
Introduction
Anti-leukemia immunity plays an important role in protecting against the development of leukemia cells. Graft-versus-leukemia effects have been proven to have the power to eliminate minimal residual disease (MRD) in the setting of allogeneic stem cell transplantation (allo-SCT) and donor lymphocytes infusion (DLI) [1-3]. Other immunotherapeutic strategies under investigation include vaccination and adoptive T cell transfer, which have had some exciting results [3-5], but initial trial data have been disappointing [6]. Leukemia cells and the leukemia cell-induced immune suppressive microenvironment are likely to be responsible for failure in these immunotherapeutic approaches. For example, downregulation or overexpression of costimulation or activation molecules can interfere with the function and survival of immune cells, besides, exceptional secreted cytokines and exosomes from leukemia environment can disturb the metabolism and/or differentiation of immune cells [7-10]. In addition, several immune suppressive cells have been found to be increased in leukemia patients, such as regulatory T cells (Treg) [11] and myeloid-derived suppressor cells (MDSCs) [12, 13], and there is an imbalance in the Th1/Th2 ratio. Therefore, a better understanding of the impact of leukemia cells on the host immune system is critical for developing successful immunotherapy strategies.
CML is a clonal hematopoietic stem cell disorder characterized by the translocation t(9;22)(q34;q11.2), which results in the creation of the novel BCR-ABL fusion gene with abnormal tyrosine kinase activity [14-17]. Tyrosine kinase inhibitors (TKIs) such as imatinib mesylate (IM) is a targeted molecular drug that serves as frontline therapy for all phases of CML that works by binding to the tyrosine kinase domain of BCR-ABL and inhibiting its function [18]. TKIs are often well tolerated; however, TKI-based therapy is considered to be lifelong because rapid disease relapse often occurs upon drug discontinuation. As tyrosine kinases are key regulators of immune responses, long-term off-target effects on normal cells and tissues may raise concerns that immunosuppressive effects may occur by continued treatment with TKIs. Studies from other groups have shown that IM can reversibly inhibit T cell proliferation in vitro; however, the distribution of lymphocyte subsets and the proliferation and activation status of lymphocytes from imatinib-treated patients were similar to healthy controls in a study by Rohon et al. with the exception of a lower number of γδ T cells and a larger amount of CD45RO positive CD4+ T cells in PB [19]. It is well known that an adequate number of naïve and memory cells must exist to achieve lifelong protection against pathogens, particularly for T memory cells, which have the ability to rapidly transition from a quiescent state to a highly proliferative cytolytic population of effector cells upon antigen reexposure [20, 21]. Recently, a new subset of human memory T cells has been identified based on expression of the surface markers CD95 and CD122. These memory T cells possess a stem cell-like capacity to self-renew and differentiate into all subsets of memory and effector cells; thus, these cells were termed stem cell memory T cells (TSCM), and they have been revealed to be quite important for T cell immune reconstitution after HSCT and long-term immune surveillance [22].
Based on the importance of T cell memory and homeostasis in human immunity, in this study, we assayed the distribution of all of the memory T cell subsets in patients with CML and those who achieved CR by treatment with TKIs to assess the impact of this disease and TKIs on the patient T cell subsets as well.
Results and discussion
To compare the proportion of peripheral CD3+, CD4+, and CD8+ T cells in CML patients with those from HIs, as well as samples from CML-CR patients treated with TKIs, we used eight types of antibodies for analysis. The gating strategy is shown in Figure 1A. CD45high lymphocytes expressing CD3 and CD4, or CD3 and CD8 were gated as CD3+, CD4+, and CD8+ T cells, respectively, and then divided into four populations by the expression of CD45RO and CCR7. Naïve T cells (TN) and TSCM cells were defined as CD45RO-CCR7+, TCM cells were CD45RO+CCR7+, TEM cells were CD45RO+CCR7-, and TEF cells were CD45RO-CCR7-. The TSCM cells were further gated from the CD45RO-CCR7+ population by CD95 and CD28 expression, and with the exception of the TSCM subset, the other subsets could be further analyzed for their expression of CD28 [23, 24].
