such as type II diabetes, insulin resistance, cardiovascular diseases and some cancer [2]. Thus, human health problems caused by obesity are attracting more attention. Importantly, pig is anatomically and physiologically similar to human, so it can be a good medical model for studying lipid metabolic disease. Better clarification of the molecular basis of fat deposition in pigs will deepen our understanding of diseases associated with fat metabolism. Previous research has shown that adipogenic differentiation and lipid metabolism are regulated by transcription factors and signaling pathways [3, 4]. The transcription factors include bone morphogenetic protein 4 (BMP4), peroxisome proliferator activated receptor gamma (PPARγ), CCAAT/enhancer binding proteins (C/EBPs), adipocyte determination and differentiation dependent factor 1 (ADD1), Kruppel-like factors (KLFs) and zinc finger protein 423 (ZFP423). The signaling pathways include PPAR, mitogen-activated protein kinase (MAPK), transforming growth factor β (TGF β) and Insulin. Several genes have been identified to regulate adipogenesis and lipid metabolism in pig, including KLF13 [5], fibroblast growth factor (FGF21) [6], lipin 1 (LPIN1) [7] and stearoyl-CoA desaturase (SCD) [8]. However, the regulatory mechanisms have not yet been elucidated completely.
LncRNAs, non-protein coding transcripts longer than 200 nucleotides, have important regulatory functions. RNA-seq has high sensitivity in identifying differentially expressed genes as well as comprehensively characterizing and quantitatively analyzing transcriptome [9, 10]. It has been widely applied in the identification and functional analysis of lncRNAs and mRNAs of adipose tissues in pig [8, 11], chicken [12], sheep [13, 14] and cattle [15]. Ramayo et al [16] identified 270 and 186 lncRNAs in the liver tissues of female pigs with high and low intramuscular fatty acid contents, respectively. Zhou et al [11] analyzed the methylation of lncRNA and found the expression level of linc-sscg3623 varied with pig breeds and developmental stages, affecting fat synthesis. Zhang et al [12] analyzed the lncRNA and mRNA expression profiles of preadipocytes at different stages in abdominal adipose tissue of Jinghai Yellow chicken. They found that the target genes of lncRNAs were significantly enriched in MAPK and PPAR signaling pathway associated with adipocyte differentiation. These studies indicate that lncRNAs might determine fat deposition and fatty acid composition, and regulate adipogenic differentiation and lipid metabolism in livestock and poultry. However, there is limited research on the expression profiles and the functions of lncRNAs in subcutaneous adipose tissue of pig breeds with significant differences in fat deposition.
Laiwu (LW) pig, an excellent fatty breed of North China, is characterized by high quality meat with bright color, high water-holding capacity and especially high intramuscular fat content (10.32%) [17]. Compared with Erhualian, Laiwu and Lulai Black pigs [18, 19], Large White (LY) pig, the most widely distributed lean-type pig breed, has lower contents of subcutaneous and intramuscular fat. Thus, LW and LY pigs provide good resources for studying fat deposition, adipogenic differentiation and diseases associated with fat metabolism.
In this study, RNA-seq technology was used to comparatively analyze the gene expression profiles of subcutaneous adipose tissue between LW and LY pigs. Then, key lncRNAs and genes associated with adipogenesis and lipid metabolism were identified. Furthermore, with the help of Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Co-expression and protein-protein interaction (PPI) network analyses, we investigated the molecular mechanism of differentially expressed lncRNAs and genes regulating fat deposition. The results can provide useful information for studying fat deposition regulated by lncRNAs in porcine adipose tissue. In addition, this study is useful for studying lipid metabolic disease and breeding livestock and poultry with high quality meat.
RESULTS
Total RNA sequencing and mapping
To comprehensively understand the transcriptomes of subcutaneous fat tissues in LW and LY pigs, total RNA was isolated and sequenced by Illumina sequencing platform. Approximately 11 Gb sequencing data per sample were obtained. After pre-processing and low-quality trimming of the sequencing data, about 90 million clean reads were obtained for each sample. About 71.27–76.90% clean reads were mapped to the reference genome of pig and 60% were uniquely mapped (Table 1). By assembly and reconstruction, a total of 92,508 transcripts (FPKM ≥ 0.01) were obtained. Among them, 21,644 were protein-coding transcripts.
Table 1: Summary of raw reads after quality control and mapping to the reference genome
Sample |
Raw reads |
Clean reads |
Valid ratio (%) |
Q30 (%) |
GC content (%) |
Total mapped |
Uniquely mapped |
|---|---|---|---|---|---|---|---|
LY1 |
94810516 |
90927088 |
95.84% |
94.61% |
51.50% |
69921634 (76.9%) |
57700161 (63.46%) |
LY2 |
92647174 |
88999976 |
96.00% |
94.73% |
51.50% |
68031369 (76.44%) |
56194896 (63.14%) |
LY3 |
95069640 |
91532758 |
96.22% |
94.78% |
55.00% |
68970804 (75.35%) |
54923704 (60%) |
LW1 |
92860394 |
89189416 |
95.98% |
95.28% |
51.00% |
66219222 (74.25%) |
54266624 (60.84%) |
LW2 |
94815260 |
91416676 |
96.34% |
95.43% |
52.00% |
65157231 (71.27%) |
55934087 (61.19%) |
LW3 |
92647706 |
88944438 |
95.94% |
95.18% |
50.00% |
65611080 (73.77%) |
54756763 (61.56%) |
Identification and characterization of lncRNAs
A total of 1904 lncRNA candidates were identified, including 1503 lincRNAs, 166 anti-sense lncRNAs, 137 sense lncRNAs and 98 intronic lncRNAs in the six libraries (Figure 1A). LincRNA was the most common type of lncRNAs and its proportion was also the largest, which were consistent with current research. Among all the lncRNAs, 771 (about 40.49%) were mapped to ALDB database. As shown in Figure 1B, most lncRNAs had two exons (an average of 3.01 exons), which were significantly less than the exons of protein-coding gene (up to 118 exons, an average of 9.62 exons). In addition, the amount of exons in lncRNAs in subcutaneous adipose tissue of pig was consistent with that in thyroid [20] and testicular tissue [21] of pig. Overall, the distributions of lncRNA and protein-coding gene lengths were consistent, and the proportion of relatively long mRNA transcripts was higher than that of lncRNAs (Figure 1C). The average length of lncRNAs and mRNAs was 2314 and 2225 nt, respectively.
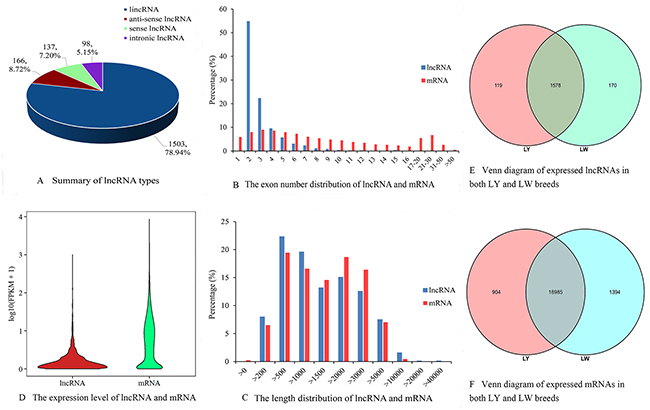
Figure 1: LncRNA characterization and gene expression. (A) Summary of lncRNA types. (B) The exon number distribution of lncRNA and mRNA. (C) The length distribution of lncRNA and mRNA. (D) The expression level of lncRNA and mRNA. (E) Venn diagram of expressed lncRNAs in both LY and LW breeds. (F) Venn diagram of expressed mRNAs in both LY and LW breeds.
