
Introduction
Enterohemorrhagic Escherichia coli (EHEC) belongs to the family of Shiga toxins (Stxs)-producing E. coli, many of whose members carry the eae gene and can cause severe disease [1]. As A-B-type toxins, Stxs are considered to be major virulence factors in the pathogenesis of haemolytic uremic syndrome and hemorrhagic colitis [2]. Similar to enteropathogenic E. coli (EPEC), EHEC causes attaching and effacing lesions, which represent another major virulence factor in colonization of the EHEC in the colon [3]. Enterohemorrhagic Escherichia coli (EHEC) was first isolated in 1977 by Konowalchuk et al [4]. In 1982, the bacterium first caused an outbreak of hemorrhagic enteritis in North America and then was named EHEC [5]. In Japan, the EHEC infection broke out in 1996. More than 9000 people were infected, resulting in 11 deaths. The event has attracted worldwide attention [6]. In United States, an outbreak of EHEC infection occurred in 2006. It was found that the EHEC contaminated spinach. 117 people were eventually infected and 3 died [7]. The outbreak of EHEC in developed countries such as the United States, Japan, Canada and the United Kingdom showed a trend of increasing year by year.
The most significant sources of EHEC infection in humans are food and water [8-10], the primary source of EHEC infection is the cattle in ruminants [11]. With the increased use of rabbits as a food source and in research, as well as their increased activity in large wildlife populations, the risk and likelihood of interspecies pathogen transmissions have also risen [12]. For one, EHEC transmission can occur between cattle and wild rabbits [13, 14]. Rabbits are a new reservoir host of EHEC that may pose a zoonotic risk for humans [15]. In response to the heightened risk, several animal models of EHEC-induced disease have been developed. When the newly emergent pathogenic E. coli was first studied more than 30 years ago, experiments involved EHEC O157:H7 using piglets [16, 17], and the oral administration of EHEC caused diarrhea as well as attaching and effacing lesions in the ileum, cecum, and colon of rabbits [18]. In the use of rabbits infected with rabbit-origin E. coli as experimental models of human disease, rabbits orally inoculated with EPEC strain RDEC-1 emerged as a good model for human EPEC infection [19], which simulates the development of infectious gastroenteritis in children [20]. Other studies have shown that natural and experimental infection with EHEC strains caused hemorrhagic diarrhea, cecal colitis, nephropathy, and hemolytic uremic syndrome in Dutch belted rabbits [1, 15, 21].
Stxs can induce classical mitogen-activated protein kinases and IL-8 when co-administered with TNF-α [22] and flagellin [23]. During the innate immune response process, pro-inflammatory cytokines induce immune cells differentiated from white blood cells that migrate to inflammatory sites, thereby eliminating pathogenic microorganisms. In turn, immune cells secrete pro-inflammatory cytokines such as IL-1β, IL-2, IL-6, IL-8, IFN-γ, and TNF-α. Different types of white blood cells, including natural killer cells and mononuclear macrophages, secrete cytokines and defensins via MyD88- and NF-κB-mediated signal pathways [24, 25] that promote lymphocyte proliferation and differentiation, as well as participate in the development of inflammation.
Antimicrobial peptides not only directly inhibit various microorganisms, but also can recruit immature dendritic cells and macrophages to reach the skin and mucosal tissues of microbial invasion through chemokine receptors, thus inducing specific immunity of pathogenic bacteria. Since their production is more than 100 times faster than that of IgM, they are also the first line of host defense [26]. Members of a large, diverse family of cationic antimicrobial peptides, defensins are strongly induced by inflammation or tissue damage. For example, the expression of human β-defensin-2 is rapidly induced in response to infection by intestinal pathogenic bacteria, and human β-defensin-3 is strongly upregulated in inflammatory diseases such as Crohn’s disease [27, 28]. Along with the direct killing of microorganisms, antimicrobial peptides can recruit and promote other elements of host immunity, particularly innate immunity [29, 30]. In other research, drosophila has responded to infection by synthesizing antimicrobial peptides (e.g., defensins) that, secreted into the hemolymph, exhibit antibacterial activity [24].
Although diseases in rabbits caused by EHEC have been reported [1, 15], research on the pathogenicity of EHEC and the role of innate immunity in disease processes remains scarce. What is the innate immune mechanism of rabbit resistance to EHEC infection? To investigate the inflammatory and anti-bacterial response, our research detected the major inflammatory cytokines and defensins. The purpose of this study is to systematically describe clinical symptoms and the histopathological analysis of E. coli content in tissues and innate immune responses in EHEC-infected rabbits.
Results
Isolation of EHEC
The bacterial colony of E. coli was bluish green with a metallic luster on the eosin-methylene blue medium. As shown in Table 2, the eae, Stx1, Stx2, and hlyA genes were all amplified from the MY strain; however, in the TS strain, the Stx2 gene was absent (Table 2). In addition, after 4 h of incubation, the number of E. coli MY rapidly reached 1.46 × 108 CFU/mL, while that of TS was 5.99 × 107 CFU/mL (p < 0.05). After 6 h of incubation, the number of E. coli MY and TS reached 1.96 × 109 CFU/mL and 2.08 × 109 CFU/mL, respectively. During the stable phase of bacterial growth, the highest number of E. coli TS reached 6.46 × 109 CFU/mL, which was higher than that of MY 2.29 × 109 CFU/mL at 14 h (p < 0.05). MY strain grew at a faster rate than the TS stain during the logarithmic growth phase, although its greatest concentration was lower than that of the TS stain (Figure 1).
Table 1: Primers used in this study.