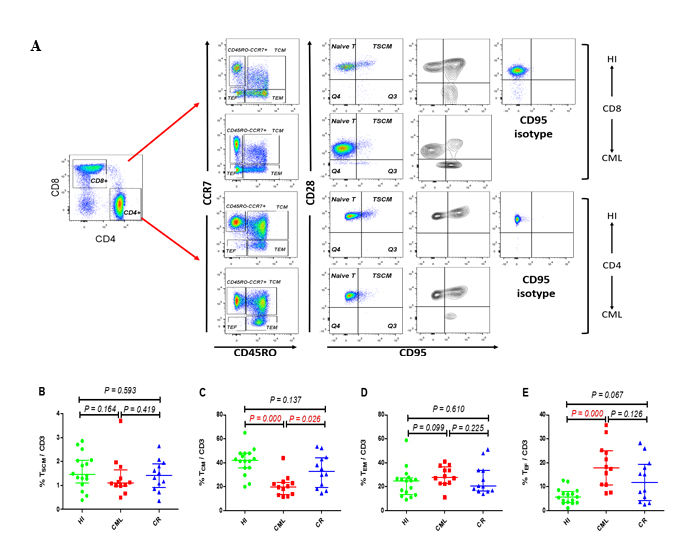
Figure 1: Gating strategy for identifying the CD4 and CD8 T cell subsets and the frequency of TSCM, TCM, TEM and TEF cells within CD3+ population. (A) Gating strategy for the identification of CD4 and CD8 T cell subsets in one HI and one CML patient. CD45RO and CCR7 were used to divide the T cells into four subsets, and TSCM cells were then further gated from the CD45RO- CCR7+ population by CD95 and CD28 expression, and separation of the TSCM cells from CD95− TN cells was performed with the help of gating CD95 on the CD4 or CD8 population and the CD95 FMO control; (B - E). Frequency of TSCM, TCM, TEM, and TEF among CD3+ T cells from16 HIs, 9 patients with CML, and 9 cases with CML-CR. HI: healthy individuals, CML: chronic myeloid leukemia, CML-CR: CML achieved complete remission.
We first compared the CD3+ TSCM, TCM, TEM, and TEF subsets distribution in the three groups, and the results demonstrated that there was no difference between the groups for TSCM and TEM cells, while there was a distinct decreased proportion in the TCM subset in the CML group (20.0%, p = 0.000); however, there was no significant difference in the CML-CR group (32.8%, p = 0.163) compared with HIs (42.1%) (Figure 1C). Accordingly, there was a significantly increased proportion of TEF cells in the CML group (17.9%, p = 0.000) compared with HIs (5.6%) but not in the CML-CR group, which was examined at the same time (Figure 1E). Due to the different functions of the CD4+ and CD8+ T cells in immunity, we further analyzed the distribution of the different subsets in the CD4+ and CD8+T cell populations. Similarly, there was no difference in the proportion of CD4+ and CD8+ TN cells between the CML or CML-CR groups and HIs. With the development of multicolor flow cytometry and high throughput sequencing, human memory T cells could be further classified into four subsets, and TSCM and TCM cells have the most potential for adoptive therapy because they can rapidly differentiate into the TEM and TEF subsets after antigen restimulation to control the invasion and spread of pathogens [25]. In this study, we found a dramatically decreased CD8+ TSCM subset in the CML group (0.7%, p = 0.007) compared with HIs (2.3%). While this change was recovered in CML-CR patients (2.2%, p = 0.472), the CD4+ TSCM population also demonstrated the same pattern, but the difference was not statistically significant (Figure 2A and 2E). For the TCM proportion, both CD4+ and CD8+ TCM cells were sharply decreased in the CML group (CD4+: 39.2%, p = 0.002; CD8+: 5.5%, p = 0.000), but they were nearly normalized in CR patients (CD4+: 50.4%, p = 0.227; CD8+:11.3% p = 0.164), when compared with HIs (CD4+: 55.1%; CD8+: 14.7%) (Figure 2B and 2F).
