
INTRODUCTION
Colorectal cancer is the second leading cause of cancer-related deaths in the U.S. [1], and the prognosis for individuals with advanced colorectal cancer remains poor [2, 3]. Accumulating evidence indicates that colorectal cancer is a heterogeneous group of diseases, and that responsiveness to treatment varies from patient to patient [4–7]. This heterogeneity necessitates precision medicine approaches that take tumor molecular characteristics into account in order to predict an individual’s response to a specific agent.
Aspirin (acetylsalicylic acid) is one of the most commonly used medications worldwide as an effective analgesic, antipyretic and cardiovascular prophylactic agent. Several observational studies and randomized controlled trials have shown that regular use of aspirin after a diagnosis of colorectal cancer is associated with a superior clinical outcome [8–14]. Randomized trials of aspirin for prevention of cardiovascular and cerebrovascular events also suggest that regular aspirin use has the potential to reduce colorectal cancer incidence and mortality [15, 16]. Recently, the U.S. Preventive Services Task Force (USPSTF) recommended the use of aspirin for prevention of colorectal cancer in individuals at a 10% increased risk for cardiovascular disease compared with the general population [17], while cautioning against the potential harms associated with regular aspirin use including gastrointestinal pain and bleeding [18]. Therefore, it is of particular interest to identify subgroups of individuals who are most likely to benefit from aspirin-based therapeutic strategies through the development of informative tumor biomarkers [14].
In line with this precision medicine approach, multiple observational studies of colorectal cancer patients have indicated that the beneficial effects of aspirin may be stronger for PIK3CA-mutant colorectal cancer than for PIK3CA-wild-type tumors [12, 13, 19], though other studies have shown alternate findings [20, 21]. PIK3CA mutations are observed in approximately 15% to 20% of human colorectal carcinomas, and are associated with proximal tumor location [22–25] and KRAS mutations [26–28]. PIK3CA mutations can activate multiple oncogenic pathways, including the phosphatidylinositol-4,5-bisphosphonate 3-kinase (PI3K)/AKT/MTOR, WNT/CTNNB1, and NFKB signaling pathways [29–31]. Studies have shown that PI3K upregulation also enhances prostaglandin-endoperoxide synthase-2 (PTGS2, also known as cyclooxygenase-2) activity and prostaglandin E2 synthesis [32], which promotes cancer cell proliferation and inhibits apoptosis [33, 34]. Therefore, we hypothesized that the anti-cancer effects of aspirin might be stronger for PIK3CA-mutant colon carcinoma cell lines than for PIK3CA-wild-type colon carcinoma cell lines.
To test this hypothesis, we assessed the effect of aspirin treatment on cell cycle arrest and apoptosis using 13 colon cancer cell lines with known PIK3CA mutation status and isogenic colon cancer cells with knock-in PIK3CA-activating mutations. Additionally, our study identified cell models that may serve as ideal in vitro models to further clarify the molecular mechanisms of aspirin’s anti-tumor effects in PIK3CA-mutant colorectal cancer.
RESULTS
Strong cell viability reduction by aspirin in PIK3CA-mutant colon cancer cells
To test the hypothesis that aspirin’s anti-cancer effects might differ by PIK3CA mutation status, we investigated the anti-proliferative activity of aspirin at physiologically attainable concentrations in seven PIK3CA-mutant and six PIK3CA-wild-type colon cancer cell lines (Table 1) using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H- tetrazolium inner salt (MTS) assay. Initially, we examined the effect of a variety of aspirin doses (0, 0.5, 1, 2, 4, 6, 8, 10, and 12 mM) on cell proliferation after exposure to aspirin for 12 to 96 hours in two PIK3CA-mutant (HCT15 and HCT116) and PIK3CA-wild-type (SW480 and SW620) colon cancer cell lines. We found that the most prominent effects of aspirin on proliferation occurred between 48 and 72 hours of exposure. Therefore, all subsequent experiments were performed after treatment with aspirin for 48 and 72 hours. The dose-response curves of PIK3CA-mutant and PIK3CA-wild-type colon cancer cell lines were generated (Figure 1) and the half maximal inhibitory concentration (IC50) values of each PIK3CA subgroup were compared (Supplementary Figure 1A). PIK3CA-mutant colon cancer cell lines were considerably more sensitive to the anti-proliferative effects of aspirin than PIK3CA-wild-type cells (Figure 1A). Aspirin did not show differential effects according to BRAF or KRAS mutation status (Figure 1B). Next, in order to validate that PIK3CA-mutant colon cancer cell lines were specifically sensitized to aspirin but not to common DNA damaging chemotherapeutic agents, we investigated the IC50 values of 5-fluorouracil and cisplatin in our thirteen colon cancer lines from a compound sensitivity database of the Cancer Cell Line Encyclopedia (CCLE) [35]. However, no significant differences were found between PIK3CA-mutant and PIK3CA-wild-type colon cancer cell lines (Supplementary Figure 1B).