Primer name |
Sequence(5’-3’) |
Purpose |
eae F |
tgcggcacaacaggcggcga |
Gene cloning |
eae R |
cggtcgccgcaccaggattc |
|
Stx1B F |
cccggatccatgaaaaaaacattattaatagc |
Gene cloning |
Stx1B R |
cccgaattcagctattctgagtcaacg |
|
Stx2A and variants F |
atcctattcccgggagtttacg |
Gene cloning |
Stx2A and variants R |
gcgtcatcgtatacacaggagc |
|
hlyA F |
ggtgcagcagaaaaagttgtag |
Gene cloning |
hlyA R |
tctcgcctgatagtgtttggta |
|
IL-1β F |
tggcacgtatgagctgaaag |
RT-PCR |
IL-1β R |
ggccacaggtatcttgtcgt |
|
IL-4 F |
cactccggcagttctacctc |
RT-PCR |
IL-4 R |
gcagaggttcctgtcgagtc |
|
IL-6 F |
ctgaagacgaccacgatcca |
RT-PCR |
IL-6 R |
aaggacacccgcactccat |
|
IL-8 F |
ctctcttggcaaccttcctg |
RT-PCR |
IL-8 R |
ttgcacagtgaggtccactc |
|
IL-10 F |
aaaagctaaaagccccagga |
RT-PCR |
IL-10 R |
cgggagctgaggtatcagag |
|
IFN-γ F |
ctcgaatttcggtggatgat |
RT-PCR |
IFN-γ R |
agcgtctgactcctttttcg |
|
TNF-α F |
cacttcagggtgatcggc |
RT-PCR |
TNF-α R |
tgcgggtttgctactacg |
|
DEFB114 F |
taccagccacatgctctttg |
RT-PCR |
DEFB114 R |
cctgtcgacacagcaaatct |
|
DEFB124 F |
gcaccaagcaagagtccttc |
RT-PCR |
DEFB124 R |
acgccagagccagctactta |
|
DEFB125 F |
cgtgctgcatctccttaaca |
RT-PCR |
DEFB125 R |
gcgaagcagaaaattgatcc |
|
DEFB127 F |
cccacagtaaccgagcaact |
RT-PCR |
DEFB127 R |
gctgaggcagcagtatctcc |
|
DEFB128 F |
gggctcaaggctttctcttt |
RT-PCR |
DEFB128 R |
aaatctcgcctagcttgcac |
|
DEFB134 F |
agcctgtctgcctggagtag |
RT-PCR |
DEFB134 R |
gatgaggagaggcttcatgg |
|
DEFB135 F |
gctgcatctccaaatccaat |
RT-PCR |
DEFB155 R |
tagtgggatggtgcaactga |
|
NP 5 F |
aggcaggcgtgttctgtact |
RT-PCR |
NP 5 R |
ggtctccacgcaaataagga |
|
GAPDH F |
aggtcatccacgaccacttc |
RT-PCR |
GAPDH R |
gtgagtttcccgttcagctc |
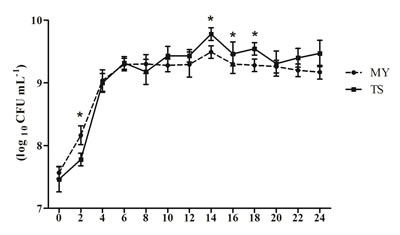
Figure 1: Growth curve of E. coli TS and MY strains. After inoculation bacteria with 1% (v/v) seed liquid of the E. coli TS or MY, E. coli content was calculated every 2h. The data shows means ± SDs of three independent experiments. Significant differences are indicated with an *.
Survival rate and E. coli content
The incidence of diarrhea in rabbits infected with the MY strain was 80%, while that of rabbits infected with TS strain was 66.7%. When rabbits were infected with the MY strain, the number of deaths peaked at 1 d postinfection (dpi) dropped at 2 and 3 dpi, after which none of the rabbits died. By contrast, among rabbits infected with TS strain, the number of deaths peaked at 2 dpi (27.8%), and none of the rabbits died after 4 dpi. Altogether, the mortality of rabbits infected with the MY strain was 77.8%, which was greater than that of rabbits infected with TS strain (55.6%) (p < 0.05, Figure 2A).
Table 2: Amplification of virulence factors.
Strain |
Eae |
Stx1 |
Stx2 |
HlyA |
TS |
+ |
+ |
- |
+ |
MY |
+ |
+ |
+ |
+ |
E. coli content in the heart, liver, spleen, brain, and cecal contents was tested at 1 and 3 dpi. At 1 dpi, E. coli rapidly replicated in the tested tissues with elevated content, and E. coli content in the heart, liver, and brain infected with the TS strain were greater than those infected with the MY strain (Figure 2B). Especially, E. coli content in brain infected with the TS strain was 4.09 × 107 CFU/mL, significantly higher than that of MY strain (2.03 × 106 CFU/mL) at 1 dpi (p < 0.05, Figure 2B). At 3 dpi, the amount of E. coli in tested tissues had declined since 1 dpi. E. coli content in heart, brain, and cecal contents infected with the TS strain was greater than those infected with the MY strain at 3 dpi (p < 0.05, Figure 2C). However, after MY strain infection, the number of bacteria in the spleen was 1.89 × 106 CFU/mL, higher than that of infected TS strain (2.60 × 105 CFU/mL) at 3 dpi (p < 0.05, Figure 2C). Taken together, both E. coli could cause systemic infection in rabbits and spread to multiple organs, although the MY strain can do so more efficiently than the TS strain.
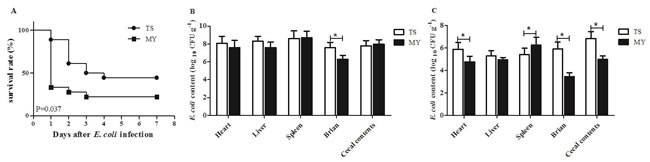
Figure 2: Rabbits were inoculated intraperitoneally with 1.0 mL of E. coli TS and MY bacterial suspension (108 CFU/mL). A. The survival rate of rabbits after infection with the TS and MY strains (n = 10); B. E. coli content of infected rabbits at 1 dpi (log10 CFU/g); and C. E. coli content of infected rabbits at 3 dpi (log10 CFU/g). Bars represented the means ± SDs of three independent experiments (five rabbits per experiment). Significant differences are indicated with an *.
