
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide, resulting in approximately 550,000 diagnoses and 300,000 deaths a year [1]. Risk factors for HNSCC include tobacco and alcohol use, involved in 75% of the cases, and infection with human papilloma virus (HPV), associated with 40–60% of the oropharyngeal cancers [2, 3]. Standard treatment for HNSCC is a combination of surgery, radiotherapy (RT) and chemotherapy (CT) as most HNSCC are locally advanced at time of diagnosis. [4]. The five year survival rate of 40–50% is relatively poor despite advances in surgical techniques, chemo- and radiotherapy (cRT) [5]. Unfortunately, there is an inability to further intensify the current therapy due to unacceptable toxicity and morbidity [2]. Taking into account the high relapse rate and the limited therapeutic options, it is of utmost importance to underpin the molecular mechanisms of resistance.
The significant local relapse rate in HNSCC is mainly due to the high DNA repair capacity of cancer cells. Cells rely on the DNA damage response (DDR) to signal the presence of DNA damage so this can be repaired in order to survive. The key DNA repair pathways are: base excision repair (BER), nucleotide excision repair (NER) and mismatch repair (MMR) for single strand DNA breaks (SSBs), and homologous recombination (HR) and non-homologous end joining (NHEJ) for double strand breaks (DSBs). The exact mechanism of these pathways is beyond the scope of this review, but are clearly explained in the following papers [6–9]. Cancer cells often have deficiencies in these DNA repair pathways enabling the tumor cells to accumulate genetic alterations which attribute to their aggressive phenotype [9]. On the other hand, cancer cells rely on the remaining proficient DNA repair pathways to survive. DNA targeted agents try to exploit these backup DNA repair processes to generate synthetic lethality [8]. Cetuximab, a recombinant monoclonal antibody against the epidermal growth factor receptor (EGFR), is the only targeted agent for HNSCC to date that was approved by the Federal Food and Drug Administration (FDA). Because of the high resistance and low response rates, the search for suitable alternatives is urging. Modulating DNA repair after cRT with targeted agents is a promising technique to increase the therapeutic efficacy and decrease normal tissue toxicity. This can benefit a large number of HNSCC patients and is even applicable for a wide range of tumors [7]. In this review several possible DNA targeted therapies for HNSCC are discussed (see Figure 1).
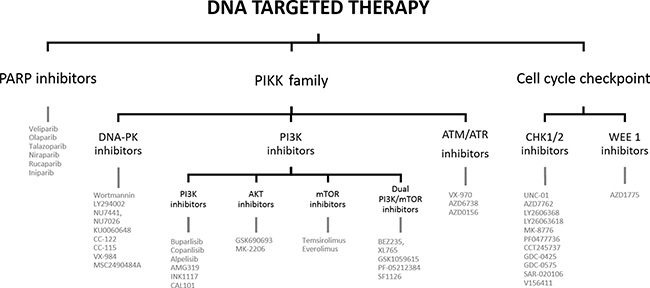
Figure 1: Outline of the review.
PARP INHIBITORS
Poly ADP-ribose polymerase (PARP) enzymes have a variety of important functions: DNA damage repair, cell cycle regulation and replication which influence tumor growth and progression [10–12]. PARP1 is an important sensor for SSBs and plays a critical role in BER [9, 13]. When DDR is activated, PARP1 synthetizes PAR polymers via autoPARylation to reseal and repair the DNA damage [14]. Recently it has been shown that PARP1 is also a regulator of HR, that it is involved in the alternative NHEJ and is required for restarting stalled/collapsed replication forks [9, 14–16]. Therefore PARP inhibition is interesting for both the regulation of transcription and inhibiting its function in DNA repair [13].