Table 1: Mutational status of PIK3CA, BRAF, and KRAS in colon cancer cell lines used in the current study
Cell line |
Mutation status* |
||
|---|---|---|---|
PIK3CA |
BRAF |
KRAS |
|
HCT15 |
c.1633G>A (p.E545K), |
WT |
c.38G>A (p.G13D) |
HCT116 |
c.3140A>G (p.H1047R) |
WT |
c.38G>A (p.G13D) |
HT-29 |
c.1345C>A (p.P449T) |
c.1799T>A (p.V600E) |
WT |
LS174T |
c.3140A>G (p.H1047R) |
WT |
c.35G>A (p.G12D) |
RKO |
c.3140A>G (p.H1047R) |
c.1799T>A (p.V600E) |
WT |
SW948 |
c.1624G>A (p.E542K) |
WT |
c.182A>T (p.Q61L) |
T84 |
c.1624G>A (p.E542K) |
WT |
c.38G>A (p.G13D) |
CaCo-2 |
WT |
WT |
WT |
COLO205 |
WT |
c.1799T>A (p.V600E) |
WT |
LOVO |
WT |
WT |
c.38G>A (p.G13D), c.41C>T (p.A14V) |
SW48 |
WT |
WT |
WT |
SW480 |
WT |
WT |
c.35G>T (p.G12V) |
SW620 |
WT |
WT |
c.35G>T (p.G12V) |
*Mutation according to the American Type Culture Collection. WT, wild-type.
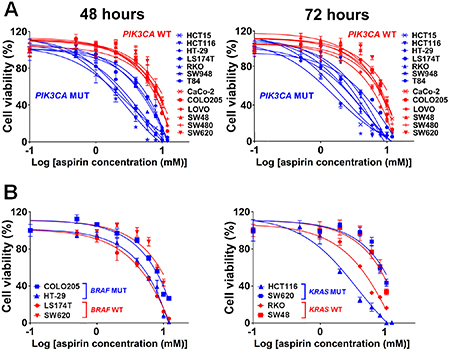
Figure 1: Aspirin causes decreased cell viability in PIK3CA-mutant human colon cancer cells. (A) Dose-response curves of PIK3CA-mutant (blue lines) or PIK3CA-wild-type (red lines) human colon cancer cells after treatment with increasing concentrations of aspirin (0, 0.5, 1, 2, 4, 6, 8, 10 and 12 mM) for 48 and 72 hours. (B) Dose-response curves of human colon cancer cells with BRAF-mutant (blue lines) or BRAF-wild-type (red lines) as well as KRAS-mutant (blue lines) or KRAS-wild-type (red lines) after treatment with increasing concentrations of aspirin (0, 0.5, 1, 2, 4, 6, 8, 10 and 12 mM) for 48 hours. Percent cell viability is relative to that of DMSO-treated control cells. The data shown represent mean ± standard deviation of three replicates. DMSO, dimethyl sulfoxide; WT, wild-type; MUT, mutation.
Aspirin induces apoptosis and G0/G1 cell cycle arrest in PIK3CA-mutant cells
We evaluated the effects of aspirin on induction of apoptosis and cell cycle distribution in three PIK3CA-mutant cell lines (HCT15, HCT116, and SW948) that were particularly sensitive to aspirin exposure in the MTS assay and three PIK3CA-wild-type colon cancer cell lines (COLO205, SW480, and SW620) that were resistant to aspirin exposure. Cells were treated with 0, 1, 2.5, and 5 mM aspirin for 48 and 72 hours after plating. Aspirin treatment led to a more profound induction of apoptosis in colon cancer cells harboring a PIK3CA mutation than in those with wild-type PIK3CA (Figure 2). We next assessed the percentage of cells in each phase of the cell cycle within these PIK3CA mutant and wild-type colon cancer cells (Figure 3). All PIK3CA-mutant cell lines underwent a dose-dependent increase in G0/G1 cell cycle arrest after aspirin exposure for 48 and 72 hours. This effect was not observed in PIK3CA-wild-type colon cancer cell lines.
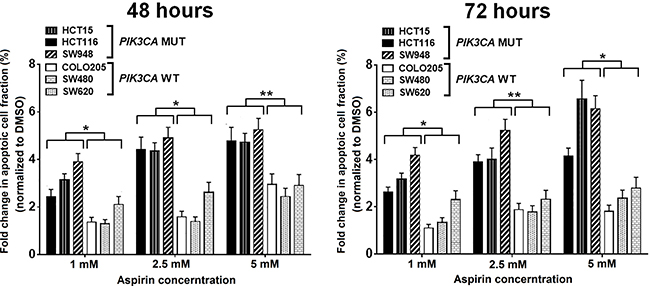
Figure 2: Aspirin treatment results in more apoptosis in PIK3CA-mutant human colon cancer cell lines. PIK3CA-mutant human colon cancer cell lines (HCT15, HCT116, and SW948) and PIK3CA-wild-type colorectal cancer cell lines (COLO205, SW480, and SW620) were incubated with aspirin (1, 2.5, and 5 mM) or DMSO for 48 and 72 hours. Apoptosis analysis was performed by flow cytometry with Annexin V-FITC/PI staining. The percentage of Annexin V-FITC/PI positive cells after aspirin treatment at each dose and time point for all cell lines were normalized by DMSO group and compared according PIK3CA status. Student’s t-test was performed to determine significance. The data shown represent mean ± standard deviation of three replicates. *P value < 0.05. **P value < 0.01. DMSO, dimethyl sulfoxide; MUT, mutation; WT, wild-type.