Expression level of genes and differential expression analysis
Violin plot of FPKM values for the transcripts indicated that the expression level of mRNAs was relatively higher than that of lncRNAs (Figure 1D), which was consistent with the research on lncRNAs in pig testicular tissue [21], mouse [22] and human [23]. FPKM values for most lncRNAs were less than 10, whereas those for mRNAs were evenly distributed. A total of 119 lncRNAs and 904 mRNAs were expressed specifically in subcutaneous adipose tissues of LY pig, and 170 lncRNAs and 1394 mRNAs were expressed specifically in subcutaneous adipose tissues of LW pig (Figure 1E and 1F). Differential expression analysis ((LW vs LY) indicated that 40 lncRNAs were up-regulated and 14 were down-regulated in LW pig (Supplementary Table 1). A total of 482 known differentially expressed mRNAs were identified. Of these, 223 mRNAs were up-regulated in LW pig and the rest were down-regulated (Supplementary Table 2).
GO annotation of differentially expressed genes
A total of 211 differentially expressed genes were annotated in GO terms (Supplementary Table 3). As shown in Figure 2, in the category of biological process, differentially expressed genes were mainly enriched in “response to oxygen-containing compound”, “fat cell differentiation”, “regulation of lipid metabolic process”, “fatty acid metabolic process”, “response to steroid hormone”, etc. In the category of molecular function, differentially expressed genes were mainly enriched in “ubiquitin-like protein ligase binding”, “transcription regulatory region sequence-specific DNA binding”, “lipase activity”, etc. In the category of cellular component, differentially expressed genes were mainly enriched in “mitochondrial envelope”, “side of membrane”, etc. The differentially expressed genes in LY and LW pigs should mainly regulate adipocyte differentiation and lipid metabolism.
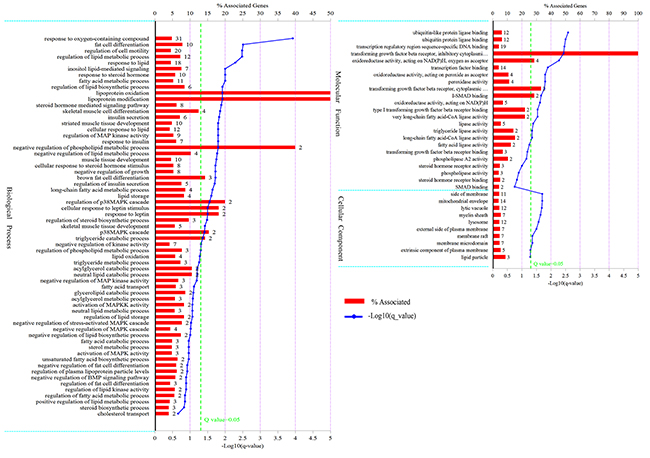
Figure 2: GO analysis of differentially expressed genes. The figure is composed of three parts: biological processes, molecular functions, and cellular components. The significance level of enrichment was set at corrected P value (Q value) < 0.05.
KEGG enrichment analysis of differentially expressed genes
KEGG enrichment analysis demonstrated that 115 differentially expressed genes were enriched in 63 signaling pathways. Among these signaling pathways, five were significantly enriched (Supplementary Table 4). PPAR signaling pathway (Q value=0.0097) and fluid shear stress and atherosclerosis (Q value=0.012) were the two most significantly enriched signaling pathways. In addition, differentially expressed genes were also enriched in signaling pathways associated with lipid metabolism, such as regulation of lipolysis in adipocytes, fatty acid metabolism and phospholipase D signaling pathway (Figure 3). These results suggested that the differentially expressed genes in subcutaneous adipose tissues in LY and LW pigs might be associated with lipid metabolism and adipocyte differentiation. PPAR signaling pathway was the key pathway regulating adipocyte differentiation in LY and LW pigs. Thus, the genes enriched in PPAR signaling pathway might regulate subcutaneous fat deposition and adipocyte differentiation in LW and LY pigs.
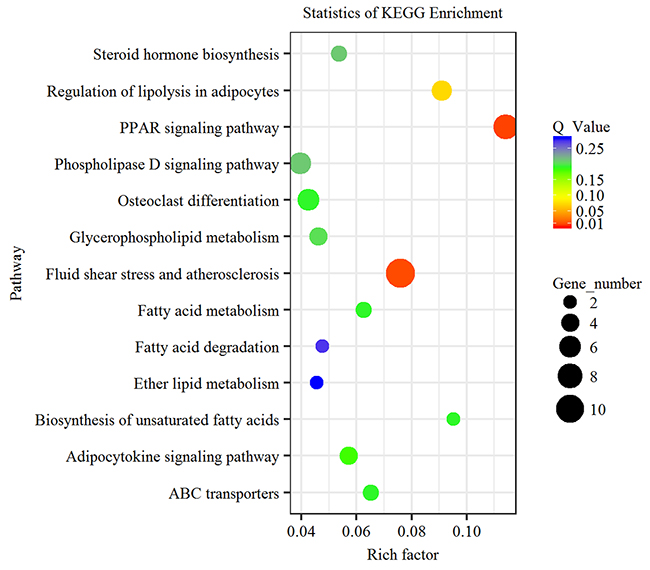
Figure 3: KEGG pathway analysis of differentially expressed genes. Advanced bubble chart shows enrichment of differentially expressed genes in signaling pathways. Y-axis label represents pathway, and X-axis label represents rich factor (rich factor = amount of differentially expressed genes enriched in the pathway/amount of all genes in background gene set). Size and color of the bubble represent amount of differentially expressed genes enriched in the pathway and enrichment significance, respectively.
PPI network for differentially expressed genes
PPI network for the differentially expressed genes related to lipid metabolism was constructed. On this basis, the candidate genes regulating fat deposition in LY and LW pigs were identified (Supplementary Table 5). In fact, some researchers previously identified candidate genes related to intramuscular fat deposition in cattle by using PPI network, which provided useful information for our study [24]. Gene-encoded proteins including early growth response (EGR1), leptin (LEP), CD36 molecule (CD36), SMAD6 family member 6 (SMAD6), SMAD6 family member 7 (SMAD7), nuclear receptor subfamily 4 group 3 (NR4A3), NADPH oxidase heavy chain subunit (CYBB), nuclear receptor subfamily 1 group D member 2 (NR1D2), dynamin 1 (DNM1), activin receptor 1C (ACVR1C) were at hub positions in the PPI network (Figure 4). Thus, they might be important for regulating subcutaneous fat metabolism and adipogenic differentiation in pigs.