TEM and TEF cells are more differentiated subsets that can continually secrete cytokines (CD4) or cytotoxic molecules (CD8) to protect against infections and tumor cells when exposed to antigens [20, 25]. Here, we reported that the CD4 TEM and TEF cell subsets increased in the CML group (TEM: 22.9%, p = 0.003; TEF: 3.5%, p = 0.046) compared with HIs (TEM: 14.1%, TEF: 0.2%), but there was no significant difference between the CML-CR group and HIs (Figure 2C and 2D). Similar to the CD4 subset, the proportion of the CD8 TEM and TEF cell population also increased in the CML group (Figure 2G and 2H); however, the changes were only significant for the CD8 TEF subset (CML: 22.8%, HI: 12.0%, p = 0.039). The TEM and TEF subsets were nearly normal in CR patients; however, some of the CR patients still had a higher level of the CD4 and/or CD8 TEM and TEF subsets, which may be associated with the degree of remission in these patients. It appears that the inclination toward more differentiated T cell subsets is not a unique characteristic of CML patients because circulating CD8+ T cells skewing from naïve to TEF and TEM subsets accompanying the shrunken pool of TCM was also reported in head and neck squamous cell carcinoma (HNSCC) patients; moreover, activated TEM and TEF cells were dramatically increased in patients with advanced-stage disease [20, 21]. These results indicated that T cells in CML patients have the capacity to more quickly differentiate into effector; however, it remains unclear whether these cells maintain full immune function. Moreover, TKI treatment can recover T cell subsets.
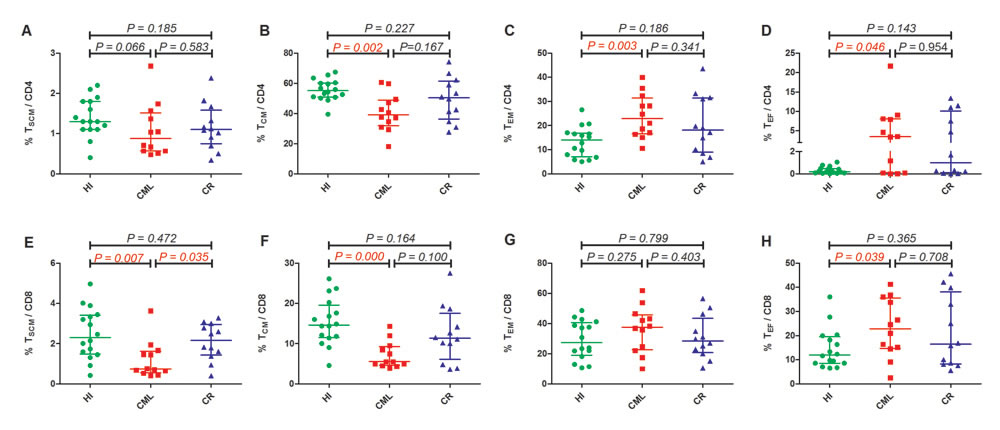
Figure 2: Frequency of TSCM, TCM, TEM and TEF among the CD4+ ( Top ) and CD8+ (Below). (A). TSCM/CD4+, (B). TCM/CD4+, (C). TEM/CD4+, (D). TEF/CD4+, (E). TSCM/CD8+, (F). TCM/CD8+, (G). TEM/CD8+, (H). TEF/CD8+. HI (n=16), CML (n=12), CML-CR (n=12). HI: healthy individuals, CML: chronic myeloid leukemia, CML-CR: CML achieved complete remission
It has been reported that most of the increased TEF and TEM cells have a CD28 negative phenotype, and these CD28− TEF cells demonstrate a lack of TCRζ chain expression or overexpression of early apoptosis markers. After tumor excision or treatment with chemotherapy, the skewed T cell subsets and CD28 expression have been reported to recover to normal [21]. We also found that there was an increased percentage of CD28- population in total CD8 (CML: 46.3%, HI: 26.7%, p = 0.024) (Figure 3A) but not in CD4 (CML: 7.1%, HI: 3.7%, p = 0.186) T cell subset of CML patients, further analysis revealed that those increased percentage of CD28 negative population mainly distributed in the TN, TCM subsets (Figure 3B to 3D), meanwhile, CD28- population distribution in CR group showed almost the same with HI group, not matter in the total CD8 subset or the CD8+ memory T cell subsets (Figure 3A to 3D). Recently, CD8+CD28− cells have been defined as CD8+ Treg cells and have been observed to have an immune regulatory function in patients who have undergone successful organ transplantation or alloanergized HLA mismatched bone marrow grafts [26] or who were suffering from autoimmune diseases and cancers [27-29]. These cells can mediate immune suppression by directly contacting antigen presenting