Clinical symptoms and gross lesions
At 1 dpi, rabbits infected with either the MY or TS strain showed typical clinical symptoms, including mental depression, anorexia, and inactivity, as well as diarrhea (Figure 3A and 3B). At 3 dpi, they also showed typical gross lesions, including fibrinous pericarditis (Figure 3D and 3E), endocardial hemorrhage (arrow in Figure 3G and 3H), and liver and abdominal cavity surfaces with white cellulose attachments (Figure 3M and 3N). The Livers exhibited hemorrhage (arrow in Figure 3J and 3K), and the spleens were enlarged with fibrinous exudate and severe hemorrhage (Figure 3P and 3Q). No clinical symptoms were observed in the control group (Figure 3C, 3F, 3I, 3L, and 3O).
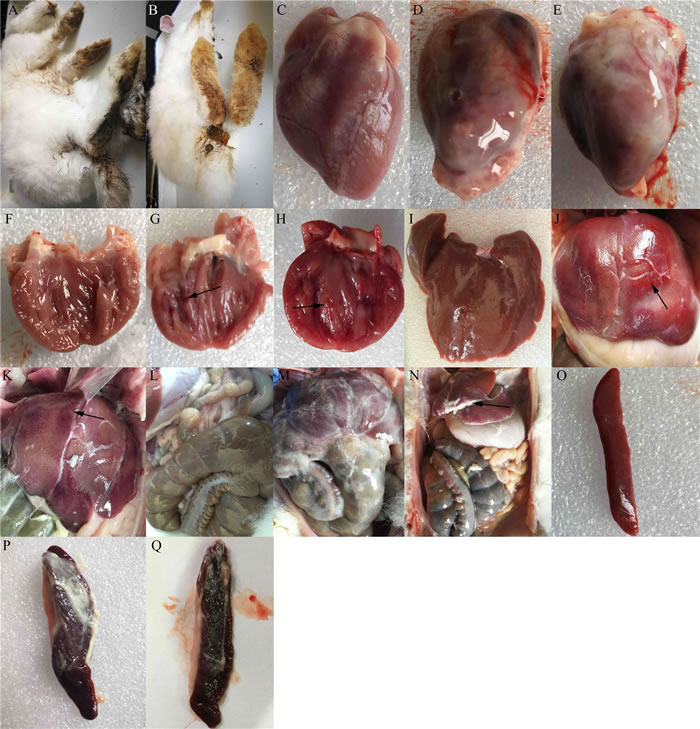
Figure 3: Clinical symptoms and gross lesions of rabbits after infection with enterohemorrhagic Escherichia coli. Rabbits infected with the TS strain showed A. diarrhea, D. fibrinous pericarditis, G. endocardial hemorrhage, J. spotted hemorrhage of liver, M. abdominal cavity surface with white cellulose attachment, and P. spleen with cellulose attachment, splenomegaly, and hemorrhage. Rabbits infected with the MY strain showed B. diarrhea, E. fibrinous pericarditis, H. endocardial hemorrhage, F. spotted hemorrhage of the liver, N. a slight fibrinous exudate in the abdominal cavity, and Q. spleen with cellulose attachment, splenomegaly, and hemorrhage. Images C, F, I, L, and O show the epicardium, endocardium, liver, abdominal cavity, and spleen of the control group, respectively.
Histopathological analysis
At 1 and 3 dpi, pathological changes occurred in various tissues in infected rabbits. Lesions in the heart, liver, and colon increased with time. Pathological changes in rabbits infected with the MY strain were more serious than those infected with the TS strain. Slight myocardial hemorrhage appeared in rabbits infected with the TS strain at 1 dpi (Figure 4B), and had grown severe by 3 dpi (Figure 4D). After being infected with the MY strain, rabbits clearly showed myocardial necrosis, rupture, and hemorrhage (Figure 4C and 4E). Hepatocyte hemorrhage was observed in rabbits infected with the TS strain (Figure 4G), whereas hepatocyte necrosis was also observed in rabbits infected with the MY strain at 1 dpi (Figure 4H). At 3 dpi, vacuolar degeneration, necrosis, and hemorrhage were significant in the livers of rabbits in both experimental groups (Figure 4I and 4J). At 1 dpi, the spleens of rabbits infected with TS strain exhibited massive lymphocyte loss with diffuse hemorrhaging (Figure 4L), but the reduction of lymphocytes with hemorrhage had decreased significantly by 3 dpi (Figure 4N). Similarly, infected with the MY strain, rabbits showed massive lymphocyte loss with severe hemorrhage at 1 dpi (Figure 4M), which had decreased by 3 dpi (Figure 4O). Intestinal villus rupture was observed in rabbits infected with the TS strain (Figure 4Q), whereas goblet cell enlargement and muco-enteritis were salient in rabbits infected with the MY strain at 1 dpi (Figure 4R). At 3 dpi, intestinal villus shedding and structural destruction had occurred (Figure 4S and 4T). By total contrast, the control group showed no obvious pathological change whatsoever (Figure 4A, 4F, 4K, and 4P).