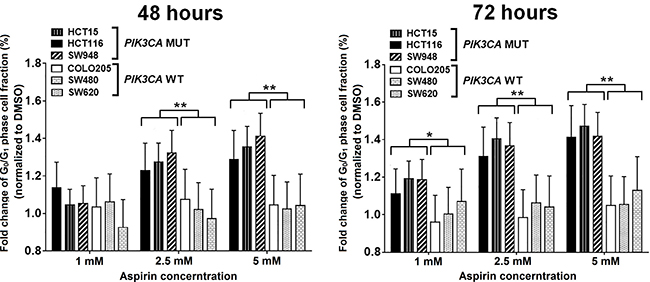
Figure 3: Aspirin leads to a higher proportion of cells in G0/G1 phase arrest in PIK3CA-mutant human colon cancer cell lines. PIK3CA-mutant human colon cancer cell lines (HCT116, HCT15, and SW948) and PIK3CA-wild-type colorectal cancer cell lines (COLO205, SW620, and SW480) were incubated with aspirin (0, 1, 2.5, and 5 mM) for 48 and 72 hours. Cell cycle analysis was performed by flow cytometry with PI staining. The percentage of G0/G1 phase cells of each aspirin treatment point was normalized by DMSO group and was compared according PIK3CA status. Student’s t-test was performed to determine significance. The data shown represent mean ± standard deviation of three replicates. *P value < 0.05. **P value < 0.01. DMSO, dimethyl sulfoxide; MUT, mutation; WT, wild-type.
Mutations in PIK3CA oncogene sensitize colon cancer cells to aspirin
The PIK3CA c.3140A>G (p.H1047R) and c.1633G>A (p.E545K) somatic mutations are commonly found in colorectal carcinoma, and were present in the colon cancer cells (HCT15 and HCT116, respectively) included in the published studies [36, 37]. In order to further confirm our findings and ensure that the observed PIK3CA-mutant-specific effect was not confounded by the genomic backgrounds of the selected cell lines, we investigated whether activation of PI3K via knock-in of the c.3140A>G (p.H1047R) or c.1633G>A (p.E545K) mutation could sensitize a PIK3CA wild-type colon cancer cell line to aspirin treatment. Two isogenic cell lines were derived from parental SW48 cells, each of which carried constitutively active mutant alleles with mutations at either c.3140A>G [SW48 (PIK3CA c.3140A>G/+)] or c.1633G>A [SW48 (PIK3CA c.1633G>A/+)]. The IC50 values of each group showed that aspirin treatment of isogenic SW48 cells carrying either PIK3CA mutation resulted in a statistically significant loss of cell viability of up to 47% relative to parental SW48 cells (P = 0.031) (Figure 4).
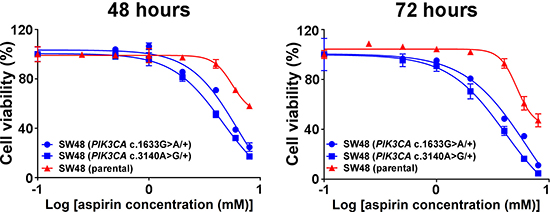
Figure 4: Knock-in of PIK3CA mutations sensitizes colon cancer cells to aspirin. Dose-response curves of parental SW48 cells (red) and individual knock-in of PIK3CA-activating mutations at either alleles c.3140A>G (p.H1047R) or c.1633G>A (p.E545K) of SW48 cells (blue) were treated with aspirin (0, 0.5, 1, 2, 4, 6 and 8 mM) for 48 and 72 hours. Percent cell viability is relative to that of DMSO-treated control cells. The data shown represent mean ± standard deviation of three replicates. DMSO, dimethyl sulfoxide; MUT, mutation; WT, wild-type.
DISCUSSION
We conducted this study to provide in vitro experimental evidence supporting the recent molecular pathological epidemiology studies that suggest tumor PIK3CA mutation status as a biomarker to predict benefits from aspirin therapy for colorectal cancer [9, 12, 13, 21, 38]. In this study, we have shown that physiologically attainable concentrations of aspirin can exert stronger anti-cancer effects on PIK3CA-mutant colon cancer cells relative to PIK3CA-wild-type colon cancer cells.
Our results are consistent with a recent report by Zumwalt et al [39] that clarified the relationship of PIK3CA mutations in colorectal cancer cells and aspirin-induced chemoprevention in vitro and in vivo. In that study, eight colon cancer cell lines and engineered HCT116 cells with PIK3CA kinase domain mutant allele knockout were used to assess the effects of aspirin on cell proliferation and cell-cycle distribution. Our current study further investigated 13 commonly used colon cancer cell lines and two isogenic cell lines with heterozygous knock-in of either of PIK3CA mutations c.3140A>G (p.H1047R) and c.1633G>A (p.E545K), both of which are commonly observed in colorectal carcinoma. In addition, aspirin treatment of breast cancer cells carrying mutations in PIK3CA at either exon 9 (c.1633G>A) or exon 20 (c.3140A>G) also resulted in a significant decrease of cell viability [40].
Although multiple lines of evidence indicate that aspirin use after colorectal cancer diagnosis is beneficial, the toxicities of therapy, particularly in older patients, have limited its use in routine clinical practice [14, 19, 41]. In 2016, the USPSTF gave a “B” recommendation (high certainty that the net benefit is moderate or moderate certainty that the net benefit is moderate or substantial) for routine aspirin use for colorectal cancer prophylaxis in U.S. adults between the ages of 50 and 59 with a greater than 10% 10-year risk of cardiovascular events [17]. The USPSTF recommendations also highlight the need to clarify the mechanisms by which aspirin prevents development of colorectal cancer [18]. There are currently several clinical trials underway (including ADD-ASPIRIN [42], ASCOLT [43], and ASPIRED [44]) that are attempting to clarify the association between aspirin use and survival after diagnosis of colorectal cancer, as well as distinguishing the subgroups of individuals for whom the benefits outweigh the harm. Thus, it is essential to confirm the efficacy of biomarkers used to predict benefits from aspirin therapy through multifaceted approaches, including clinical trials and in vitro or in vivo experimental models [14]. The present study serves this purpose by improving our understanding of the use of tumor PIK3CA mutation status for prediction of patient response to aspirin therapy.