cells (APCs) and upregulating inhibitory receptors (ILT3 and ILT4) on APCs or by secreting IL-10 [30]. Importantly, CD8+CD28− T cells isolated from healthy donors do not show immunosuppressive properties [30]. Hence, it would appear that CD8+CD28− Treg cells are induced in the periphery following disturbances in immune homeostasis [31], and in vitro experiments have revealed that the CD28− T cell population can be induced by the continued stimulation of CD28+ T cells with tumor cells, which may be related to improper co-stimulation signals provided by tumor cells. Similarly, Rifca Le Dieu reported an increase in the absolute number of CD3+CD56+ cells ( though not natural killer T (NK-T) cells) in the blood of patients with acute myeloid leukemia (AML) compared with healthy controls, and these cells more frequently expressed CD57 and less frequently expressed CD28 [22].
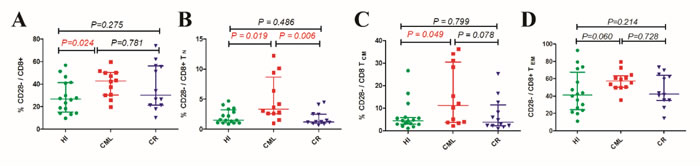
Figure 3: Increased CD28- T cell in CD8 population and their increased expression in CD8 T cell subsets. (A). Frequency of CD28- cells in total CD8+ population. (B). CD28- cell frequency in CD8+TN subsets. (C). CD28- cell frequency in CD8+ TCM subsets. (D). CD28- cell frequency in CD8+ TEM subsets. HI (n=16), CML (n=12), CML-CR (n=12). HI: healthy individuals, CML: chronic myeloid leukemia, CML-CR: CML achieved complete remission.
In this study, we report for the first time the characteristics of the skewed T cell subsets in the PB of CML patients with leukemia burden, which is similar to that found in HNSCC and AML patients, and the abnormal distribution pattern nearly recovered after CR by IM treatment. This discovery is also similar with the finding that excising a tumor could restore the TLI subset distribution in HNSCC. These results suggest that direct contact and stimulation with tumor cells will result in a T cell response and differentiation into the TEF and TEM populations; however, the continued imbalanced in T cell distribution yields protection potential for naïve T cells and a long-term protective capacity for the TSCM and TCM subsets. Moreover, chronic tumor-associated antigen stimulation may result in a dysfunction in the TEF and TEM population similar to that found with chronic virus infection. Although we do not have direct evidence of T cell dysfunction in present study, this evidence may be indirectly provided in our previous studies and other reports that have shown low TCR ζ chain expression and high expression of program death 1 (PD-1) on the T cells of CML patients [23-28]. Further investigation will focus on determining the ways in which dysfunction occurs in the different T cell subsets and revealing the mechanisms underlying it. In addition, we also assayed for the first time the TSCM distribution in the PB of CML patients in this study, and our results indicate that CD8+ TSCM combined with TCM loss may greatly contribute to patient infection susceptibility, especially for fungus and virus infection, the speculation is based on TSCM and TCM playing an important role in the adaptive immune response upon reinfection, as well as that CML patients showed more susceptibility to Cryptococcus and other infection diseases compare with healthy individuals [22, 25, 32, 33]