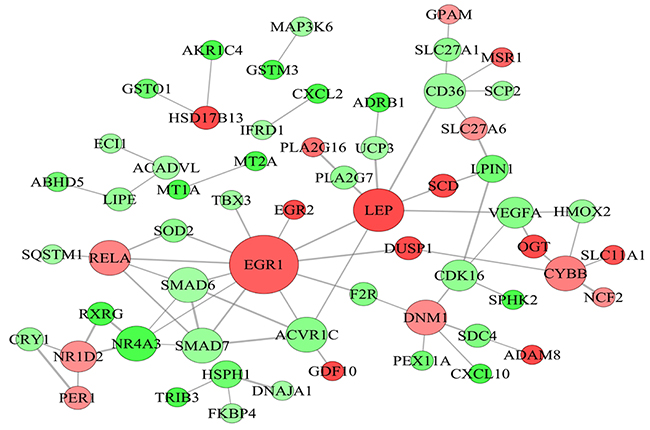
Figure 4: Protein-protein interaction network of differentially expressed genes related to lipid metabolism. Node represents protein, edge represents interaction between proteins, size of the node is proportional to degree of this node (degree of the node is defined as amount of proteins that interact with this node), and color of node represents Log2FoldChange in expression levels of differentially expressed genes between Laiwu and Large White pigs.
Target genes of lncRNAs and functional analysis
By correlation analysis of the expression levels of lncRNAs and mRNAs, 425 differentially expressed mRNA were found to co-express with differentially expressed lncRNAs (Supplementary Table 6). According to the results of GO and KEGG analyses, the differentially expressed genes related to lipid metabolism, fat-associated diseases and adipocyte differentiation were investigated to identify key lncRNAs regulating fat deposition. LncRNA-mRNA co-expression network with the threshold |PCC|≥0.9 was constructed (Supplementary Table 7). As showed in Figure 5, some lncRNAs were at the central positions of the network. Using Degree (Degree Centrality) ≥ 30 as the threshold, 14 key lncRNAs were selected. They were XLOC_013639, XLOC_053194, XLOC_028962, XLOC_019518, XLOC_042912, XLOC_ 048197, XLOC_007718, XLOC_062285, XLOC_017637, XLOC_078946, XLOC_053859, XLOC_065384, XLOC_ 043153 and XLOC_025524. These could serve as a basis for further research on lncRNA function.
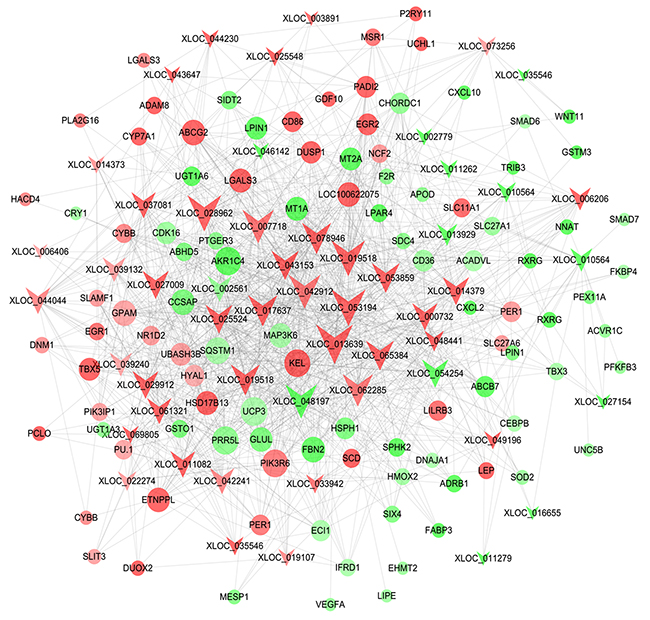
Figure 5: Co-expression network of the differentially expressed lncRNAs and genes related to lipid metabolism. Note that color denotes differential expression levels. Red represents up-regulation and green represents down-regulation. Size represents the importance of a node (Degree). The edge denotes the interaction strength. Circles and inverted triangles represent genes and lncRNAs, respectively.
There were only 5 lncRNAs that were predicted to cis-regulate target genes (Supplementary Table 8). There were many target genes that were trans-regulated by lncRNAs (Supplementary Table 9). Considering genes related to lipid metabolism, four pairs of lncRNA-mRNA were selected (Table 2). By trans-target gene analysis, three key lncRNAs were identified. Combined with the 14 key lncRNAs obtained by co-expression analysis, there were a total of 17 key lncRNAs.
Table 2: Trans-target genes related to lipid metabolism of differentially expressed lncRNAs
lncRNA id |
lncRNA expression |
gene id |
mRNA expression |
|---|---|---|---|
XLOC_014379 |
up |
HSD17B13 |
up |
XLOC_014379 |
up |
SCD |
up |
XLOC_011279 |
down |
LPIN1 |
down |
XLOC_064871 |
down |
TRIB3 |
down |
The target genes of lncRNAs were also enriched in PPAR signaling pathway and biological processes including adipogenic differentiation, fatty acid metabolism and lipid metabolism. This indicated that the differentially expressed lncRNAs might regulate adipogenic differentiation and lipid metabolism by targeting key genes involved in above signaling pathway and biological processes. To further explore the function of key lncRNAs, GO annotation and KEGG enrichment analysis were performed for the target genes of each lncRNA. LncRNAs whose target genes were significantly enriched in PPAR signaling pathways, fat cell differentiation and fatty acid metabolic process were selected for the construction of lncRNA-mRNA-signaling pathway network (Figure 6). According to the network, the regulatory patterns of key lncRNA-mRNA were explored.
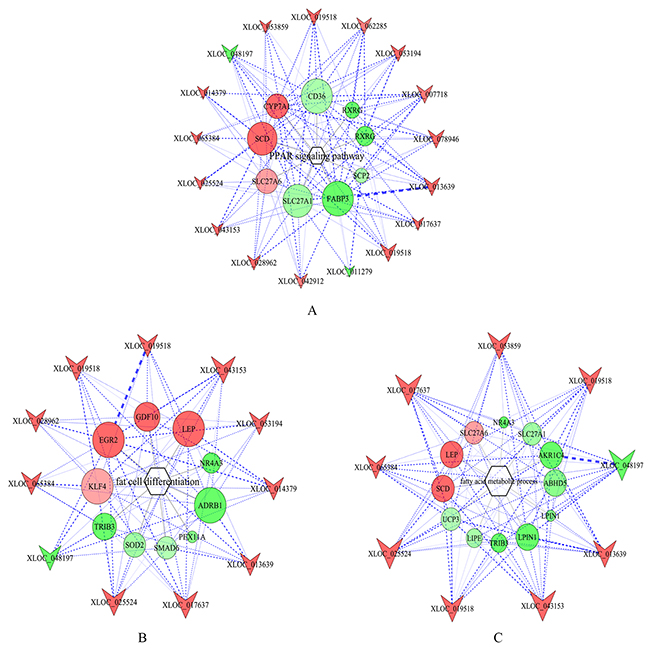
Figure 6: LncRNA-gene-pathway network related to lipid metabolism. (A) lncRNA-gene-PPAR signaling pathway network. (B) lncRNA-gene-fat cell differentiation network. (C) lncRNA-gene-fatty acid metabolism process network. Hexagons, circles and inverted triangles represent pathways, genes and lncRNAs, respectively. Note that color denotes differential expression levels. Red represents up-regulation, and green represents down-regulation. Size represents the importance of a node (Degree). The edge denotes the interaction strength.