PIK3CA mutations (or PIK3CA amplifications [45]) may result in constitutive activation of PI3K and the downstream AKT pathway, enhancing PTGS2 activity and prostaglandin E2 synthesis and leading to inhibition of apoptosis in colorectal cancer cells [11]. Recent in vitro and in vivo studies have suggested that aspirin may suppress cancer cell growth and induce apoptosis through activation of protein kinase A (PRKA, also referred to as AMPK), inhibition of MTOR downstream signaling, and inhibition of PI3K-induced prostaglandin E2 synthesis [46–48]. The present study was designed to detect the potency of aspirin therapy in colon cancer cell lines according to major somatic driver mutations such as PIK3CA, BRAF, and KRAS mutations. The results demonstrate that aspirin may selectively cause G0/G1 cell cycle arrest, induce apoptosis, and inhibit cell growth in human colon cancer cells with PIK3CA mutations in vitro. Our findings in the apoptotic and cell cycle assays are consistent with the data on the previous in vitro studies [39, 40, 47, 48]. The reduced viability of PIK3CA-mutant colon cancer cells was also confirmed in colon cancer cells containing the heterozygous PIK3CA knock-in mutations c.3140A>G (p.H1047R) and c.1633G>A (p.E545K). Previous retrospective analyses investigating aspirin use in colon cancers according to PIK3CA mutation status did not specify which PIK3CA mutations were included [10, 12]. The present study is the first to show that the isogenic conversion of a PIK3CA-wild-type colon cancer cell into a PIK3CA-mutant cell line is sufficient to promote sensitization to aspirin. PIK3CA c.3140A>G (p.H1047R) and c.1633G>A (p.E545K) mutations are common in colorectal carcinomas. Heterozygous knock-in c.3140A>G and c.1633G>A mutations in the PIK3CA gene were found to activate multiple oncogenic pathways and promote cell growth and invasion in vitro for human breast cancer and colon cancer cell lines [37, 49]. Notably, breast cancer cells carrying a c.3140A>G (p.H1047R) mutation at exon 20 were less sensitive to aspirin than those carrying a c.1633G>A (p.E545K) mutation at exon 9 [40]. This phenomenon may be attributed to differences in the aberrant activation of signaling pathways. However, few, if any, differences in phenotype were observed according to PIK3CA mutation variants.
The relationship between somatic or germline genetic variants and the chemopreventive use of aspirin is not limited to patients with PIK3CA-mutant colorectal cancer. Lizaka and colleagues utilized a genome-wide complementary DNA microarray containing 23,040 genes to analyze the time-dependent alteration of gene expression in response to two nonsteroidal anti-inflammatory drugs (NSAIDs), sulindac and aspirin, in SNU-C4, SW480, and SW948 colon cancer cell lines [50]. Interestingly, they found that PIK3C2A, which belongs to the PIK3 family, was down-regulated only in aspirin-sensitive cells. We previously performed a genome-wide analysis of the interactions between single nucleotide polymorphisms (SNPs) and aspirin use in relation to colon cancer risk, which identified two SNPs on chromosomes 12 and 15 that differentially correlate with aspirin and NSAID effects on colon cancer prevention [51]. One, on chromosome 12, is located upstream from PIK3C2G, which belongs to the PIK3 family. Combined, these findings further support the use of PIK3CA mutation status as a biomarker for precision aspirin chemoprevention and adjuvant therapy strategies for colon cancer.
Our current study has limitations. As our colon cancer cell panel did not include all possible activating alterations in PIK3CA, we were unable to elucidate the potency of aspirin treatment on colon cancer cells with PIK3CA alterations other than the ones examined. In addition, our current cancer cell panel was limited to immortalized colon cancer cell lines due to the lack of reasonable PIK3CA-mutant and PIK3CA-wild-type rectal cancer cell line pairs in the American Type Culture Collection (ATCC). Taking into account the potential heterogeneity of tumor behavior between colon and rectal carcinomas, anti-cancer effects of aspirin according to PIK3CA mutation status in rectal cancer cells and human primary cells should be examined in future studies. In the present study, the aspirin concentrations utilized in apoptosis and cell cycle analyses are relatively high (as high as 5 mM) compared to typical plasma concentrations of salicylate (0.19 to 0.63 mM), a major metabolite derived from aspirin. Thus, the doses may not be attainable through administration of standard doses (81 or 325 mg) of aspirin. However, the dose-dependent nature of the response to aspirin treatment suggests that some effects may be experienced in vivo even at low doses. Furthermore, Turturro et al. reported that the serum levels of aspirin could safely reach concentrations of up to 10 mM [40], indicating that the 5 mM aspirin dose is physiologically achievable in humans. Our in vitro models did not take into account the critical role of the tumor stromal microenvironment. Accumulating evidence indicates that regular aspirin use may synergize with other immunomodulatory pathways including the immune checkpoint blockade in colorectal cancer [52–56]. Further investigations are warranted to analyze interactions of multiple cell types that exist in the tumor microenvironment, and to examine the dual blockade of the PTGS2 and immunosuppressive pathways. Another drawback is that our current study did not utilize functional assays to investigate potential molecular mechanisms through which aspirin may exert stronger anti-cancer effects on PIK3CA-mutant colon cancer cells than PIK3CA-wild-type cells. Nonetheless, our study hypothesis was based on human clinical studies [8–13, 57, 58]. It is important to perform functional studies to explore potential mechanisms underlying our findings in the future. We point out an important synergism of experimental research and molecular pathological epidemiology (MPE) research [59–61] as replicating biological complexities of human tumors in experimental models is a substantial challenge. In addition, it is of particular interest to investigate modifying effects of other endogenous and exogenous factors (such as diet, lifestyle, microbiota, germline genetics, and immunity) on medications (such as aspirin) in relation to tumor molecular characteristics [4, 6, 7] in integrative pharmaco-MPE research [62, 63] in the future.