The popularity of PARP inhibitors (PARPi) is based on several studies in breast and ovarian cancer showing that PARPi could selectively kill HR-deficient cancer cells [9, 10, 15]. This led to the FDA-approval of three PARP inhibitors for ovarian cancer, namely Olaparib, Rucaparib and recently Niraparib [17]. The precise mechanism of action remains a matter of active debate, although it is thought that the cytotoxicity to HR-deficient cells is due to the accumulation of SSBs in the absence of PARP1. This leads to replication fork collapse and DSBs, which need HR to be repaired [18, 19]. As BRCA1 and BRCA2 are essential for HR, these DSBs cannot be repaired, which will eventually lead to cell death [20]. Also PARP trapping contributes significantly to the synthetic lethality [21]. More mechanisms of action of PARP inhibitors are suggested, although these two are the most apparent and reported ones. These mechanisms show that the combination of HR-deficient cells with PARP1 inhibition is synthetically lethal [8, 13, 15].
PARP inhibitors investigated in HNSCC
In the development of DNA damage response agents, PARPi have advanced the furthest. Several small molecule inhibitors are in clinical development: Niraparib, Talazoparib, Olaparib, Veliparib, Rucaparib and Iniparib [22]. Combining DDR inhibitors with DNA damaging agents has been the natural first step in combination strategies [21].
In vitro studies showed that HR-deficient HNSCC cells are hypersensitive to PARPi as they are unable to repair radiation-induced SSBs and that PAR induction by RT is probably prevented [18, 23–25]. Moreover, it is expected that PARPi would also work in HR-proficient cells since replication-dependent conversion of SSBs to DSBs focusses on rapidly proliferating cells more than on normal cells [22]. This hypothesis was confirmed in other studies where both HR-deficient and HR-proficient HNSCC cells were radiosensitized by Olaparib [24, 25]. As expected, lower concentrations were needed in HR-deficient cells to obtain the same radiosensitizing effect [22]. In the study of Weaver et al., HPV-positive cells had an increased sensitivity to PARPi as these cells were unable to recruit DNA-dependent protein kinase (DNA-PK) and BRCA2 to repair crosslinking damage [26]. These findings show HR-deficiency in HPV-positive cells as was earlier shown in the study of Dok et al. [27]. Recently, the idea was postulated to combine PARPi with HR inhibiting agents to broaden the applicability of PARPi. Possible agents to do so, are inhibitors of checkpoint kinase 1 (CHK1), WEE1, phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), EGFR or heat shock protein 90 (HSP90) [28].
When PARPi were combined with CT and RT, potentiating effects were seen. The chemopotentiating effect of PARPi was demonstrated in oral cavity cancer cells as Cisplatin attenuates the repair of radiation-induced DSBs [7, 22, 29]. PARPi sensitizes cancer cells to platinum-based drugs, Temozolomide and topoisomerase poisons. These promising synergistic effects are further tested in multiple ongoing clinical trials, which combine PARPi with RT, CT or Cetuximab (see Table 1).
Table 1: Ongoing clinical trials evaluating PARP inhibitors in head and neck cancer patients
Compound |
Combination |
Phase |
State |
NCT number |
|---|---|---|---|---|
Olaparib |
/ |
1 |
Not yet recruiting |
NCT02686008 |
RT |
1 |
Recruiting |
NCT01460888 |
|
RT |
1 |
Recruiting |
NCT02229656 |
|
RT + Cisplatin |
1 |
Recruiting |
NCT02308072 |
|
RT+ Cisplatin |
1 |
Withdrawn |
NCT01491139 |
|
RT+ Cisplatin |
1 |
Recruiting |
NCT01562210 |
|
Cisplatin |
2 |
Recruiting |
NCT02882308 |
|
RT + Cetuximab |
1 |
Recruiting |
NCT01758731 |
|
Veliparib |
Chemotherapy |
1/2 |
Suspended |
NCT01711541 |
Talazoparib |
/ |
1 |
Not yet recruiting |
NCT02567396 |
RT: Radiotherapy, NCT: Number clinicaltrials.gov identifier.