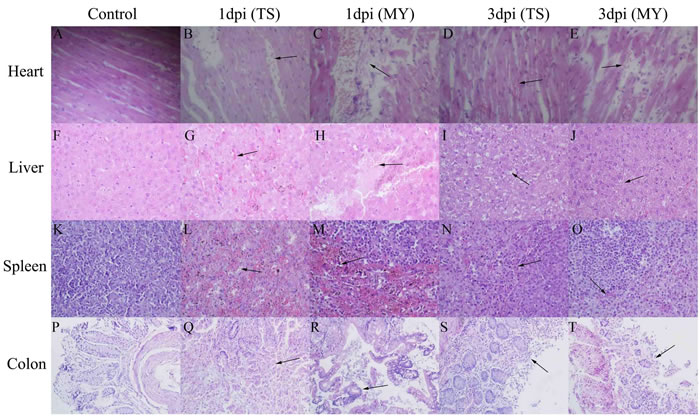
Figure 4: Pathological changes of the enterohemorrhagic Escherichia coli-infected rabbits at 1 and 3 d postinfection. Image B. shows slight myocardial hemorrhage at 1 dpi, C. myocardial necrosis and hemorrhage at 1 dpi, D. myocardium hemorrhage at 3 dpi, E. hemorrhage, rupture, and necrosis of the myocardium at 3 dpi, G. hepatocyte hemorrhage at 1 dpi, H. hepatocyte necrosis with hemorrhage at 1 dpi, I. vacuolar degeneration, necrosis, and slight hemorrhage of hepatocytes at 3 dpi, J. vacuolar degeneration, necrosis, and hemorrhage of hepatocytes at 3 dpi, L. massive lymphocyte loss with diffuse hemorrhage at 1 dpi, M. massive lymphocyte loss with severe hemorrhage at 1 dpi, N. lymphocyte disappearance with hemorrhage at 3 dpi, O. a slight reduction of lymphocytes and hemorrhage at 3 dpi, Q. intestinal villus structure destruction at 1 dpi, R. intestinal villus fracture and goblet cell enlargement at 1 dpi, S. intestinal villus shedding at 3 dpi, and T. intestinal villus shedding at 3 dpi. Images A, F, K, and P show the heart, liver, spleen, and colon of rabbits from the control group, respectively; magnification of the heart, liver, and spleen tissues was 400×, whereas colon tissue was magnified 200×.
Expression of innate immune-related genes in the spleens of infected rabbits
To investigate the induction of innate immune response in rabbits infected with E. coli, the expression of genes involved in such response was examined in the spleen at 1 and 3 dpi. Expressions of pro-inflammatory cytokines IL-1β and IL-8 displayed no significant difference at 1 dpi, yet had significantly upregulated by 3 dpi. For example, at 3 dpi, the expression of IL-1β was upregulated in rabbits infected with the MY and TS strains by 32.21- and 10.22-fold, respectively (p < 0.05, Figure 5A), whereas IL-8 was upregulated by 3.94- and 21.07-fold in each respective experimental group (p < 0.05, Figure 5D). At 1 dpi, the expression of IL-6 in rabbits infected with the MY and TS strains was also significantly upregulated by 20.06- and 56.80-fold, respectively (p < 0.05), yet decreased at 3 dpi by 5.72- and 12.24-fold, also respectively (p < 0.05, Figure 5C). By contrast, anti-inflammatory cytokines IL-4 and IL-10 were downregulated at 1 dpi, but upregulated at 3 dpi by 38.86-fold (p < 0.05) and 4.86-fold (p < 0.05) in rabbits infected with the MY and TS strains, respectively, to a greater extent in the former group than in the latter (Figure 5B and 5E). The expression of IFN-γ was significantly upregulated by 381.83- and 560.38-fold (p < 0.05, Figure 5F) in rabbits infected with the MY and TS strains, respectively, whereas at 3 dpi TNF-α showed increases of only 10.81- and 19.62-fold (p < 0.05, Figure 5G) in each respective group.
At 1 dpi, the expression of α-defensin (i.e., NP5) and β-defensin (i.e., DEFB114, DEFB124, DEFB125, DEFB127, DEFB128, DEFB134, and DEFB135) displayed no significant changes in rabbits infected with either the MY or TS strain. However, at 3 dpi, those genes showed greater expression in rabbits infected with the MY strain than those in the control group and those infected with the TS strain. At 3 dpi, NP5, DEFB 114, DEFB124, and DEFB128 showed a relatively low expression (Figure 5H, 5I, 5J, and 5M), whereas DEF125, DEFB127, DEFB134, and DEFB135 displayed a change of 3.26-23.62-fold (Figure 5K, 5L, 5N, and 5O). At 3 dpi, the expression of DEFB127 and DEFB134 was especially upregulated, by 491.24- and 382.11-fold, in rabbits infected with the MY strain (p < 0.05; Figure 5L and 5O).
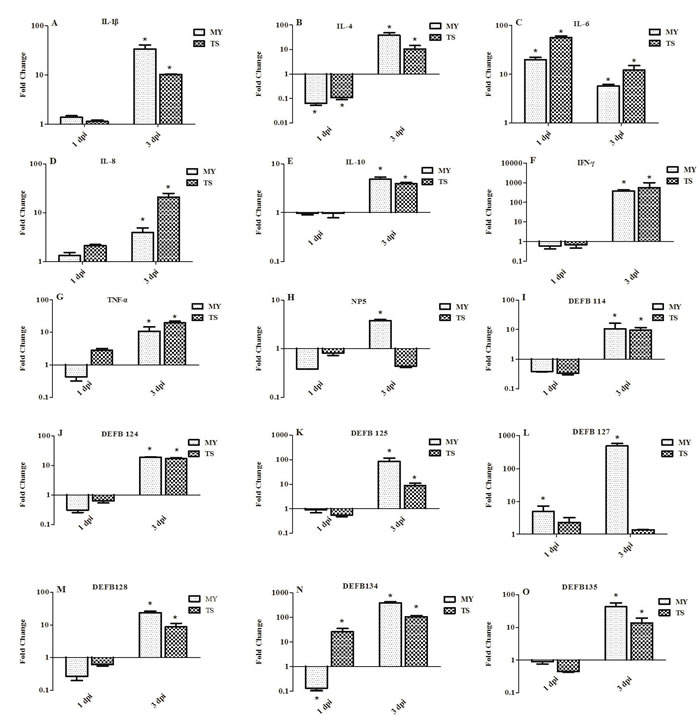
Figure 5: Expression profiles of immune-related genes in the spleen of rabbits at 1 and 3 d postinfection. Image A. shows IL-1β, B. IL-4, C. IL-6, D. IL-8, E. IL-10, F. IFN-γ, G. TNF-α, H. NP5, I. DEFB114, J. DEFB124, K. DEFB125, L. DEFB127, M. DEFB128, N. DEFB134, and O. DEFB135. Fold change was calculated from the gene expression of two groups of infected rabbits compared with that of control group. Bars represented the means ± SDs of three independent experiments (five rabbits per experiment); significant differences are indicated with an *.