qRT-PCR verification
qRT-PCR is considered as the golden standard for quantitative analysis of genes [25]. RNA-Seq data are significantly correlated to qRT-PCR results (R2 = 0.97) [26]. Thus, ten differentially expressed genes were randomly chosen for the verification of RNA-seq results (Supplementary Table 10). As shown in Figure 7, tribbles pseudokinase 3 (TRIB3), LPIN1, XLOC_054254, XLOC_011279 and XLOC_064871 were up-regulated in subcutaneous adipose tissue in LY pig. Solute carrier family 27 member 6 (SLC27A6), SCD, XLOC_019518, early growth response 2 (EGR2) and XLOC_014379 were up-regulated in subcutaneous adipose tissue in LW pig. These results were consistent with the sequencing results, implying the reliability of sequencing results.
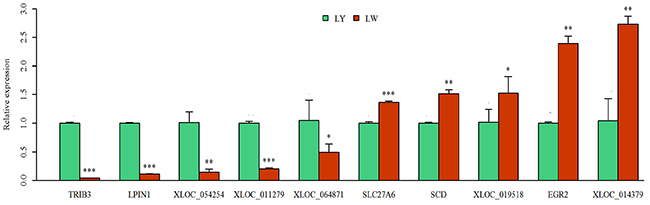
Figure 7: qRT-PCR verification of the differentially expressed genes. The differential expression of genes in subcutaneous adipose tissues between Laiwu and Large White pigs was verified by qRT-PCR. *: P<0.05; **: P<0.01; ***: P<0.001.
DISCUSSION
Fat content in animals is determined by preadipocyte differentiation, adipocyte differentiation and proliferation, and fat synthesis. Fat deposition is closely related to the genetic background, developmental stage and nutritional level of animals and is regulated by many transcription factors, key genes and signaling pathways. In this study, RNA-seq technology was used to analyze and compare the gene expression profiles of subcutaneous adipose tissues between LW and LY pigs. GO and KEGG enrichment analysis indicated that the differentially expressed genes were mainly involved in PPAR signaling pathway and biological processes including fatty acid metabolism and adipocyte differentiation. Analysis of differential lncRNA-mRNA co-expression patterns and functional analysis of target genes indicated that lncRNAs regulated subcutaneous fat metabolism and deposition by regulating genes associated with above-mentioned signaling pathway and biological processes. In sum, this study explored the role of lncRNAs in regulating subcutaneous fat deposition in pig and might provide a basis for studying the molecular mechanism of lncRNAs regulating adipogenic differentiation and lipid metabolism.
PPAR signaling pathway is a key pathway closely related to fatty acid and sterol metabolism as well as adipogenic differentiation [27]. In this study, PPAR signaling pathway was significantly enriched with eight differentially expressed genes: CD36, cytochrome P450 family 7 subfamily A polypeptide 1 (CYP7A1), fatty acid binding protein 3 (FABP3), RXRG, SCD, sterol carrier protein 2 (SCP2), solute carrier family 27 member 1 (SLC27A1) and SLC27A6. These 8 differentially expressed genes were regulated by 17 lncRNAs. CD36 that positively correlates with the expression of PPARγ can facilitate cellular uptake of LCFA and promote adipocyte differentiation and fat biosynthesis [28]. CYP7A1 is a key rate-limiting enzyme for transformation of cholesterol to bile acid, and its activity is negatively correlated with the levels of plasma low-density lipoprotein and cholesterol in rodents and humans [29, 30]. SCP2, by regulating protein and lipid compositions in lipid droplets [31], can inhibit HDL-mediated efflux of sterol from lipid droplets [32]. The lipid deposition in mice with the absence of SCP2 is significantly increased [33], indicating that SCP2 is conducive to lipid maintenance. SLC27A1 and SLC27A6 are genes encoding proteins belonging to fatty acid transport protein (SLC27 protein) family. SLC27 proteins are key regulator of fatty acid metabolism and can help maintain lipid homeostasis and promote uptake of LCFA and lipid deposition [34].
SCD is a key enzyme transforming saturated fatty acid to endogenous oleic acid in food and regulating unsaturated fatty acid biosynthesis, and it can promote lipid deposition [35]. In this study, SCD was up-regulated in subcutaneous adipose tissue of LW pig, which was consistent with previous study showing that SCD was highly expressed in subcutaneous adipose tissue of pigs with thick backfat [8, 36]. The backfat thickness of LW was higher than that of LY, implying that SCD promoted subcutaneous fat deposition in LW. Meanwhile, XLOC_014379 might target SCD and involve in PPAR signaling pathway, thus regulating fatty acid metabolism. Previous study indicated that the expression level of FABP3 in subcutaneous adipose tissue was positively correlated with the backfat thickness in cattle [37]. In this study, however, FABP3 was down-regulated in subcutaneous adipose tissue in LW, implying that the mechanisms of FABP3 regulating fat deposition in pig and cattle might be different. The above results indicated that the genes enriched in PPAR signaling pathway were mainly involved in fatty acid and sterol metabolism, which could influence lipid deposition. LncRNAs such as XLOC_013639 (Figure 6) might regulate PPAR signaling pathway by regulating these genes, thus playing an important role in fatty acid metabolism and lipid deposition in subcutaneous adipose tissue in LW and LY pigs. Especially, XLOC_014379 might target SCD and play a crucial role.