PARP inhibitor biomarkers
BRCA1 and BRCA2 mutations are recognized as molecular targets for PARPi in several tumor types [11]. HR mutations rarely occur in HNSCC, with only 6% of the patients having BRCA1/2 mutations [30]. Other processes affecting HR might influence the cellular response to PARPi as there is evidence that a larger group of HNSCC is HR-deficient [18]. Alternative HR repair genes that sensitize cancer cells to the inhibition of PARP, often referred as 'BRCAness' have been identified [14]. Defects in the Fanconi anemia (FA) pathway and mutations in ataxia telangiectasia (ATM), ataxia telangiectasia and Rad3-related (ATR) or phosphatase and tensin homolog (PTEN) are suggested to contribute to the HR-deficient phenotype [10, 15, 19, 21, 31, 32]. Therefore, other accurate biomarkers than BRCA1 and BRCA2 are necessary to indicate HR-deficiency. Different HR-deficiency assays are available, but must be tested in prospective clinical trials. For example, the BROCA assay can identify mutations in 13 HR-genes via next generation sequencing. Secondly, functional assays can provide real-time information about DNA repair, like RAD51 focus formation assay for the activation of HR machinery and γH2AX foci to reflect DSBs, but cannot be implemented in clinical practice due to technical reasons [33].
Besides testing for HR-deficiency, multiple tests are available to predict PARPi response, such as loss of heterozygosity (LOH) which is gaining popularity as surrogate for HR and to predict PARPi response [34]. LOH is a large chromosomal event that results in the loss of an entire gene together with the surrounding chromosomal region. These genomic scars can be identified by the Myriad Genetics HR assay, which is now tested in prospective clinical trials in breast cancer [11]. According to Stover et al. LOH profiling might be more predictive for PARPi response than HR-deficiency assays in early disease stage [19]. Besides LOH, the combination of p53-binding protein 1 (53BP1) and BRCA1 was recently suggested as biomarker for PARPi sensitivity, as 53BP1-deficiency would impair sensitivity to PARPi [35]. Finally, PARP1 enzyme levels seem to be a logic biomarker for PARPi activity, but this does not correlate with the clinical response. So in conclusion, a variety of assays are available to assess HR-deficiency and to predict PARPi response although the optimal, clinically feasible assays need to be validated in clinical trials [11, 19, 21]. Two ongoing clinical trials in HNSCC have foreseen to do laboratory biomarker analyses (NCT02567396 and NCT01711541).
Future challenges with PARP inhibitors
PARP inhibitors are the cornerstone of DNA repair targeted therapies [36]. The efficacy of PARP inhibition was demonstrated in BRCA1/2 mutated ovarian cancers. Although PARPi are well tolerated in ovarian cancer treatment, careful monitoring for long-term toxicity is mandatory [37]. Moreover, majority of patients will develop resistance eventually. Three mechanisms of resistance are identified so far. First, the HR function can be restored by secondary mutations in the BRCA1/2 gene together with loss of 53BP1 [9]. Secondly, an upregulation of polyglycoprotein 1 (PgP) pumps can cause efflux of the drugs. Finally, loss of PARP1 expression can lead to 100-fold resistance [14, 21]. Continued identification of resistance mechanisms is critical for further clinical use of PARPi.
HR-deficient HNSCC cells have shown hypersensitivity to PARPi, although the applicability of PARPi could be broadened to HR-proficient cells as Olaparib showed radiosensitization in both. These promising preclinical results are translated in ten ongoing clinical trials in the HNSCC field. Challenges for PARP inhibitor research remain to optimize the clinical efficacy and widen the utility of PARPi as most preclinical work is now focused on HR-deficient cells, leaving the question how PARPi sensitizes HR-proficient HNSCC cells unanswered [22, 38]. Furthermore, combination schedules with PARPi need to be further investigated where concomitant induced toxicities and pharmacological drug-drug interactions are important challenges. Therefore, optimization of the dose regimen and sequence becomes the key factor in successful clinical trial design, accompanied by accurate biomarkers that enable patient selection [14, 21, 22].