Expression of innate immune-related genes in the livers of infected rabbits
In the livers of rabbits in both groups at 1 dpi, the expression of IL-1β and IL-6 showed elevated expression (Figure 6A and 6C). At 3 dpi, the expression of IL-1β was significantly upregulated, by 54.29- and 37.20-fold, in rabbits infected with the MY and TS strains, respectively (Figure 6A). The expressions of IL-4 and IL-10 in rabbits infected with the MY strain were greater than in rabbits infected with the TS strain (Figure 6B and 6E); the expression of IL-4 was particularly upregulated, by 29.69- and 55.11-fold, in rabbits infected with the MY strain at 1 and 3 dpi, respectively (p < 0.05, Figure 6B). IL-8 showed slight variation at 1 dpi, but was significantly upregulated, by 83.12- and 165.82-fold, in rabbits infected with the MY and TS strains, respectively, at 3 dpi (p < 0.05, Figure 6D). Furthermore, at 3 dpi, the expression of IFN-γ was upregulated by 179.43- and 272.5-fold in rabbits infected with the MY and TS strains, respectively (p < 0.05, Figure 6F). However, at 1 dpi, the expression of TNF-α was upregulated by only 7.34-fold (p < 0.05) in rabbits infected with the TS strain, and its expression showed no significant changes in that group of rabbits at 3 dpi (Figure 6G).
Both α- and β-defensin showed greater expressions in rabbits infected with the TS strain than the MY strain, except regarding DEFB135 (Figure 6O). At 1 and 3 dpi, the expression of NP5 was significantly upregulated, by 17.89- and 17.52-fold, respectively, in rabbits infected with the TS strain (p < 0.05, Figure 6H). The expression of DEFB114, DEFB127, and DEFB128 were downregulated at 1 dpi and upregulated at 3 dpi (Figure 6I, 6L, 6M). However, at 3 dpi, the expression of DEFB128 was significantly upregulated, by 321.28-fold (p < 0.05), in rabbits infected with the TS strain and to a far greater extent than that of other defensins (Figure 6M). At 3 dpi, the expression of DEFB124, DEFB125, and DEFB134 reached 68.16-, 45.61-, and 23.57-fold in rabbits infected with the TS strain (p < 0.05, Figure 6J, 6K, 6N).
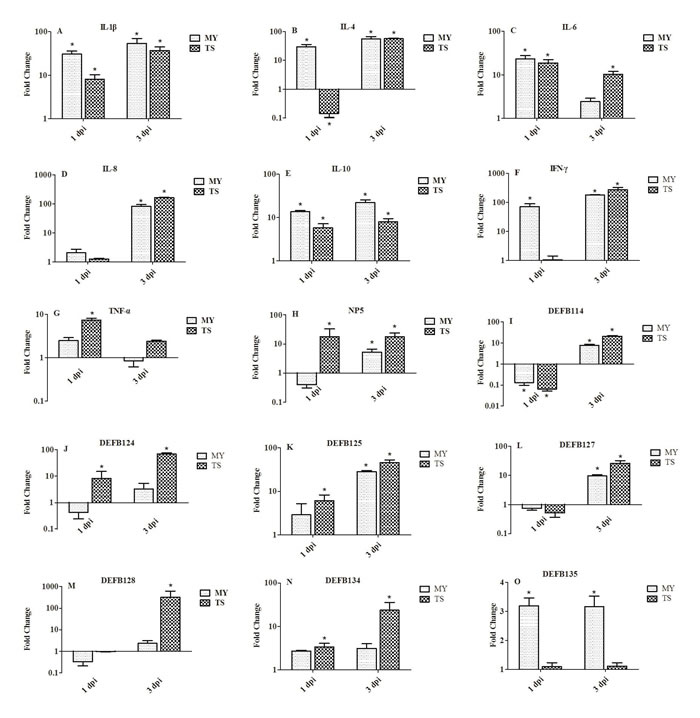
Figure 6: Expression profiles of immune-related genes in the liver of rabbits at 1 and 3 d postinfection. Image A. shows IL-1β, B. IL-4, C. IL-6, D. IL-8, E. IL-10, F. IFN-γ, G. TNF-α, H. NP5, I. DEFB114, J. DEFB124, K. DEFB125, L. DEFB127, M. DEFB128, N. DEFB134, and O. DEFB135. Fold change was calculated from the gene expression of two groups of infected rabbits compared with that of control group. Bars represented the means ± SDs of three independent experiments (five rabbits per experiment); significant differences are indicated with an *.
Discussion
Increasingly used as a food source, as experimental animals, and as household pets, rabbits nevertheless suffer from diarrhea and even rapid death due to EHEC infection during breeding. In response, clinical symptoms, histopathological aspects, E. coli content in tissues, and host innate immune in rabbits were systematically examined in the present study.
MY and TS strains carry different toxin genes, MY strain carries Stx2 and TS strain does not. Therefore, MY and TS strains might have different ability to produce toxin. The survival rate of rabbits infected with the MY strain was lower than those infected with the TS strain. The clinical pathology of diarrhea and sudden death of rabbits was consistent with findings from previous research [31]. Pathological changes in rabbits infected with the MY strain were more serious than those infected with the TS strain at 1dpi and 3dpi. Although the pathogenicity of the MY strain was stronger than that of the TS, E. coli content in tissues infected with the TS strain was greater than those infected with the MY strain, except for the spleen. The possible reason is that MY strain grew at a faster rate than the TS strain during the logarithmic growth phase in vitro. MY strain can damage the tissues and organs of the host more quickly. The amount of E. coli had decreased by 3 dpi, and the decreased level was higher in rabbits infected with the MY strain. This could explain why rabbits infected with the MY strain no longer died after 3 dpi. Bacteria in the heart, liver, spleen, and cecal contents of both E. coli-infected rabbits were capable of reaching 108 CFU/g and thus causing extensive clinical symptoms and histopathological lesions, thereby indicating that EHEC had broad tissue tropism, especially in the spleen and liver. Moreover, the E. coli content in spleen which is the main immune organ infected with the MY strain was greater than that infected with the TS strain. We hypothesized that the MY strain would cause stronger immunosuppression.