In summary, we characterized for the first time a skewed distribution of the CD3 T cell subsets (decreased TCM combined with increased TEF) in CML patients, but this became normalized in patients with CML-CR after TKI therapy. Further analysis of the CD4 and CD8 sub-populations revealed that the reduced T cell subsets were CD8+ TSCM cells and CD4 and CD8 TCM cells. In contrast, the accumulated T cell subsets were CD4 TEM and CD4 and CD8 T EF subsets. The imbalance in memory CD4+ and CD8+ T cell subsets is schematically shown in Figure 4. Based on the discovered T cell dysfunction in CML patients reported by previous studies, we hypothesize that most of the accumulated TEM and TEF cells were exhausted to some degree, while the different degree of accumulation and exhaustion may have a direct impact on the patient response to TKIs and prognoses, which is worth further investigation. Overall, the findings suggested that these responsive T cells might gradually become exhausted due to the continued existence of leukemia cells and their environment; therefore, T cell activation using a different approach remains a key point for enhancing global T cell immunity in CML patients, even for those with CR status.
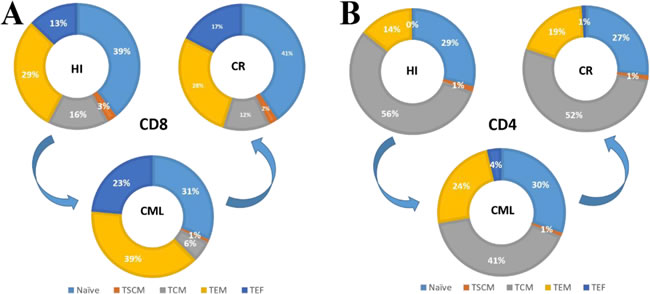
Figure 4: Pie charts summarize the distribution changes in the CD4 and CD8 T cell subsets. A. The CD8+ T cell subsets changes from HIs to CML patients and then recovers in CML-CR patients; B. CD4+ T cell subsets change from HIs to CML patients and then recovers in CML-CR patients.
Materials and methods
Samples
PB samples were obtained with consent from 12 CML patients, including 7 cases in chronic phase (CP), 2 cases in acceleration phase (AP) and 3 cases in blast crisis (BC), (7 males and 5 females with a median age of 37 years); 12 CML patients who achieved complete remission (4 males and 8 females with a median age of 41 years) by treating with Imatinib (200 to 400 mg/day), the length of treatment was different for each patient, from 1 month to 5 years, there were 3 CR patients also assayed at the time of diagnosis which included in the CML group, the response degree in each patient was different, which from complete hematologic response (CHR) to Major molecular response (MMR); 16 healthy volunteers (7 males and 9 females with a median age of 36.5 years). The characteristics of the patients and healthy volunteers are summarized in Table 1. All of the procedures were conducted according to the guidelines of the Medical Ethics Committees of the Health Bureau of the Guangdong Province in China, and ethical approval was obtained from the Ethics Committee of the Medical School of Jinan University.
Table 1: Characteristics of healthy volunteers, patients with CML, and CML patients in CR treated with TKIs
Variable* |
HI group |
CML group |
CR group |
|
Age (Year) |
36.5 (21~52; 16) |
37 (19~61; 12) |
41 (19~61; 12) |
|
Male/Female |
7/9 |
7/5 |
4/8 |
|
WBC (×109/L) |
/ |
170 (33~523; 12) |
6 (3~9; 12) |
|
PLT (×109/L) |
/ |
389 (52~853; 12) |
179 (84~573; 12) |
|
Blast and Promyelocytes in blood (%) |
/ |
CP |
1 (0~7; 7) |
/ |
AP+BC |
10 (1~67; 5) |
|||
WBC = white blood cell, PLT= platelet, CP = chronic phase, AP = acceleration phase, BC = blast crisis. *Continuous variables are expressed as median (range; number of cases).