PIKK family
The phosphatidylinositol 3-kinase-related kinases (PIKK) family is a serine/threonine kinase family that consists of six members: DNA-PK, PI3K, mechanistic target of rapamycin (mTOR), ATR, ATM and human suppressor of morphogenesis in genitalia-1 (hSMG-1). Family members DNA-PK, ATM and ATR play an important role in DDR, along with PI3K and mTOR that are involved in cell growth and differentiation. Due to the conserved kinase domain, inhibitors are often active against multiple members of the PIKK family [39].
DNA-PK
DNA-PK has an extensive role in tumor associated processes including: G2/M cell cycle checkpoint regulation, genomic stability, hypoxia, metabolism, transcription support and inflammation making its inhibition an attracting therapeutic target [29, 40–42]. However, the established role of DNA-PK in innate immunity and pro-inflammatory signaling is an important aspect to consider with respect to long-term DNA-PK inhibitor use [21]. Furthermore, DNA-PK is an essential component of the NHEJ pathway, where it mediates direct ligation of broken DSBs and recruits other repair molecules [19, 21, 43, 44]. The precise mechanism how DNA-PK acts in NHEJ is beyond the scope of this review but is clearly described in the following articles [9, 21, 42–44]. In addition, there is evidence for a role of DNA-PK in HR. Though it is not definitively determined if DNA-PK promotes HR or if the failure of NHEJ promotes HR [42]. The pharmacological inhibition of DNA-PK results in inefficient repair and hypersensitivity to DSBs, hence suggesting its susceptibility to DNA-damaging agents.
DNA-PK inhibitors
Different DNA-PK inhibiting molecules target the ATP binding site of the kinase. Early inhibitors are Wortmannin and LY294002, which are both non-specific DNA-PK inhibitors that showed in vivo toxicity and off-target effects resulting in a narrow therapeutic index [21]. Modifications of LY294002 led to two highly specific molecules, NU7441 and NU7026, both showing promising preclinical results as chemo- and radiosensitizers. However, their poor water solubility and oral bioavailability must be taken into account in further clinical evaluation. These problems are addressed in KU0060648, a dual DNA-PK and PI3K inhibitor with a better oral bioavailability and pharmacokinetic profile. Other DNA-PK inhibitors under investigation are: CC-122 a pleotropic pathway modifier, CC-115 a DNA-PK and mTOR inhibitor, VX-984 and MSC2490484A.
Remarkably, all agents are focused on the kinase subunit of DNA-PK, but the inhibition of the regulatory Ku subunit could also reduce DNA-PK activity [40]. Other approaches for DNA-PK inhibition could be nucleotide or antibody based inhibitors, which showed to have significant effects in vitro [44]. These could overcome the two primary faced obstacles with DNA-PK inhibitory compounds, namely poor water solubility and short serum half-lives [44]. The development of new DNA-PK inhibitors with good ADME (absorption, distribution, metabolism and elimination) profiles will be based on the recently discovered X-ray crystal structure of DNA-PK [40, 44].
DNA-PK inhibitors investigated in HNSCC
Monotherapy with DNA-PK inhibitors has modest effects, but there is potential for antitumor synergy in combination with DNA-damaging agents [21]. Cells defective in DNA-PK are highly sensitive to RT, indicating that DNA-PK inhibition could be radiosensitizing [7]. This hypothesis was confirmed in different preclinical studies and was attributed to the fact that NHEJ is the primary pathway for the resolution of radiation-induced DSBs [26, 44]. Inhibition of DNA-PK promotes radiation-induced cell killing via mitotic catastrophe, senescence and autophagic cell death. Both NU7026 and NU7441 are proven to sensitize topoisomerase 2 inhibitors and are extreme radiosensitizers [45, 46]. Moreover, the radiosensitizing effect of NU7411 was shown in multiple cancer types: lung cancer cells, liver cells and breast cancer cells due to increased G2/M accumulation and prolonged delay in radiation-induced DSB repair [15, 41, 46–49]. The radiosensitizing effect is further increased in EGFR overexpressing cells as EGFR normally promotes NHEJ via DNA-PK [8, 50, 51]. Therefore, the effect of combining Cetuximab with DNA-PK inhibitors would be an interesting research topic.