In summary, our findings provide valuable in vitro data to further support the use of aspirin in patients with PIK3CA-mutant colorectal cancer. Further investigations into the precise molecular mechanisms associated with aspirin treatment of cancers with mutant PIK3CA are warranted.
MATERIALS AND METHODS
Cell lines and culture
A panel of 13 human colon cancer cell lines (CaCo-2, COLO205, HCT15, HCT116, HT-29, LOVO, LS174T, RKO, SW48, SW480, SW620, SW948, and T84) was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The mutational gene backgrounds of the cell lines were described previously [35, 64]. We used X-MAN™ isogenic cell lines, SW48 (PIK3CA c.1633G>A (p.E545K); Catalog # HD103-001) and SW48 (PIK3CA c.3140A>G (p.H1047R); Catalog # HD103-005) heterozygous knock-in of PIK3CA activating mutations, as well as SW48 parental cells (Catalog # HD PAR-006) (Horizon Discovery, Cambridge, MA, USA). The parental cell line SW48 was used as a control. All cells were cultured in RPMI-1640 (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco) and 100 U/ml of penicillin-streptomycin in a 5% CO2 and 95% air humidified atmosphere at 37°C. All colon cancer cell lines used in this study passed the STR (Short Tandem Repeat) authentication by manufacturers.
Cell proliferation MTS assay
Cells were seeded in 96-well microplates at a density of 4x104 cells/ml and cultured overnight; they were treated with a range of aspirin (Sigma-Aldrich, St. Louis, MO, USA) doses (0, 0.5, 1, 2, 4, 6, 8, 10 and 12 mM) in dimethyl sulfoxide (DMSO). These doses are representative of physiologically attainable plasma concentrations as previously reported [40]. Control cells received DMSO only. Cell proliferation was assessed after 48 hours and 72 hours from initial drug exposure using the MTS assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Cell proliferation is directly proportional to the absorbance at 490 nm by a formazan product that is bio-reduced from MTS in living cells. All experiments were carried out in hexaplicate and were repeated at least three times independently.
Apoptosis and cell cycle analysis
The apoptosis levels and cell cycle phase of colorectal cancer cells after aspirin exposure were assessed as previously described [65, 66]. In brief, for apoptosis detection, trypsinized cells were collected gently and stained with the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) Apoptosis Detection kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s protocol. Cell cycle analysis was performed using PI staining. Cells fixed with 80% ethanol overnight at 4°C were resuspended in phosphate-buffered saline supplemented with 0.1% Triton X-100 (Sigma-Aldrich, St Louis, MO, USA), 25 mg/ml PI (BD Biosciences) and 0.2 mg/ml RNase A (Sigma-Aldrich), then incubated for 30 minutes at room temperature in the dark before analysis. Apoptosis and cell cycle phase were measured on a FACS Ariall cytometer (BD Biosciences), and analysis was performed using FlowJo software (FLOWJO, Ashland, OR, USA).
Statistical analysis
Graphpad Prism 6.0 software (Graphpad Software, San Diego, CA, USA) was used to describe dose-response curves and perform statistical analysis. All data were represented for at least three independent experiments. The Student’s t-test was used to compare continuous measurements in two groups. A P value less than 0.05 was considered statistically significant.
Abbreviations
DMSO, dimethyl sulfoxide; IC50, half maximal inhibitory concentration; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt; NSAID, nonsteroidal anti-inflammatory drug; PI, propidium iodide; PI3K, phosphatidylinositol-4,5-bisphosphonate 3-kinase; SNP, single nucleotide polymorphism; USPSTF, the U.S. Preventive Services Task Force.
Use of standardized official symbols
We use HUGO (Human Genome Organisation)-approved official symbols (or root symbols) for genes and gene products, including AKT, BRAF, CTNNB1, KRAS, MTOR, NFKB, PIK3CA, PTGS2, and WNT; all of which are described at www.genenames.org. The official symbols are italicized to differentiate from non-italicized colloquial names that are used along with the official symbols. This format enables readers to familiarize the official symbols for genes and gene products together with common colloquial names.
Author contributions
M.Gu., C.S.F., Z.R.Q., and S.O. developed the main concept and designed the study. M.Gu., A.T.C., C.S.F., Z.R.Q., and S.O. wrote grant applications. M.Gu. and Z.R.Q. performed experiments. M.Gu., C.S.F., Z.R.Q., and S.O. performed data analysis and interpretation. M.Gu. and Z.R.Q. drafted the manuscript. All authors discussed data and contributed to manuscript review and revision.
CONFLICTS OF INTEREST
A.T.C. previously served as a consultant for Bayer Healthcare, Pfizer Inc., and Aralez Pharmaceuticals. This study was not funded by Bayer Healthcare, Pfizer Inc. or Aralez Pharmaceuticals. No other conflict of interest exists. The other authors declare that they have no conflicts of interest.