Researches have suggested that the innate immune system is fixed in the genome, and can rapidly mobilize and identify pathogenic microorganisms that induce conserved molecular patterns [32, 33]. When stimulated with pathogenic microorganisms, innate immune responses are implemented and ready to activate natural antibodies and the alternative complement system. Innate immunity relates closely to antimicrobial effector functions provided by local inflammation [34]. In response, the expression of immune-related genes was explored in the spleen and liver of rabbits infected with EHEC in the present study.
Pro-inflammatory cytokines IL-1β and IL-6 cause inflammatory events in order to eliminate pathogenic microorganisms. Previous research has indicated that Stxs can induce the production of IL-8 in HCT-8 cells, since Stxs-mediated ERK1/2 activation is necessary for Stx1-mediated IL-8 expression and can promote the systemic absorption of Stxs, thereby promoting systemic disease [35, 36]. Results of the present study revealed the significant upregulation of IL-8 expression, by 83.12- and 165.82-fold, in the liver during infection with the MY and TS strains, respectively, possibly due to the stimulation of Stxs. Produced following lipopolysaccharide stimulation, TNF-α induces IL-1β, IL-6, and IL-8 secretion. IL-1β at once enhances the expression of IL-6, IL-8, and TNF-α [37]. The results of the present study showed that IL-1β, IL-6, and TNF-α were all significantly induced by MY and TS strains in the spleen at 3 dpi, thereby indicating the activation of the host innate immune response. IL-1β, IL-6, IL-8, and TNF-α promote with each other to form a network of interactions that maintain an appropriate immune response and prevent excessive host immune defense.
In various defense mechanisms, antimicrobial peptides are important evolutionarily conserved components of innate immunity that exist widely in nearly all species, including bacteria, fungi, insects, tunicates, amphibians, crustaceans, birds, fish, and mammals, including humans. Roughly three decades ago, antimicrobial peptides were initially isolated from the lymph of insects, skin of frogs, and granules of mammalian neutrophils and demonstrated the ability to kill bacteria in vitro [38]. Our results showed that the α- and β-defensin showed no obvious change induced by MY and TS strains at 1 dpi. It is possible that some pathogenic factors of bacteria may have prevented the expression and activation of defensins. However, the expressions of α- and β-defensin were significantly upregulated, and bacteria in tissues and deaths of rabbits had both declined by 3 dpi. Moreover, the pathological changes in spleen were alleviated at 3 dpi. That phenomenon is consistent with the antibacterial ability of defensins, which inhibit the further reproduction of bacteria in the host. Notably, the expression of α- and β-defensin induced by the MY strain was greater than that induced by the TS strain in the spleen. However, the expression of α- and β-defensin induced by the TS strain was greater than that induced by the MY strain in the liver. Such results could stem from the circumstance that E. coli content in spleens infected by the MY strain exceeded that of spleens infected with the TS strain and that E. coli content in livers infected with the TS strain was greater than in those infected by the MY strain. In short, more E. coli induced the production of defensins.
Altogether, the findings show that pathogenicity of the MY strain was greater than that of the TS strain in rabbits. Two E. coli could replicate efficiently in many tissues of rabbits, and pro-inflammatory cytokines IL-1β, IL-6, IL-8, IFN-γ, and TNF-α were all induced in the spleen and liver by the MY and TS strains. The expressions of α- and β-defensin induced by the MY and TS strains differed in the spleen and liver. As such, the present study has pioneered the systematic exploration of the expression of immune-related genes in EHEC-infected rabbits. Its findings also provide useful information about the relationship between EHEC pathogenicity and host immune responses in rabbits.
Materials and methods
Ethics statement
This study was carried out in accordance with the recommendations of Shandong Agricultural University Animal Care and Use Committee (no. SDAUA-2015-005).
Bacterial strains
The EHEC TS and MY strains were isolated from intestine of clinically infected rabbits that developed acute diarrhea. Using eosin-methylene blue medium as the selective medium, and suspected colonies were subjected to staining microscopy and biochemical tests. Then bacterial strains were grown in nutrient broth medium at 37°C. Overnight cultures of bacteria were harvested for DNA extraction using the TIANamp Bacteria DNA Kit (Transgen Biotech Co., Ltd., Beijing, China) according to the manufacturer’s instructions. Following those tests, 16S rDNA sequencing was performed with universal primers 27F (5’-AGAGTTTGATCCTGGCTCAG-3’) and 1492R (5’-GGTTACCTTGTTACGACTT-3’), and the sequences were blasted in NCBI. To further amplify the classical virulence genes of EHEC, the primer pairs of eae, Stx1B, Stx2A and variants, as well as hlyA genes [1] are listed in Table 1.
Growth curve
Overnight cultures of TS and MY strains in amounts of 1 mL (109 CFU) were introduced into 100 mL nutrient broth medium. The new liquid medium were incubated at 37°C for 24 h, and calculations of E. coli content every 2 h were used to develop a growth curve.