The promising chemopotentiating and radiosensitizing effects of DNA-PK inhibitors are translated in multiple ongoing clinical trials in solid tumors, although none are listed in HNSCC specifically (see Table 2). CC-115 was well tolerated in a phase 1 trial with preliminary antitumor effects [21]. These promising in vitro results suggest it would be interesting to combine CC-115 with platinum-based chemotherapy in HR-deficient tumors [9].
Table 2: Ongoing clinical trials with DNA-PK inhibitors in solid tumors
Compound |
Subject |
Phase |
State |
NCT number |
|---|---|---|---|---|
CC-115 |
Advanced solid tumors |
1 |
Not yet recruiting |
NCT01353625 |
MSC2490484A+RT |
Advanced solid tumors |
1 |
Recruiting |
NCT02316197 |
Solid tumors |
1 |
Recruiting |
NCT02516813 |
|
C-122 |
Advanced solid tumors |
1 |
Recruiting |
NCT01421524 |
VX-984 |
Healthy volunteers |
1 |
Recruiting |
NCT02644278 |
RT: Radiotherapy, NCT: Number clinicaltrials.gov identifier.
DNA-PK inhibitor biomarkers
The DNA-PK inhibitor KU0060648 showed synthetic lethality in ATM-defective cells [9]. It has also been proven in the study of Shaheen et al. that ATM-deficient cells are addicted to DNA-PK for survival [15]. This suggests that HR-deficiency in general is associated with NHEJ addiction [9]. Mutations in HR genes (BRCA1/2, RAD51 and ATM) can predict sensitivity to KU0060648. These genes could be used as biomarker, but this has not been studied sufficiently. Biomarkers that are already used in preclinical research are similar as for the PARPi: γH2AX and RAD51 foci [52]. Biomarkers for NHEJ inhibitors are not yet validated [19].
Future challenges with DNA-PK inhibitors
The radiosensitizing effect of DNA-PK inhibitors is proven in multiple cancer types, even though the effect as monotherapy is small. Unfortunately research in the HNSCC field is confined and there is only one clinical trial ongoing for the moment (NCT02516813). However, HNSCC could be a very attractive target for the application of DNA-PK inhibitors as p53 is often mutated or deregulated by HPV and HR-deficient cells are addicted to NHEJ, so synthetic lethality can be induced [19, 26]. Since KU0060648 efficiently radiosensitized ATM-deficient cells and these cells are addicted to DNA-PK for survival, it can be a promising approach to combine PARPi with DNA-PK inhibitors. Moreover, there appears to be a relationship between PARP and DNA-PK as PARP inhibited cells are susceptible to DNA-PK inhibition via the alternative NHEJ pathway [16, 50]. In comparison with PARPi, biomarkers for DNA-PK inhibitors are limited to HR-deficiency assays and are not validated for NHEJ inhibition. These two gaps in the area of DNA-PK inhibitory agents encourage research in the HNSCC domain as many promising results are awaited.
PI3K inhibitors
The PI3K/AKT/mTOR pathway is heavily implicated in tumorigenic processes such as proliferation, invasion, apoptotic resistance, angiogenesis and metastases [53, 54]. The PI3K pathway, which is downstream of EGFR, is activated by growth factors and cytokines [55]. Important players in this pathway are protein kinase B (AKT), a second messenger, and the central mTOR complex which will promote cell growth via downstream effectors. This cascade is negatively regulated by the tumor suppressor gene PTEN. The RAS/RAF/MEK/ERK pathway is closely linked to the PI3K/AKT/mTOR pathway as there is crosstalk between PI3K and RAS [56, 57]. The precise mechanism of the pathway is beyond the scope of this review, but is discussed in the following articles [53, 54].