FUNDING
This work was supported by U.S. National Institutes of Health (NIH) grants (P50 CA127003 to C.S.F.; R01 CA137178 to A.T.C.; K24 DK098311 to A.T.C.; R35 CA197735 to S.O.; and K07 CA190673 to R.N.); Nodal Award (to S.O.) from the Dana-Farber Harvard Cancer Center; and the Project P Fund. M.Gu. was supported by a scholarship grant from Chinese Scholarship Council, National Natural Science Foundation of China grants (81673607, 81303235) and a fellowship from Zhejiang Chinese Medical University. K.K. was supported by a grant from Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers from Japanese Society for the Promotion of Science. Z.R.Q. was supported by Medical Oncology Translational Grant Program from Department of Medical Oncology, Dana-Farber Cancer Institute and Harvard Medical School.
REFERENCES
1. Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RG, Barzi A, Jemal A. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017; 67:177–93.
2. Dienstmann R, Salazar R, Tabernero J. Personalizing colon cancer adjuvant therapy: selecting optimal treatments for individual patients. J Clin Oncol. 2015; 33:1787–96.
3. Linnekamp JF, Wang X, Medema JP, Vermeulen L. Colorectal cancer heterogeneity and targeted therapy: a case for molecular disease subtypes. Cancer Res. 2015; 75:245–9.
4. Kudryavtseva AV, Lipatova AV, Zaretsky AR, Moskalev AA, Fedorova MS, Rasskazova AS, Shibukhova GA, Snezhkina AV, Kaprin AD, Alekseev BY, Dmitriev AA, Krasnov GS. Important molecular genetic markers of colorectal cancer. Oncotarget. 2016; 7:53959–83. https://doi.org/10.18632/oncotarget.9796.
5. Patil H, Saxena SG, Barrow CJ, Kanwar JR, Kapat A, Kanwar RK. Chasing the personalized medicine dream through biomarker validation in colorectal cancer. Drug Discov Today. 2017; 22:111–19.
6. Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci. 2013; 14:16365–85.
7. Kocarnik JM, Shiovitz S, Phipps AI. Molecular phenotypes of colorectal cancer and potential clinical applications. Gastroenterol Rep (Oxf). 2015; 3:269–76.
8. Tougeron D, Sha D, Manthravadi S, Sinicrope FA. Aspirin and colorectal cancer: back to the future. Clin Cancer Res. 2014; 20:1087–94.
9. Nishihara R, Lochhead P, Kuchiba A, Jung S, Yamauchi M, Liao X, Imamura Y, Qian ZR, Morikawa T, Wang M, Spiegelman D, Cho E, Giovannucci E, et al. Aspirin use and risk of colorectal cancer according to BRAF mutation status. JAMA. 2013; 309:2563–71.
10. Li P, Wu H, Zhang H, Shi Y, Xu J, Ye Y, Xia D, Yang J, Cai J, Wu Y. Aspirin use after diagnosis but not prediagnosis improves established colorectal cancer survival: a meta-analysis. Gut. 2015; 64:1419–25.
11. Paleari L, Puntoni M, Clavarezza M, DeCensi M, Cuzick J, DeCensi A. PIK3CA Mutation, Aspirin Use after Diagnosis and Survival of Colorectal Cancer. A Systematic Review and Meta-analysis of Epidemiological Studies. Clin Oncol (R Coll Radiol). 2016; 28:317–26.
12. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, Sun R, Nosho K, Meyerhardt JA, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012; 367:1596–606.
13. Frouws MA, van Herk-Sukel MP, Maas HA, Van de Velde CJ, Portielje JE, Liefers GJ, Bastiaannet E. The mortality reducing effect of aspirin in colorectal cancer patients: Interpreting the evidence. Cancer Treat Rev. 2017; 55:120–27.
14. Drew DA, Cao Y, Chan AT. Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer. 2016; 16:173–86.
15. Rothwell PM, Fowkes FGR, Belch JFF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011; 377:31–41.
16. Rothwell PM, Wilson M, Price JF, Belch JFF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012; 379:1591–601.
17. Bibbins-Domingo K, and U.S. Preventive Services Task Force. Aspirin Use for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2016; 164:836–45.
18. Dehmer SP, Maciosek MV, Flottemesch TJ, LaFrance AB, Whitlock EP. Aspirin for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: A Decision Analysis for the U.S. Preventive Services Task Force. Ann Intern Med. 2016; 164:777–86.
19. Domingo E, Church DN, Sieber O, Ramamoorthy R, Yanagisawa Y, Johnstone E, Davidson B, Kerr DJ, Tomlinson IP, Midgley R. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J Clin Oncol. 2013; 31:4297–305.
20. Gray RT, Cantwell MM, Coleman HG, Loughrey MB, Bankhead P, McQuaid S, O’Neill RF, Arthur K, Bingham V, McGready C, Gavin AT, Cardwell CR, Johnston BT, et al. Evaluation of PTGS2 Expression, PIK3CA Mutation, Aspirin Use and Colon Cancer Survival in a Population-Based Cohort Study. Clin Transl Gastroenterol. 2017; 8:e91.
21. Murphy C, Turner N, Wong HL, Sinnathamby M, Tie J, Lee B, Desai J, Skinner I, Christie M, Hutchinson R, Lunke S, Waring P, Gibbs P, et al. Examining the impact of regular aspirin use and PIK3CA mutations on survival in stage 2 colon cancer. Intern Med J. 2017; 47:88–98.
22. Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, Liao X, Waldron L, Hoshida Y, Huttenhower C, Chan AT, Giovannucci E, Fuchs C, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut. 2012; 61:847–54.