GO annotation demonstrated that differentially expressed genes were mainly associated with fatty acid metabolism and adipocyte differentiation. There were 9 and 11 key lncRNAs in fatty acid metabolism network and adipocyte differentiation network, respectively. Thus, the function of these lncRNAs should be closely related to fatty acid metabolism and adipogenic differentiation. After excluding the genes associated with PPAR signaling pathway, 14 genes including abhydrolase domain containing 5 (ABHD5), AKR1C4, lipase E (LIPE), LPIN1, NR4A3, TRIB3, uncoupling protein 3 (UCP3), peroxisomal biogenesis factor 11 alpha (PEX11A), SMAD6 and superoxide dismutase 2 (SOD2) were up-regulated in subcutaneous adipose tissue in LY pig, whereas LEP, EGR2, growth differentiation factor 10 (GDF10) and KLF4 were up-regulated in subcutaneous adipose tissue in LW pig. In vitro study indicates that ABHD5 can activate adipose triglyceride lipase (ATGL) and promote lipid decomposition [38]. LPIN1 is important for adipocyte differentiation and lipid metabolism [39] and is a candidate gene regulating fat deposition in pig [7]. Some research demonstrates that LPIN1 mRNA level is positively correlated with insulin sensitivity in subcutaneous adipose tissue of human [40]. In this study, XLOC_011279 might involve in subcutaneous adipogenesis, lipid metabolism and insulin sensitivity by targeting LPIN1. NR4A3 belongs to nuclear hormone receptor (NR) family that can regulate lipid and carbohydrate metabolism [41]. NR4A3, a target of beta-adrenergic signaling, play a key role in regulating fatty acid oxidation in skeletal muscle in mice, and muscle-specific NR4A3 expression can reduce deposition of white and brown fat [42, 43]. Another research suggests NR4A3 could promote the differentiation of myocytes into myotubes in biceps of pig [44]. In this study, NR4A3 was up-regulated in subcutaneous adipose tissue in LY pig, indicating it might inhibit adipogenic differentiation. However, PPI network demonstrated that NR1D2, another member in the NR family, was highly expressed in subcutaneous adipose tissue of LW pig. NR1D2 can induce the expression of PPARs, CEBPα and FABP4 genes and promote adipogenic differentiation in dwarf chickens [45]. Thus, NR1D2 might promote adipogenic differentiation in subcutaneous adipose tissue of LW pig. TRIB3, a kind of pseudokinase, can inhibit synthesis of fatty acid by combining with E3 ubiquitin ligase and protect against diet-induced obesity by stimulating fatty acid oxidation in adipose of mice during fasting [46]. In addition, TRIB3 can negatively regulate the transcriptional activation of PPARγ and inhibit adipogenesis in 3T3-L1 cells [47]. TRIB3 is expressed in porcine adipose tissue and skeletal muscle, and is related to meat quality and production performance [48]. These indicate that TRIB3 might inhibit subcutaneous fat deposition in LY pig and be closely related to obesity-associated diseases. In this study, XLOC_064871 can trans-regulate TRIB3, indicating that XLOC_064871 might also play a vital role in adipocyte differentiation and fatty acid metabolism in pig. UCP3 is a negative regulator of energy balance and lipid metabolism. By inhibiting triglycerides storage in cytoplasm, UCP3 gene can affect lipid metabolism and inhibit fat deposition [49, 50]. Polymorphism analysis indicates that UCP3 gene can help determine backfat thickness and intramuscular fat content of pig [51] and cattle [52]. Thus, the high expression level of UCP3 gene might negatively regulate subcutaneous fat deposition in LY pig. SMAD6 and SMAD7 are mainly associated with TGFβ signaling pathway. In 3T3-F442A preadipocytes, over-expression of SMAD6/7 can inhibit early differentiation of adipocytes and thus negatively regulate adipogenesis [53]. Accordingly, the high expression level of SMAD6/7 might inhibit pig fat deposition. On above basis, all these genes that were highly expressed in subcutaneous adipose tissue of LY pig might negatively regulate adipocyte differentiation and lipogenesis. In other words, these genes may inhibit subcutaneous fat deposition in LY pig, leading to lower backfat thickness than that of LW pig.
Compared with genes in subcutaneous adipose tissue in LY pig, those in LW pig were highly expressed, mainly promoting adipogenesis in LW pig. EGR2 and KLF4 are important zinc finger transcription factors promoting early adipocyte differentiation, and can stimulate adipogenesis by trans-activating C/EBPβ in 3T3-L1 preadipocytes [54, 55]. In LY pigs with different backfat thicknesses, EGR2 is highly expressed in pig with thicker backfat [36], which is consistent with our results. Taken together, subcutaneous adipocytes in LW pig might have greater differentiation ability than those in LY pig, leading to significant difference in subcutaneous fat deposition between the two pig breeds. This was consistent with the results of Zhang et al [56]. In sum, key lncRNAs played an important role in regulating subcutaneous adipocyte differentiation and affecting fat deposition in LW and LY pigs. XLOC_011279 and XLOC_064871 can target LPIN1 and TRIB3, respectively, and might play a crucial role in adipogenic differentiation. In addition, they could be potential targets for therapy of obesity-associated diseases.
MATERIALS AND METHODS
Ethic statement
All the procedures involving animals were approved by the Animal Care and Use Committee at Institute of Animal Sciences, Chinese Academy of Agricultural Sciences where the experiments were conducted. All the experiments were performed in accordance with relevant guidelines and regulations set by the Ministry of Agriculture of the People’s Republic of China.
Animal and sample preparation
In this study, three healthy female Large White pigs (LYs) and three healthy female Laiwu pigs (LWs) fed a diet formulated to meet current nutritional requirements were selected and raised at Daqian Farming Co. Ltd. of Laiwu (Laiwu city, Shangdong Province, China). All animals had free access to water and food under natural lighting. At slaughter age (180 days old), the mean bodyweight of pigs was 108 ± 2 kg. After slaughter, the subcutaneous adipose tissue of longissimus dorsi muscle was sampled into 5 mL tubes within 30 min, frozen in liquid nitrogen immediately and then transferred to refrigerator at -80° for long-term preservation and further total RNA extraction.
Isolation of total RNA and quality control
The same amount of adipose tissue samples were taken for total RNA isolation. mirVanaTM RNA Isolation Kits (#AM1561, Ambion, USA) were used to isolate total RNA according to the manufacturer’s instruction. The isolated total RNA from each sample was preserved at -80°. NanoDrop 2000 spectrophotometer (Thermo Scientific, USA) was used to determine RNA concentration and OD260 nm/OD280 nm absorption ratio, which was controlled in the range of 1.9–2.1. Bioanalyzer 2100 (Agilent, Santa Clara, CA) was used to evaluate the quality of total RNA (RIN>=7 and 28S/18S>=0.7). RNase-free DNase I (Ambion Inc., Texas, USA) was used to eliminate potential genomic DNA contamination.
cDNA library construction and RNA sequencing
Approximately 1 μg of total RNA from each sample was used for the construction of cDNA libraries (including LW1, LW2 and LW3; LY1, LY2 and LY3) according to Illumina® TruSeq™ RNA Sample Preparation Guide. The process includes (1) separation, enrichment and purification of mRNA using oligo (dT) magnetic beads, (2) enzymatic fragmentation of RNA, (3) synthesis of cDNA, (4) sequencing adapter ligation and (5) PCR amplification. Agilent DNA 1000 Kits on Agilent 2100 Bioanalyzer (Agilent technologies, Santa Clara, CA) was used to determine the size and purity of cDNA libraries. ABI StepOnePlus Real-time fluorescence-based quantitative PCR system was used to accurately determine the effective concentration of cDNA libraries (> 2 nmol/L), thus ensuring the quality of cDNA library. Illumina HiSeqTM2500 platform was used for paired-end sequencing of cDNA libraries and raw reads were obtained.
Reference genome mapping and transcriptome assembly
Clean reads were obtained by removing reads containing adapter contamination, reads containing more than 10% poly-N, and low-quality reads (containing more than 15% bases with Q-scores no higher than 20) using fastx_toolkit software (v0.0.14). TopHat [57, 58] (v2.0.12) was used to align clean reads of each sample to reference genome Sscrofa10.2 (ftp://ftp.ensembl.org/pub/release-87/fasta/sus_scrofa/dna/). Reads were located and mismatch was set to 2 (all other parameters were set to their default values). Cufflinks [58] (v2.1.1) was used for transcriptome assembly and reconstruction. Then, the transcripts were aligned to reference annotation files Sscrofa10.2.87.chr.gtf (ftp://ftp.ensembl.org/pub/release-87/gtf/sus_scrofa). The known lncRNA and mRNA transcripts were thus identified. Meanwhile, the position of transcripts was obtained.