The PI3K/AKT/mTOR pathway is the most frequently deregulated pathway in HNSCC both on genomic and proteomic level [54]. Activating mutations of PI3K catalytic subunit 110α (PIK3CA) are seen in 56% of HPV-positive HNSCC and 39% of HPV-negative HNSCC according to The Cancer Genome Atlas (TCGA) [58–60]. Other deregulating mutations are possible in the regulatory subunit of PI3K, p85, which lead to activation of the PI3K pathway without growth factor stimulation. Furthermore, mutations in PTEN, AKT and the mTOR complex can result in the constitutive activation of the pathway leading to cell growth and proliferation in the absence of nutrients. Activation of this pathway is an important mechanism in resistance to EGFR inhibitors and cRT promoting tumor progression [55].
PI3K inhibitors investigated in HNSCC
Therapeutic agents can tackle this pathway and promising in vitro and in vivo results show less proliferation, more apoptosis and sensitization to therapy. However, PI3K inhibition alone can trigger compensatory feedback via the RAS/MEK/ERK pathway or EGFR which induces resistance. Combination therapy with other therapeutic agents or DNA damaging agents can achieve synergistic effects [54, 61, 62]. RT activates EGFR and other prosurvival pathways like PI3K. When PI3K is inhibited, this causes downregulation of BRCA1/2, which are important in HR. Eventually this leads to inhibition of radiation-induced DDR [22]. Different types of PI3K inhibitors are developed and tested in preclinical research.
PAN-PI3K inhibitors
Pan-PI3K agents inhibit more than one isoform of PI3K and are directed to tumors with PIK3CA mutations that are addicted to the PI3K pathway for growth and survival. Currently used inhibitors in clinical trials for HNSCC are Buparlisib and Copanlisib.
Buparlisib, also known as BKM120, is an oral reversible PI3K inhibitor competing to ATP [63, 64]. The anti-proliferative and pro-apoptotic effects of BKM120 are proven in tumor cells, irrespective the PIK3CA status [65]. However the half maximum inhibitory concentration (IC50) of BKM120 to fully block all PI3K forms as monotherapy is high, leading to cellular toxicity via tubulin [66]. The combination of BKM120 with Cetuximab showed synergistic effects in PIK3CA-mutant and wildtype HNSCC cell lines via the downregulation of AKT and induces apoptosis when BKM120 was given after Cetuximab [54, 67]. When these drugs are given in the opposite order an antagonistic effect is achieved since BKM120 downregulates the PI3K/AKT/mTOR pathway. In addition cells probably activate other pathways like the RAS/RAF/MEK/ERK pathway to rescue cell survival leading to treatment failure. These synergistic results were tested in vivo by Bozec et al. in an orthotopic HNSCC model [58]. Here, the synergistic effect of BKM120 with Cetuximab was seen as a significant inhibition of tumor growth, Ki67 reduction and mitogen-activated protein kinase (MAPK) pathway inhibition. Interestingly, RT alone normally upregulates the MAPK pathway as a compensatory mechanism and is implicated in tumor repopulation. This repopulation is a major challenge in the treatment of cancers but strikingly, the triple combination of RT with Cetuximab and BKM120 suppresses this radiation-induced activation of the MAPK pathway and combines anti-proliferative with pro-apoptotic effects [58].
The combination of BKM120 with Cetuximab is tested in multiple ongoing clinical trials (see Table 3). Results of a phase 2 study combining BKM120 with Paclitaxel showed improved clinical efficacy with a manageable safety profile, indicating an effective second-line treatment for metastatic HNSCC [68].
Table 3: Ongoing clinical trials with PAN-PI3K inhibitors in head and neck cancer patients
Compound |
Combination |
Phase |
State |
NCT number |
|---|---|---|---|---|
Buparlisib |
/ |
2 |
Unknown |
NCT01527877 |
/ |
2 |
Recruiting |
NCT01737450 |
|
Cisplatin + IMRT |
1b |
Recruiting |
NCT02113878 |
|
Paclitaxel |
2 |
Not yet recruiting |
NCT01852292 |
|
Cetuximab |
2 |
Not yet recruiting |
NCT01816984 |
|
Cisplatin/ Carboplatin |
1b |
Not yet recruiting |
NCT02439489 |
|
Copanlisib |
Cetuximab |
1+2 |
Recruiting |
NCT02822482 |
IMRT: Intensity-modulated radiation therapy, NCT: Number clinicaltrials.gov identifier.