23. Rosty C, Young JP, Walsh MD, Clendenning M, Sanderson K, Walters RJ, Parry S, Jenkins MA, Win AK, Southey MC, Hopper JL, Giles GG, Williamson EJ, et al. PIK3CA activating mutation in colorectal carcinoma: associations with molecular features and survival. PLoS One. 2013; 8:e65479.
24. Vogelaar F, Van Erning F, Reimers M, Van Der Linden J, Pruijt J, Van Den Brule A, Bosscha K. The prognostic value of Microsatellite Instability, KRAS, BRAF and PIK3CA mutations in stage II colon cancer patients. Mol Med. 2015; 17:1–26.
25. Phipps AI, Ahnen DJ, Cheng I, Newcomb PA, Win AK, Burnett T. PIK3CA Somatic Mutation Status in Relation to Patient and Tumor Factors in Racial/Ethnic Minorities with Colorectal Cancer. Cancer Epidemiol Biomarkers Prev. 2015; 24:1046–51.
26. Imamura Y, Morikawa T, Liao X, Lochhead P, Kuchiba A, Yamauchi M, Qian ZR, Nishihara R, Meyerhardt JA, Haigis KM, Fuchs CS, Ogino S. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin Cancer Res. 2012; 18:4753–63.
27. Manceau G, Marisa L, Boige V, Duval A, Gaub MP, Milano G, Selves J, Olschwang S, Jooste V, le Legrain M, Lecorre D, Guenot D, Etienne-Grimaldi MC, et al. PIK3CA mutations predict recurrence in localized microsatellite stable colon cancer. Cancer Med. 2015; 4:371–82.
28. Imamura Y, Lochhead P, Yamauchi M, Kuchiba A, Qian ZR, Liao X, Nishihara R, Jung S, Wu K, Nosho K, Wang YE, Peng S, Bass AJ, et al. Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: cohort study and literature review. Mol Cancer. 2014; 13:135.
29. Cathomas G. PIK3CA in Colorectal Cancer. Front Oncol. 2014; 4:35.
30. Leystra AA, Deming DA, Zahm CD, Farhoud M, Olson TJ, Hadac JN, Nettekoven LA, Albrecht DM, Clipson L, Sullivan R, Washington MK, Torrealba JR, Weichert JP, et al. Mice expressing activated PI3K rapidly develop advanced colon cancer. Cancer Res. 2012; 72:2931–6.
31. Samuels Y, Diaz LA Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005; 7:561–73.
32. Papadatos-Pastos D, Rabbie R, Ross P, Sarker D. The role of the PI3K pathway in colorectal cancer. Crit Rev Oncol Hematol. 2015; 94:18–30.
33. Wang DZ, Wang HB, Shi Q, Katkuri S, Walhi W, Desvergne B, Das SK, Dey SK, DuBois RN. Prostaglandin E-2 promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004; 6:285–95.
34. Guo XN, Rajput A, Rose R, Hauser J, Beko A, Kuropatwinski K, Levea C, Hoffman RM, Brattain NG, Wang J. Mutant PIK3CA-bearing colon cancer cells display increased metastasis in an orthotopic model. Cancer Res. 2007; 67:5851–58.
35. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–7.
36. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501.
37. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304:554.
38. Mei ZB, Duan CY, Li CB, Cui L, Ogino S. Prognostic role of tumor PIK3CA mutation in colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2016; 27:1836–48.
39. Zumwalt TJ, Wodarz D, Komarova NL, Toden S, Turner J, Cardenas J, Burn J, Chan AT, Boland CR, Goel A. Aspirin-Induced Chemoprevention and Response Kinetics Are Enhanced by PIK3CA Mutations in Colorectal Cancer Cells. Cancer Prev Res (Phila). 2017; 10:208–18.
40. Turturro SB, Najor MS, Ruby CE, Cobleigh MA, Abukhdeir AM. Mutations in PIK3CA sensitize breast cancer cells to physiologic levels of aspirin. Breast Cancer Res Treat. 2016; 156:33–43.
41. Chan AT, Ladabaum U. Where Do We Stand With Aspirin for the Prevention of Colorectal Cancer? The USPSTF Recommendations. Gastroenterology. 2016; 150:14–8.
42. Coyle C, Cafferty FH, Rowley S, MacKenzie M, Berkman L, Gupta S, Pramesh CS, Gilbert D, Kynaston H, Cameron D, Wilson RH, Ring A, Langley RE, and Add-Aspirin investigators. A phase III, double-blind, placebo controlled, randomised trial assessing the effects of aspirin on disease recurrence and survival after primary therapy in common non-metastatic solid tumours. Contemp Clin Trials. 2016; 51:56–64.
43. Ali R, Toh HC, Chia WK, and ASCOLT Trial Investigators. The utility of Aspirin in Dukes C and High Risk Dukes B Colorectal cancer--the ASCOLT study: study protocol for a randomized controlled trial. Trials. 2011; 12:261.
44. Drew DA, Chin SM, Gilpin KK, Parziale M, Pond E, Schuck MM, Stewart K, Flagg M, Rawlings CA, Backman V, Carolan PJ, Chung DC, Colizzo FP 3rd, et al. ASPirin Intervention for the REDuction of colorectal cancer risk (ASPIRED): a study protocol for a randomized controlled trial. Trials. 2017; 18:50.