Immunophenotyping analysis by flow cytometry
The strategy for TSCM analysis followed the protocol of Lugli E [23]. Cell surface staining for flow cytometry was performed with the following: CD45-APC (C1.7), CD3-FITC (HIT3a), CD4-APC-H7 (RPA-T4),CD8-Percp-Cy5.5 (SK1), CCR7-BV421 (150503), CD45RO-BV510 (UCHL1), CD28-PE (CD-28.2), CD95-PE-Cy7 (DX2), BV510 Isotype Control (G155-178) and PE-Cy7 Isotype Control (MOPC-21). Extracellular staining was performed according to the manufacturer’s instructions. Five microliters of CCR7-BV421 fluorescent antibody was incubated with approximately 200µl peripheral blood without plasma at 37oC for 15 minutes in the dark. Redundant fluorescent antibodies were eliminated by washing with 2 ml Cell Staining Buffer followed by centrifugation at 350g for 5 minutes, and the supernatant was discarded. Cells were then resuspended in 50µl antibody cocktail and incubated at room temperature for an additional 15 minutes in the dark. Then, 2ml 1X Red Blood Cell (RBC) Lysis Buffer (BD, Biosciences, USA) was added to suspend the stained cells, which were then incubated in dark at room temperature for 20 minutes. After centrifugation for 5 minutes at 350 g, the supernatant was discarded. Finally, the cells were washed twice with 2 ml Cell Staining Buffer by centrifugation at 350 g for 5 minutes, the supernatant was discarded and the samples were suspended in 0.5 ml staining buffer for analysis by flow cytometry. A total of 80,000 to 100,000 CD3+ cells were analyzed with a BD Verse flow cytometer (BD, Biosciences, USA), and data analysis was performed using FlowJo software.
Statistical analyses
All data are represented as medians, and statistically significant differences between the different T cell populations as well as between CD8+CD28- T cells were analyzed by the Mann-Whitney U test for nonparametric values. Calculations were performed using GraphPad Prism version 4.00 software.
Abbreviations
CML: Chronic myeloid leukemia, CR: Complete remission, HSCT: Hematopoietic stem cell transplantation, PB: Peripheral blood, PBMCs: Peripheral blood mononuclear cells, MRD: minimal residual disease, TN : naive T cells; TCM: central memory T cell, TSCM: stem cell memory T, TEM: effector memory T cells, TEF: terminal effector T, DLI: donor lymphocytes infusion, Treg: regulatory T cells, MDSCs: myeloid-derived suppressor cells, TKIs: Tyrosine kinase inhibitors, IM: imatinib mesylate, HNSCC: head and neck squamous cell carcinoma, APCs: antigen presenting cells, NKT: natural killer T, AML: acute myeloid leukemia, PD-1: program death 1, WBC: white blood cell, PLT: platelet, CP: chronic phase, AP: acceleration phase, BC: blast crisis.
Authors’ contributions
YQL and LX contributed to the concept development and study design. DLY, LX, JXT, YKZ, SL, DML, SHL, SHC and LJY performed the laboratory studies. XFZ, XLW, JC, JL, and YHL collected the clinical data. LX and DLY participated in the manuscript and figure preparation. YQL coordinated the study and helped draft the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We want to acknowledge the Flow Facility of Biological Translational Research Institute of Jinan University as well as Yanqiong Jia ( Research assistant ) and Dr. Yan Xu from Translational Research Institute of Jinan University. We also want to thank the volunteers who donated blood for this project.
Conflicts of Interest
The authors declare that they have no competing interests.