Identification of potential lncRNA candidates
LncRNAs are non-protein coding transcripts longer than 200 nucleotides. Potential lncRNA candidates were identified according to these two characteristics. The detailed process was as follows. (1) Identification based on transcript type. The transcripts whose class code were ’i’, ’u’, ’x’ and ’o’ were screened for identification of potential lncRNA candidates, which corresponded to intronic lncRNA, lincRNA, antisense lncRNA and sense lncRNA, respectively. (2) Identification based on exon number and transcript length. Exon number ≥2 and transcript length > 200 bp were taken as the threshold. Single-exon transcripts with low credibility were filtered out. (3) Identification based on the protein-coding potential of transcripts. PLEK [59], CNCI [60], CPC [61] and Pfam [62] were applied to assess the protein-coding potential of the above-obtained transcripts and the intersection of results from these software was as the finally result. (4) Identification of known lncRNAs. ALDB (A Domestic-Animal Long Noncoding RNA Database) [63] is a database with a focus on the domestic-animal lncRNAs. BLASTN tool was used to align lncRNA candidates to lncRNAs in the database with the setting of identity=100%, mismatch=0, E-value<1e-10 and gap_opening=0. Then, the already known lncRNAs were identified.
Analysis of differentially expressed genes
The expression levels of lncRNAs and mRNAs were normalized using fragments per kilobase of transcript per million mapped reads (FPKM) algorithm, eliminating the influence of sequencing depth, gene length and sample difference on gene expression level. For the experiment with three biological replicates, identification of differentially expressed lncRNAs and protein-coding genes was performed using R package DESeq2 [64] following negative binomial distribution. Multiple hypothesis testing correction of P value was performed with Benjamini-Hochberg algorithm. If |log2FoldChange| ≥ 1 (LW vs LY) and padj (adjusted P value) ≤ 0.05, the genes and lncRNAs were considered to be differentially expressed.
GO and KEGG pathway enrichment analysis of differentially expressed genes
GO (http://www.geneontology.org/) was the international standard classification of gene function. It classifies functions along three aspects including molecular function, biological process and cellular component. Pathway enrichment analysis can help determine the main metabolic pathways and signaling pathways in which differentially expressed genes are involved. KEGG (http://www.genome.jp/kegg) database [65] is a main public database for metabolic analysis and regulatory network research. To further explore the main biological function of differentially expressed genes, CluGO [66] plugin of Cytoscape, based on hypergeometric distribution, was used to determine the GO terms and signaling pathways that differentially expressed genes were enriched in. Benjamini-Hochberg algorithm was used for P value correction. If Q value≤ (corrected P value) ≤ 0.05, the enrichment was significant.
Protein-protein interaction (PPI) network analysis for differentially expressed genes
According to STRING database (http://string-db.org/), PPI network analysis was performed for the differentially expressed genes, and the interaction among differentially expressed genes in subcutaneous adipose tissue of LW and LY were further investigated. Cytoscape was used to visualize the obtained PPI network data files, and the key genes were identified.
Prediction and functional analysis of the target genes of differentially expressed lncRNAs
The main role of lncRNAs, as a kind of non-coding RNAs, is to regulate their target genes: cis-regulating nearby protein-coding genes and trans-regulating distant protein-coding genes. If genes have same expression pattern, their functions will be highly correlated. Therefore, the target genes of lncRNAs were analyzed and key lncRNAs were identified as follows. The Pearson correlation coefficients (PCCs) of expression levels of differentially expressed lncRNAs and mRNAs were calculated. |PCC|>0.8 and P value<0.05 were the threshold according to which co-expressed lncRNA-mRNA were selected. The protein-coding gene located 300 kb upstream and downstream of lncRNA in genome was selected. If |PCC|≥0.9, the gene would be the target gene that was cis-regulated by lncRNA. According to the interaction between lncRNA and mRNA sequences, the target genes that were trans-regulated by lncRNA were predicted. RNAplex [67] was used to calculate the free energy of combination of lncRNA and mRNA sequences. If Energy<-20 and |PCC| ≥ 0.9, the gene would be the target gene that was trans-regulated by lncRNA [68, 69].
GO annotation and KEGG pathway enrichment analysis were performed for the obtained target genes of lncRNAs. Thus, the biological processes and signaling pathways that lncRNAs were enriched in were identified. Then, the functions of lncRNAs were predicted.
Real-time fluorescence-based quantitative PCR (qRT-PCR) verification
qRT-PCR was applied to verify the expression level of genes. Ten differentially expressed genes (five lncRNAs and five mRNAs) were randomly selected. Three biological replicates were employed for each gene. GeneAmp® PCR System 9700 (Applied Biosystems, USA) was used to synthesize cDNA templates via reverse transcription. Approximately 0.5 μg of each RNA sample was used. QuantiFast® SYBR® Green PCR Kit (Qiagen, Germany) and LightCycler® 480 II Real-time PCR Instrument (Roche, Swiss) were used for qRT-PCR analysis. The reaction system consisted of 1 μL of cDNA, 5 μL of 2× QuantiFast® SYBR® Green PCR Premix (Qiagen, Germany), 0.2 μL of forward primer, 0.2 μL of reverse primer and 3.6 μL of nuclease-free water. Reaction conditions were as follows: pre-denaturation at 95 ° for 5 min, followed by 40 cycles; denaturation at 95 ° for 10 s; renaturation at 60 ° and extension for 30 s. Pig β-actin (ACTB) gene was used as the internal control and 2-ΔΔCt method was used to calculate the relative expression levels of genes between samples.
Statistics analysis
All the data were presented as “means ± SDs”. When comparisons were made, a Student’s t-test was performed and P < 0.05 was considered statistically significant.
CONCLUSIONS
In this study, RNA-seq technology and bioinformatics methods were applied to identify the differentially expressed lncRNAs and genes of subcutaneous adipose tissues between Laiwu and Large White pigs. On this basis, the molecular mechanism of fat deposition was explored. Results indicated that 54 lncRNAs and 482 known genes were differentially expressed. LncRNAs can target mRNAs and then play an important role in PPAR signal transduction pathway, adipocyte differentiation and fatty acid metabolism. By this way, they regulated subcutaneous fat deposition in Laiwu and Large White pigs. XLOC_014379, XLOC_011279, XLOC_064871, XLOC_019518 and XLOC_013639, which might target SCD, LPIN1, TRIB3, EGR2 and FABP3, respectively, play key regulatory roles. This study can provide useful information for understanding molecular mechanism of fat deposition in pig. In addition, this study may help breed pig with high meat quality as well as prevent and treat disease associated with fat metabolism.