Animal experiments
A group of 120 healthy weaned rabbits 35 d old were randomly separated into three treatment groups (n = 40) and given sufficient feed and water. Challenge tests were conducted by using intraperitoneal injections of 1.0 mL of each E. coli bacterial suspension (108 CFU/mL). Rabbits in the control group were inoculated with 1.0 mL of phosphate-buffered saline. From each group, 10 rabbits were randomly chosen to provide data used to calculate the survival rate of each group. The survival rate of the rabbits was calculated at the end of the experiment. At 1 and 3 dpi, five rabbits in each group were euthanized, the hearts, livers, spleens, brains and colons were collected, and part of the tissues were fixed with 4% paraformaldehyde solution to study histopathological changes, whereas the others parts were used for RNA extraction. The fresh and sterile heart, liver, spleen, brain, and cecal content were collected to gauge E. coli content as the following: tissue samples were weighed, suspended in PBS (1ml/g). Serial 10-fold dilutions were plated onto nutrient agar to calculate CFU per gram in each organ. All rabbits were handled according to appropriate biosecurity guidelines. All experiments were performed in a biosafety level-2 laboratory.
Quantitative real-time PCR
Total RNA was extracted from the spleen and liver of rabbits using the RNeasy plus Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For quantitative real-time (qRT) PCR, total RNA (1 µg) was reverse transcribed with TransScriptR One-step gDNA Removal and cDNA Synthesis SuperMix (Transgen Biotech Co., Ltd., Beijing, China). The synthesized cDNA was stored at -20°C until further use. qRT-PCR was performed using TransStartR Tip Green qPCR SuperMix (+Dye II) (Transgen Biotech Co., Ltd., Beijing, China). Primers of IL-6 and TNF-α used for qRT-PCR have previously been reported [39]. Other primers used for qRT-PCR were designed with Primer 3 software (http://bioinfo.ut.ee/primer3-0.4.0/) based on target sequences previously reported (Table 1). qRT-PCR was performed using the 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). PCR involved a cycle at 94°C for 30 s, followed by 40 cycles at 94°C for 5 s and 60°C for 34 s. Dissociation curves of the products were ultimately developed.
Statistical analysis
The relative expressions of genes were calculated using the 2−ΔΔCt method. The housekeeping gene GAPDH was used as an endogenous control to normalize the expression of target genes. Data were analyzed with the non-parametric Mann-Whitney U test, and the survival rate of rabbits was analyzed using the Kaplan-Meier method. All statistical analyzes were performed with GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA), with statistical significance set at p < 0.05.
Author contributions
MJG and WHY designed the experiments and wrote the paper. FHW and GEH carried out most of the experiments. RL and XYW collected and analyzed data. LMW and TJC developed the idea for the study and revise the paper.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the Key Research Project of Shandong Province (No. 2016GNC110014); the National Key Research and Development Program of China (No. 2016YFD0500905); Project of Natural Science Foundation of Shandong Province (No. ZR2014CQ050); Funds of Shandong “Double Tops” Program (515_564013 and SYL2017YSTD12).
Conflicts of interest
The authors declare no competing financial interests.
References
1. García A, Marini RP, Feng Y, Vitsky A, Knox KA, Taylor NS, Schauer DB, Fox JG. A Naturally Occurring Rabbit Model of Enterohemorrhagic Escherichia coli-Induced Disease. Journal of Infectious Diseases. 2002; 186:1682-1686.
2. Karmali MA. Infection by verocytotoxin-producing Escherichia coli. Clinical Microbiology Reviews. 1989; 2:15.
3. Frankel G, Candy DC, Everest P, Dougan G. Characterization of the C-terminal domains of intimin-like proteins of enteropathogenic and enterohemorrhagic Escherichia coli, Citrobacter freundii, and Hafnia alvei. Infection & Immunity. 1994; 62:1835.
4. Konowalchuk J, Speirs JI, Stavric S. Vero response to a cytotoxin of Escherichia coli. Infection & Immunity. 1977; 18:775.
5. Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT. Hemorrhagic colitis associated with a rare Escherichia coli serotype. New England Journal of Medicine. 1983; 308:681-685.
6. Michino H, Araki K, Minami S, Takaya S, Sakai N, Miyazaki M, Ono A, Yanagawa H. Massive outbreak of Escherichia coli O157:H7 infection in schoolchildren in Sakai City, Japan, associated with consumption of white radish sprouts. American Journal of Epidemiology. 1999; 150:787.
7. Wendel AM, Johnson DH, Sharapov U, Grant J, Archer JR, Monson T, Koschmann C, Davis JP. Multistate outbreak of Escherichia coli O157:H7 infection associated with consumption of packaged spinach, August-September 2006: the Wisconsin investigation. Clinical Infectious Diseases. 2009; 48:1079-1086.
8. O’Brien AD, Melton AR, Schmitt CK, Mckee ML, Batts ML, Griffin DE. Profile of Escherichia coli O157:H7 pathogen responsible for hamburger-borne outbreak of hemorrhagic colitis and hemolytic uremic syndrome in Washington. Journal of Clinical Microbiology. 1993; 31:2799-2801.
9. Besser RE, Lett SM, Weber JT, Doyle MP, Barrett TJ, Wells JG, Griffin PM. An outbreak of diarrhea and hemolytic uremic syndrome from Escherichia coli O157:H7 in fresh-pressed apple cider. Jama. 1993; 269:2217-2220.
10. Keene WE, Mcanulty JM, Hoesly FC, Williams LP Jr, Hedberg K, Oxman GL, Barrett TJ, Pfaller MA, Fleming DW. A swimming-associated outbreak of hemorrhagic colitis caused by Escherichia coli O157:H7 and Shigella sonnei. New England Journal of Medicine. 1994; 331:579-584.
11. Rabatsky-Ehr T, Dingman D, Marcus R, Howard R, Kinney A, Mshar P. Deer Meat as the Source for a Sporadic Case of Escherichia coli O157:H7 Infection, Connecticut1. Emerging Infectious Diseases. 2002; 8:525-527.
12. Swennes AG, Buckley EM, Parry NMA, Madden CM, García A, Morgan PB, Astrofsky KM, Fox JG. Enzootic Enteropathogenic Escherichia coli Infection in Laboratory Rabbits. Journal of Clinical Microbiology. 2012; 50:2353-2358.