45. Jehan Z, Bavi P, Sultana M, Abubaker J, Bu R, Hussain A, Alsbeih G, Al-Sanea N, Abduljabbar A, Ashari LH, Alhomoud S, Al-Dayel F, Uddin S, Al-Kuraya KS. Frequent PIK3CA gene amplification and its clinical significance in colorectal cancer. J Pathol. 2009; 219:337–46.
46. Claudius AK, Kankipati CS, Kilari RS, Hassan S, Guest K, Russell ST, Perry CJ, Stark LA, Nicholl ID. Identification of aspirin analogues that repress NF-kappaB signalling and demonstrate anti-proliferative activity towards colorectal cancer in vitro and in vivo. Oncol Rep. 2014; 32:1670–80.
47. Din FV, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K, Alessi DR, Dunlop MG. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012; 142:1504–15.
48. Jung YR, Kim EJ, Choi HJ, Park JJ, Kim HS, Lee YJ, Park MJ, Lee M. Aspirin Targets SIRT1 and AMPK to Induce Senescence of Colorectal Carcinoma Cells. Mol Pharmacol. 2015; 88:708–19.
49. Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005; 65:10992–1000.
50. Iizaka M, Furukawa Y, Tsunoda T, Akashi H, Ogawa M, Nakamura Y. Expression profile analysis of colon cancer cells in response to sulindac or aspirin. Biochem Biophys Res Commun. 2002; 292:498–512.
51. Nan H, Hutter CM, Lin Y, Jacobs EJ, Ulrich CM, White E, Baron JA, Berndt SI, Brenner H, Butterbach K, Caan BJ, Campbell PT, Carlson CS, et al, and CCFR, and GECCO. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA. 2015; 313:1133–42.
52. Oda K, Okada J, Timmerman L, Rodriguez-Viciana P, Stokoe D, Shoji K, Taketani Y, Kuramoto H, Knight ZA, Shokat KM, McCormick F. PIK3CA cooperates with other phosphatidylinositol 3’-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 2008; 68:8127–36.
53. Ma X, Aoki T, Tsuruyama T, Narumiya S. Definition of Prostaglandin E2-EP2 Signals in the Colon Tumor Microenvironment That Amplify Inflammation and Tumor Growth. Cancer Res. 2015; 75:2822–32.
54. Hamada T, Cao Y, Qian ZR, Masugi Y, Nowak JA, Yang J, Song M, Mima K, Kosumi K, Liu L, Shi Y, da Silva A, Gu M, et al. Aspirin Use and Colorectal Cancer Survival According to Tumor CD274 (Programmed Cell Death 1 Ligand 1) Expression Status. J Clin Oncol. 2017; 35:1836–44.
55. Cao Y, Nishihara R, Qian ZR, Song M, Mima K, Inamura K, Nowak JA, Drew DA, Lochhead P, Nosho K, Morikawa T, Zhang X, Wu K, et al. Regular Aspirin Use Associates With Lower Risk of Colorectal Cancers With Low Numbers of Tumor-Infiltrating Lymphocytes. Gastroenterology. 2016; 151:879–92.e4.
56. Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, Sahai E, Reis e Sousa C. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015; 162:1257–70.
57. Reimers MS, Zeestraten EC, Kuppen PJ, Liefers GJ, van de Velde CJ. Biomarkers in precision therapy in colorectal cancer. Gastroenterol Rep (Oxf). 2013; 1:166–83.
58. Reimers MS, Bastiaannet E, Langley RE, van Eijk R, van Vlierberghe RL, Lemmens VE, van Herk-Sukel MP, van Wezel T, Fodde R, Kuppen PJ, Morreau H, van de Velde CJ, Liefers GJ. Expression of HLA class I antigen, aspirin use, and survival after a diagnosis of colon cancer. JAMA Intern Med. 2014; 174:732–9.
59. Ogino S, Chan AT, Fuchs CS, Giovannucci E. Molecular pathological epidemiology of colorectal neoplasia: an emerging transdisciplinary and interdisciplinary field. Gut. 2011; 60:397–411.
60. Ogino S, Nishihara R, VanderWeele TJ, Wang M, Nishi A, Lochhead P, Qian ZR, Zhang X, Wu K, Nan H, Yoshida K, Milner DA Jr, Chan AT, et al. Review Article: The Role of Molecular Pathological Epidemiology in the Study of Neoplastic and Non-neoplastic Diseases in the Era of Precision Medicine. Epidemiology. 2016; 27:602–11.
61. Rescigno T, Micolucci L, Tecce MF, Capasso A. Bioactive Nutrients and Nutrigenomics in Age-Related Diseases. Molecules. 2017; 22:e105.
62. Nishihara R, VanderWeele TJ, Shibuya K, Mittleman MA, Wang M, Field AE, Giovannucci E, Lochhead P, Ogino S. Molecular pathological epidemiology gives clues to paradoxical findings. Eur J Epidemiol. 2015; 30:1129–35.
63. Hamada T, Keum N, Nishihara R, Ogino S. Molecular pathological epidemiology: new developing frontiers of big data science to study etiologies and pathogenesis. J Gastroenterol. 2017; 52:265–75.
64. Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknaes M, Hektoen M, Lind GE, Lothe RA. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013; 2:e71.
65. Yue W, Zheng X, Lin Y, Yang CS, Xu Q, Carpizo D, Huang H, DiPaola RS, Tan XL. Metformin combined with aspirin significantly inhibit pancreatic cancer cell growth in vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and Bcl-2. Oncotarget. 2015; 6:21208–24. https://doi.org/10.18632/oncotarget.4126.
66. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, Perez-Atayde A, Wong KK, Yuan GC, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014; 159:1126–39.