Author contributions
X.Y.M. conceived and designed the study, and wrote the paper. W.L.H. performed the experiment and data analysis, and wrote the paper. X.X.Z., A.L. and L.L.X performed the experiments and interpreted the data. X.Y.M. supervised the study. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported by National High Technology Research Development Program of China (863 Program No. 2008AA10Z140), the Agricultural Science and Technology Innovation Program (No. ASTIPIAS05), the National Basic Research and Development Program of China (973 Program) (No. 2015CB943100), and the Basic Research Fund for Central Public Research Institutes of CAAS (No. 2013ywf-zd-2).
CONFLICTS OF INTEREST
The authors declare that they have no competing interest.
REFERENCES
1. Suzuki K, Irie M, Kadowaki H, Shibata T, Kumagai M, Nishida A. Genetic parameter estimates of meat quality traits in Duroc pigs selected for average daily gain, longissimus muscle area, backfat thickness, and intramuscular fat content. J Animal Sci. 2005; 83:2058-2065.
2. Kloting N, Bluher M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord. 2014; 15:277-287.
3. Otto TC, Lane MD. Adipose development: from stem cell to adipocyte. Crit Rev Biochem Mol Biol. 2005; 40:229-242.
4. Gupta RK, Arany Z, Seale P, Mepani RJ, Ye L, Conroe HM, Roby YA, Kulaga H, Reed RR, Spiegelman BM. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010; 464:619-623.
5. Jiang S, Wei H, Song T, Yang Y, Zhang F, Zhou Y, Peng J, Jiang S. KLF13 promotes porcine adipocyte differentiation through PPARgamma activation. Cell Biosci. 2015; 5:28.
6. Wang Y, Liu X, Hou L, Wu W, Zhao S, Xiong Y. Fibroblast growth factor 21 suppresses adipogenesis in pig intramuscular fat cells. Int J Mol Sci. 2015; 17.
7. He XP, Xu XW, Zhao SH, Fan B, Yu M, Zhu MJ, Li CC, Peng ZZ, Liu B. Investigation of Lpin1 as a candidate gene for fat deposition in pigs. Mol Biol Rep. 2009; 36:1175-1180.
8. Xing K, Zhu F, Zhai L, Chen S, Tan Z, Sun Y, Hou Z, Wang C. Identification of genes for controlling swine adipose deposition by integrating transcriptome, whole-genome resequencing, and quantitative trait loci data. Sci Rep. 2016; 6:23219.
9. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008; 5:621-628.
10. Miao X, Qin QL. Genome-wide transcriptome analysis of mRNAs and microRNAs in Dorset and Small Tail Han sheep to explore the regulation of fecundity. Mol Cell Endocrinol. 2015; 402:32-42.
11. Zhou ZY, Li A, Wang LG, Irwin DM, Liu YH, Xu D, Han XM, Wang L, Wu SF, Wang LX, Xie HB, Zhang YP. DNA methylation signatures of long intergenic noncoding RNAs in porcine adipose and muscle tissues. Sci Rep. 2015; 5:15435.
12. Zhang T, Zhang X, Han K, Zhang G, Wang J, Xie K, Xue Q. Genome-wide analysis of lncRNAs and mRNAs expression during the differentiation of abdominal preadipocytes in chicken. G3 (Bethesda). 2017; 7:953-966.
13. Miao X, Luo Q, Qin X, Guo Y, Zhao H. Genome-wide mRNA-seq profiling reveals predominant down-regulation of lipid metabolic processes in adipose tissues of Small Tail Han than Dorset sheep. Biochem Biophys Res Commun. 2015; 467:413-420.
14. Miao X, Luo Q, Qin X, Guo Y. Genome-wide analysis of microRNAs identifies the lipid metabolism pathway to be a defining factor in adipose tissue from different sheep. Sci Rep. 2015; 5:18470.
15. Huang W, Guo Y, Du W, Zhang X, Li A, Miao X. Global transcriptome analysis identifies differentially expressed genes related to lipid metabolism in Wagyu and Holstein cattle. Sci Rep. 2017; 7:5278.
16. Ramayo-Caldas Y, Mach N, Esteve-Codina A, Corominas J, Castello A, Ballester M, Estelle J, Ibanez-Escriche N, Fernandez AI, Perez-Enciso M, Folch JM. Liver transcriptome profile in pigs with extreme phenotypes of intramuscular fatty acid composition. BMC Genomics. 2012; 13:547.
17. Zeng QY, Wang GL, Wei SD, Wang LY, Yang HL, Cao HF, Xu YH. Studies on carcass and meat quality performance of crossbred pigs with graded proportions of Laiwu Black genes. Hereditas. 2005; 27:65-69.
18. Gao QX, Li J, Liu HL, Wang LY, Xu YX. Comparative study on lipogenic and lipolytic gene expression in intramuscular fat tissue between growing Erhualian and large white pigs. [Article in Chinese]. Yi Chuan Xue Bao. 2004; 31:1218-1225.
19. Qian Y, Zeng YQ, Du JF, Cui JX, Li H, Chen QM, Song YP, Chen W. CDS cloning and relationship between intramuscular fat content and mRNA expression of PID1 gene in pig. Hereditas. 2010; 32:1153-1158.
20. Shen Y, Mao H, Huang M, Chen L, Chen J, Cai Z, Wang Y, Xu N. Long noncoding RNA and mRNA expression profiles in the thyroid gland of two phenotypically extreme pig breeds using ribo-zero RNA sequencing. Genes. 2016; 7:34.
21. Ran M, Chen B, Li Z, Wu M, Liu X, He C, Zhang S, Li Z. Systematic identification of long noncoding RNAs in immature and mature porcine testes. Biol Reprod. 2016; 94:77.
22. Wang F, Li L, Xu H, Liu Y, Yang C, Cowley AW Jr, Wang N, Liu P, Liang M. Characteristics of long non-coding RNAs in the Brown Norway rat and alterations in the Dahl salt-sensitive rat. Sci Rep. 2014; 4:7146.
23. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011; 25:1915-1927.
24. Lim D, Kim NK, Park HS, Lee SH, Cho YM, Oh SJ, Kim TH, Kim H. Identification of candidate genes related to bovine marbling using protein-protein interaction networks. Int J Biol Sci. 2011; 7:992-1002.
25. Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol. 2014; 32:896-902.
26. Wang C, Gong B, Bushel PR, Thierry-Mieg J, Thierry-Mieg D, Xu J, Fang H, Hong H, Shen J, Su Z, Meehan J, Li X, Yang L, et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat Biotechnol. 2014; 32:926-932.
27. Farmer SR. Regulation of PPARgamma activity during adipogenesis. Int J Obes. 2005; 29:S13-S16.
28. Christiaens V, Van Hul M, Lijnen HR, Scroyen I. CD36 promotes adipocyte differentiation and adipogenesis. Biochim Biophys Acta. 2012; 1820:949-956.
29. Wang J, Freeman DJ, Grundy SM, Levine DM, Guerra R, Cohen JC. Linkage between cholesterol 7alpha-hydroxylase and high plasma low-density lipoprotein cholesterol concentrations. J Clin Invest. 1998; 101:1283-1291.
30. Li T, Matozel M, Boehme S, Kong B, Nilsson LM, Guo G, Ellis E, Chiang JY. Overexpression of cholesterol 7alpha-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology. 2011; 53:996-1006.