Funding
This study was supported by grants from the National Natural Science Foundation of China (Nos. 91642111, U1301226 and 81570143), the Guangdong Provincial Basic Research Program (No. 2015B020227003), the Guangdong Provincial Applied Science and Technology Research & Development Program (Grant No.: 2016B020237006), the Guangdong Natural Science Foundation (No. S2013020012863), the Foundation for High-level Talents in Higher Education of Guangdong, China (No. [2013]246-54) and the Guangzhou Science and Technology Project (No. 201510010211).
References
1. Miyamoto T, Fukuda T, Nakashima M, Henzan T, Kusakabe S, Kobayashi N, Sugita J, Mori T, Kurokawa M, Mori SI. Donor Lymphocyte Infusion for Relapsed Hematological Malignancies after Unrelated Allogeneic Bone Marrow Transplantation Facilitated by the Japan Marrow Donor Program. Biol Blood Marrow Transplant. 2017; 23: 938-44.
2. Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood. 2004; 103: 767-76.
3. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, Kriley IR, Rosenberg SA. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016; 375: 2255-62.
4. Garber HR, Mirza A, Mittendorf EA, Alatrash G. Adoptive T-cell therapy for Leukemia. Mol Cell Ther. 2014; 2: 25.
5. Zha X, Xu L, Chen S, Yang L, Zhang Y, Lu Y, Yu Z, Li B, Wu X, Zheng W, Li Y. Generation of V α13/β21+T cell specific target CML cells by TCR gene transfer. Oncotarget. 2016; 7:84246-84257. https://doi.org/10.18632/oncotarget.12441.
6. Rouce RH, Sharma S, Huynh M, Heslop HE. Recent advances in T-cell immunotherapy for haematological malignancies. Br J Haematol. 2017; 176: 688-704.
7. Bachireddy P, Hainz U, Rooney M, Pozdnyakova O, Aldridge J, Zhang W, Liao X, Hodi FS, O’Connell K, Haining WN, Goldstein NR, Canning CM, Soiffer RJ, et al. Reversal of in situ T-cell exhaustion during effective human antileukemia responses to donor lymphocyte infusion. Blood. 2014; 123: 1412-21.
8. Corrado C, Saieva L, Raimondo S, Santoro A, De Leo G, Alessandro R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J Cell Mol Med. 2016; 20: 1829-39.
9. Schnorfeil FM, Lichtenegger FS, Emmerig K, Schlueter M, Neitz JS, Draenert R, Hiddemann W, Subklewe M. T cells are functionally not impaired in AML: increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J Hematol Oncol. 2015; 8: 93.
10. Ok CY, Young KH. Checkpoint inhibitors in hematological malignancies. J Hematol Oncol. 2017; 10: 103.
11. Zahran AM, Badrawy H, Ibrahim A. Prognostic value of regulatory T cells in newly diagnosed chronic myeloid leukemia patients. Int J Clin Oncol. 2014; 19: 753-60.
12. Giallongo C, Parrinello N, Brundo MV, Raccuia SA, Di Rosa M, La Cava P, Tibullo D. Myeloid derived suppressor cells in chronic myeloid leukemia. Front Oncol. 2015; 5: 107.
13. Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010; 184: 3106-16.
14. Apperley JF. Chronic myeloid leukaemia. Lancet. 2015; 385: 1447-59.
15. Li Y, Yang L, Chen S, Li R, Zhang Y, Lu Y, Luo G. Clonal expansion T cells identified in acute monoblastic leukemia by CDR3 size analysis of TCR V beta repertoire using RT-PCR and genescan. Chin Med J (Engl). 2002; 115: 69-71.
16. Xu L, Lu Y, Lai J, Yu W, Zhang Y, Jin Z, Xu Y, Chen J, Zha X, Chen S, Yang L, Li Y. Characteristics of the TCR Vbeta repertoire in imatinib-resistant chronic myeloid leukemia patients with ABL mutations. Sci China Life Sci. 2015; 58: 1276-81.
17. Ma W, Ma N, Chen X, Zhang Y, Zhang W. An overview of chronic myeloid leukemia and its animal models. Sci China Life Sci. 2015; 58: 1202-8.
18. Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, Niederwieser D, Resta D, Capdeville R, Zoellner U, Talpaz M, Druker B, Goldman J, et al, and International STI571 CML Study Group. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002; 346: 645-52.
19. Rohon P, Porkka K, Mustjoki S. Immunoprofiling of patients with chronic myeloid leukemia at diagnosis and during tyrosine kinase inhibitor therapy. Eur J Haematol. 2010; 85: 387-98.
20. Kondo T, Morita R, Okuzono Y, Nakatsukasa H, Sekiya T, Chikuma S, Shichita T, Kanamori M, Kubo M, Koga K, Miyazaki T, Kassai Y, Yoshimura A. Notch-mediated conversion of activated T cells into stem cell memory-like T cells for adoptive immunotherapy. Nat Commun. 2017; 8: 15338.
21. Sander FE, Rydström A, Bernson E, Kiffin R, Riise R, Aurelius J, Anderson H, Brune M, Foà R, Hellstrand K, Thorén FB, Martner A. Dynamics of cytotoxic T cell subsets during immunotherapy predicts outcome in acute myeloid leukemia. Oncotarget. 2016; 7:7586–96. https://doi.org/10.18632/oncotarget.7210.
22. Xu L, Zhang Y, Luo G, Li Y. The roles of stem cell memory T cells in hematological malignancies. J Hematol Oncol. 2015; 8: 113.
23. Lugli E, Gattinoni L, Roberto A, Mavilio D, Price DA, Restifo NP, Roederer M. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc. 2013; 8: 33-42.
24. Crucian B, Dunne P, Friedman H, Ragsdale R, Pross S, Widen R. Alterations in levels of CD28-/CD8+ suppressor cell precursor and CD45RO+/CD4+ memory T lymphocytes in the peripheral blood of multiple sclerosis patients. Clin Diagn Lab Immunol. 1995; 2: 249-52.
25. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013; 31: 137-61.
26. Barbon CM, Davies JK, Voskertchian A, Kelner RH, Brennan LL, Nadler LM, Guinan EC. Alloanergization of human T cells results in expansion of alloantigen-specific CD8(+) CD28(-) suppressor cells. Am J Transplant. 2014; 14: 305-18.
27. Li YQ, Yang LJ, Chen SH, Zhang YP, Zhang XL, Luo GX. T cell receptor Vbeta repertoire usage and clonal expansion of T cells in chronic myelogenous leukemia. Chin Med J (Engl). 2004; 117: 840-3.
28. Filaci G, Fenoglio D, Fravega M, Ansaldo G, Borgonovo G, Traverso P, Villaggio B, Ferrera A, Kunkl A, Rizzi M, Ferrera F, Balestra P, Ghio M, et al. CD8+ CD28- T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. J Immunol. 2007; 179: 4323-34.
29. Zelle-Rieser C, Thangavadivel S, Biedermann R, Brunner A, Stoitzner P, Willenbacher E, Greil R, Johrer K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol. 2016; 9: 116.
30. Chang CC, Ciubotariu R, Manavalan JS, Yuan J, Colovai AI, Piazza F, Lederman S, Colonna M, Cortesini R, Dalla-Favera R, Suciu-Foca N. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol. 2002; 3: 237-43.
31. Vuddamalay Y, van Meerwijk JP. CD28- and CD28lowCD8+ Regulatory T Cells: Of Mice and Men. Front Immunol. 2017; 8: 31.
32. Chandran R, Hakki M, Spurgeon S. Infections in Leukemia. Sepsis - An Ongoing and Significant Challenge. 2012. https://doi.org/10.5772/50193.
33. Hsu JW, Hiemenz JW, Wingard JR, Leather H. Viral Infections in Patients with Hematological Malignancies. In: Wiernik PH, Goldman JM, Dutcher JP, Kyle RA, editors. Neoplastic Diseases of the Blood. New York (NY): Springer New York; 2013. pp. 1193–239.