13. Pritchard GC, Williamson S, Carson T, Bailey JR, Warner L, Willshaw G, Cheasty T. Wild rabbits—a novel vector for verocytotoxigenic Escherichia coli O157. Veterinary Record. 2001; 149:567.
14. Scaife HR, Cowan D, Finney J, Kinghornperry SF, Crook B. Wild rabbits (Oryctolagus cuniculus) as potential carriers of verocytotoxin-producing Escherichia coli. Veterinary Record. 2006; 159:175-178.
15. García A, Fox JG. The Rabbit as a New Reservoir Host of Enterohemorrhagic Escherichia coli. Emerging Infectious Diseases. 2003; 9:1592-1597.
16. Tzipori S, Hogg J. The pathogenesis of hemorrhagic colitis caused by Escherichia coli O157:H7 in gnotobiotic piglets. The Journal of Infectious Diseases. 1986; 154:712-716.
17. Francis DH, Collins JE, Duimstra JR. Infection of gnotobiotic pigs with an Escherichia coli O157:H7 strain associated with an outbreak of hemorrhagic colitis. Infection & Immunity. 1986; 51:953.
18. Potter ME, Kaufmann AF, Thomason BM, Blake PA, Iii JJF. Diarrhelil Due to Escherichia coli O157:H7 in the Infant Rabbit. Journal of Infectious Diseases. 1985; 152:1341-1343.
19. Cantey JR, Blake RK. Diarrhea due to Escherichia coli in the rabbit: a novel mechanism. Journal of Infectious Diseases. 1977; 135:454.
20. Ulshen MH, Rollo JL. Pathogenesis of Escherichia coli gastroenteritis in man-another mechanism. New England Journal of Medicine. 1980; 302:99.
21. García A, Bosques CJ, Wishnok JS, Feng Y, Karalius BJ, Butterton JR, Schauer DB, Rogers AB, Fox JG. Renal Injury Is a Consistent Finding in Dutch Belted Rabbits Experimentally Infected with Enterohemorrhagic Escherichia coli. Journal of Infectious Diseases. 2006; 193:1125-1134.
22. Thorpe CM, Hurley BP, Lincicome LL, Jacewicz MS, Keusch GT, Acheson DW. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infection & Immunity. 1999; 67:5985.
23. Jandhyala DM, Rogers TJ, Kane A, Paton AW, Paton JC, Thorpe CM. Shiga Toxin 2 and Flagellin from Shiga-Toxigenic Escherichia coli Superinduce Interleukin-8 through Synergistic Effects on Host Stress-Activated Protein Kinase Activation. Infection and immunity. 2010; 78:2984.
24. Imler JL, Tauszig S, Jouanguy E, Forestier C, Hoffmann JA. LPS-induced immune response in Drosophila. J Endotoxin Res. 2000; 6:459-462.
25. Matsuguchi T, Musikacharoen T, Ogawa T, Yoshikai Y. Gene expressions of Toll-like receptor 2, but not Toll-like receptor 4, is induced by LPS and inflammatory cytokines in mouse macrophages. Journal of Immunology. 2000; 165:5767.
26. Jenssen H, Hamill P, Hancock RE. Peptide antimicrobial agents. Clinical microbiology reviews. 2006; 19:491-511.
27. Wehkamp J, Harder J, Weichenthal M, Mueller O, Herrlinger KR, Fellermann K, Schroeder JM, Stange EF. Inducible and constitutive beta-defensins are differentially expressed in Crohn’s disease and ulcerative colitis. Inflammatory Bowel Diseases. 2003; 9:215-223.
28. Zasloff M. Inducing endogenous antimicrobial peptides to battle infections. Proceedings of the National Academy of Sciences. 2006; 103:8913-8914.
29. Boman HG. Peptide Antibiotics and their Role in Innate Immunity. Annual Review of Immunology. 2003; 13:61.
30. Gudmundsson GH, Agerberth B. Neutrophil antibacterial peptides, multifunctional effector molecules in the mammalian immune system. Journal of Immunological Methods. 2000; 232:45-54.
31. Ritchie JM, Thorpe CM, Rogers AB, Waldor MK. Critical roles for stx2, eae, and tir in enterohemorrhagic Escherichia coli-induced diarrhea and intestinal inflammation in infant rabbits. Infection & Immunity. 2003; 71:7129.
32. Janeway CA Jr. Presidential Address to The American Association of Immunologists The Road Less Traveled by: The Role of Innate Immunity in the Adaptive Immune Response. Journal of Immunology. 1998; 161:539.
33. Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000; 406:782.
34. Ulevitch RJ. Regulation of Receptor-Dependent Activation of the Innate Immune Response. Journal of Infectious Diseases. 2003; 187 Suppl 2:S351-S355.
35. Thorpe CM, Smith WE, Hurley BP, Acheson DWK. Shiga Toxins Induce, Superinduce, and Stabilize a Variety of C-X-C Chemokine mRNAs in Intestinal Epithelial Cells, Resulting in Increased Chemokine Expression. Infection & Immunity. 2001; 69:6140.
36. Stone SM, Thorpe CM, Ahluwalia A, Rogers AB, Obata F, Vozenilek A, Kolling GL, Kane AV, Magun BE, Jandhyala DM. Shiga toxin 2-induced intestinal pathology in infant rabbits is A-subunit dependent and responsive to the tyrosine kinase and potential ZAK inhibitor imatinib. Frontiers in Cellular & Infection Microbiology. 2012; 2:135.
37. Orsi NM, Tribe RM. Cytokine networks and the regulation of uterine function in pregnancy and parturition. Journal of Neuroendocrinology. 2008; 20:462-469.
38. Hancock RE. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infectious Diseases. 2001; 1:156-164.
39. Liu SJ, Shi Y, Liu C, Zhang M, Zuo ZC, Zeng CJ, Zhou GB, Xian H, Song TZ. The upregulation of pro-inflammatory cytokines in the rabbit uterus under the lipopolysaccaride-induced reversible immunoresponse state. Animal Reproduction Science. 2017; 176:70-77.