31. Atshaves BP, Storey SM, McIntosh AL, Petrescu AD, Lyuksyutova OI, Greenberg AS, Schroeder F. Sterol carrier protein-2 expression modulates protein and lipid composition of lipid droplets. J Biol Chem. 2001; 276:25324-25335.
32. Atshaves BP, Starodub O, McIntosh A, Petrescu A, Roths JB, Kier AB, Schroeder F. Sterol carrier protein-2 alters high density lipoprotein-mediated cholesterol efflux. J Biol Chem. 2000; 275:36852-36861.
33. Martin GG, Atshaves BP, Landrock KK, Landrock D, Schroeder F, Kier AB. Loss of L-FABP, SCP-2/SCP-x, or both induces hepatic lipid accumulation in female mice. Arch Biochem Biophys. 2015; 580:41-49.
34. Anderson CM, Stahl A. SLC27 fatty acid transport proteins. Mol Aspects Med. 2013; 34:516-528.
35. Sampath H, Miyazaki M, Dobrzyn A, Ntambi JM. Stearoyl-CoA desaturase-1 mediates the pro-lipogenic effects of dietary saturated fat. J Biol Chem. 2007; 282:2483-2493.
36. Zambonelli P, Gaffo E, Zappaterra M, Bortoluzzi S, Davoli R. Transcriptional profiling of subcutaneous adipose tissue in Italian Large White pigs divergent for backfat thickness. Animal Genet. 2016; 47:306-323.
37. Taniguchi M, Guan LL, Basarab JA, Dodson MV, Moore SS. Comparative analysis on gene expression profiles in cattle subcutaneous fat tissues. Comp Biochem Physiol Part D Genomics Proteomics. 2008; 3:251-256.
38. Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006; 3:309-319.
39. Phan J, Peterfy M, Reue K. Lipin expression preceding peroxisome proliferator-activated receptor-gamma is critical for adipogenesis in vivo and in vitro. J Biol Chem. 2004; 279:29558-29564.
40. Miranda M, Chacon MR, Gomez J, Megia A, Ceperuelo-Mallafre V, Veloso S, Saumoy M, Gallart L, Richart C, Fernandez-Real JM, Vendrell J; Adipocyte Differentiation Study Group. Human subcutaneous adipose tissue LPIN1 expression in obesity, type 2 diabetes mellitus, and human immunodeficiency virus--associated lipodystrophy syndrome. Metabolism. 2007; 56:1518-1526.
41. Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001; 294:1866-1870.
42. Pearen MA, Myers SA, Raichur S, Ryall JG, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, a target of beta-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology. 2008; 149:2853-2865.
43. Pearen MA, Goode JM, Fitzsimmons RL, Eriksson NA, Thomas GP, Cowin GJ, Wang SC, Tuong ZK, Muscat GE. Transgenic muscle-specific Nor-1 expression regulates multiple pathways that effect adiposity, metabolism, and endurance. Mol Endocrinol. 2013; 27:1897-1917.
44. Jensen JH, Conley LN, Hedegaard J, Nielsen M, Young JF, Oksbjerg N, Hornshoj H, Bendixen C, Thomsen B. Gene expression profiling of porcine skeletal muscle in the early recovery phase following acute physical activity. Exp Physiol. 2012; 97:833-848.
45. Ye Y, Lin S, Mu H, Tang X, Ou Y, Chen J, Ma Y, Li Y. Analysis of differentially expressed genes and signaling pathways related to intramuscular fat deposition in skeletal muscle of sex-linked dwarf chickens. Biomed Res Int. 2014; 2014:724274.
46. Qi L, Heredia JE, Altarejos JY, Screaton R, Goebel N, Niessen S, Macleod IX, Liew CW, Kulkarni RN, Bain J, Newgard C, Nelson M, Evans RM, et al. TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science. 2006; 312:1763-1766.
47. Takahashi Y, Ohoka N, Hayashi H, Sato R. TRB3 suppresses adipocyte differentiation by negatively regulating PPARgamma transcriptional activity. J Lipid Res. 2008; 49:880-892.
48. Fontanesi L, Colombo M, Scotti E, Buttazzoni L, Bertolini F, Dall’Olio S, Davoli R, Russo V. The porcine tribbles homolog 3 (TRIB3) gene: identification of a missense mutation and association analysis with meat quality and production traits in Italian heavy pigs. Meat Sci. 2010; 86:808-813.
49. Azzu V, Brand MD. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci. 2010; 35:298-307.
50. Musa CV, Mancini A, Alfieri A, Labruna G, Valerio G, Franzese A, Pasanisi F, Licenziati MR, Sacchetti L, Buono P. Four novel UCP3 gene variants associated with childhood obesity: effect on fatty acid oxidation and on prevention of triglyceride storage. Int J Obes. 2012; 36:207-217.
51. Li H, Brahi OH, Zhao X, Xu N, Zhao X. Association of pig UCP3 gene mutations and back fat thickness in the sixth and seventh rib. Mol Biol Rep. 2012; 39:1823-1829.
52. Wang Y, Yang W, Gui L, Wang H, Zan L. Association and expression analyses of the Ucp2 and Ucp3 gene polymorphisms with body measurement and meat quality traits in Qinchuan cattle. J Genet. 2016; 95:939-946.
53. Choy L, Skillington J, Derynck R. Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J Cell Biol. 2000; 149:667-682.
54. Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab. 2005; 1:93-106.
55. Birsoy K, Chen Z, Friedman J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2008; 7:339-347.
56. Zhang GH, Lu JX, Chen Y, Zhao YQ, Guo PH, Yang JT, Zang RX. Comparison of the adipogenesis in intramuscular and subcutaneous adipocytes from Bamei and Landrace pigs. Biochem Cell Biol. 2014; 92:259-267.
57. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105-1111.
58. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012; 7:562-578.
59. Li A, Zhang J, Zhou Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014; 15:311.
60. Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, Liu Y, Chen R, Zhao Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013; 41:e166.
61. Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007; 35:W345-W349.
62. Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Res. 2014; 42:D222-230.
63. Li A, Zhang J, Zhou Z, Wang L, Liu Y, Liu Y. ALDB: a domestic-animal long noncoding RNA database. PLoS One. 2015; 10:e0124003.
64. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:R106.
65. Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016; 44:D457-D462.
66. Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009; 25:1091-1093.
67. Tafer H, Amman F, Eggenhofer F, Stadler PF, Hofacker IL. Fast accessibility-based prediction of RNA-RNA interactions. Bioinformatics. 2011; 27:1934-1940.
68. Li Z, Ouyang H, Zheng M, Cai B, Han P, Abdalla BA, Nie Q, Zhang X. Integrated analysis of long non-coding RNAs (LncRNAs) and mRNA expression profiles reveals the potential role of LncRNAs in skeletal muscle development of the chicken. Front Physiol. 2016; 7:687.
69. Miao X, Luo Q, Zhao H, Qin X. Co-expression analysis and identification of fecundity-related long non-coding RNAs in sheep ovaries. Sci Rep. 2016; 6:39398.