Copanlisib is another highly selective and potent PI3K inhibitor with superior antitumor activity in PIK3CA mutants. The combination of Copanlisib with Cetuximab is now evaluated in a phase 1 and 2 trial in metastatic HNSCC patients with PIK3CA mutations or amplifications and/or PTEN loss as PIK3CA mutations and amplifications are associated with resistance to Cetuximab therapy (see Table 3) [69]. Copanlisib showed good tolerability and a dose-dependent pharmacokinetic in a recent phase 1 trial in Japanese patients [70].
PI3K selective inhibitors
In contrast to pan-PI3K inhibitors, selective PI3K inhibitors focus on one specific isoform of PI3K. Mostly the 110α isoform, as this is the most commonly mutated component of PI3K [54]. Selective PI3K inhibitors are expected to produce more targeted inhibition with fewer side effects [59].
Alpelisib, also known as BYL-719, is one of the most promising selective PI3K inhibitors with a favorable safety profile. HNSCC cells proved to be sensitive and in vivo experiments showed dose-dependent growth inhibition in PIK3CA-mutated xenografts, indicating a superior effect in tumors with PIK3CA mutations [71]. However, synergistic effects were demonstrated in vitro and in vivo combining Alpelisib with Cetuximab, irrespective of the PIK3CA mutational status (69). Thanks to the encouraging preclinical results, 6 clinical trials are now ongoing (see Table 4) [54]. Early study results combining Alpelisib with Cetuximab showed promising antitumor activity in patients with recurrent HNSCC [69].
Table 4: Ongoing clinical trials in head and neck cancer with selective PI3K inhibitors
Compound |
Combination |
Phase |
State |
NCT number |
|---|---|---|---|---|
Alpelisib |
Cisplatin + IMRT |
1 |
Recruiting |
NCT02537223 |
Paclitaxel |
1 |
Completed |
NCT02051751 |
|
Cetuximab + IMRT |
1 |
Recruiting |
NCT02282371 |
|
Cetuximab |
1b |
Terminated |
NCT01602315 |
|
/ |
2 |
Not yet recruiting |
NCT02145312 |
|
Cetuximab + Cisplatin |
1/2 |
Not yet recruiting |
NCT02298595 |
|
AMG319 |
/ |
2 |
Recruiting |
NCT02540928 |
IMRT: Intensity-modulated radiation therapy, NCT: Number clinicaltrials.gov identifier.
Other selective PI3K inhibitors are on the market as well: INK1117, CAL-101 and AMG319. Cal101 and amg719 target the δ subtype, which is involved in regulatory T cells, with the purpose to break tumor-induced immune tolerance. When 110δ is inactivated by AMG319, CD8+ cytotoxic T cells are unleashed to induce tumor regression [54]. Currently, AMG319 is tested in a phase 2a trial in HPV-negative HNSCC patients (see Table 4) [54, 59]. CAL-101, also known as Idelalisib, has been approved by the FDA and the European Medicines Agency as the first-in-class PI3K inhibitor for hematological cancer therapy [72].
AKT inhibitors
The phosphorylation of AKT is a very important step in the PI3K pathway and therefore many AKT inhibitors have been developed for cancer treatment. Together with the fact that AKT activation and overexpression is often associated with chemo- or radiotherapy resistance, these drugs can have great potential in cancer treatment [54]. Two types of AKT inhibitors are possible, an ATP-competitive variant such as GSK690693 and an allosteric inhibitor represented by MK-2206. MK-2206 has proven to be highly potent in enhancing the activity of anticancer agents in vitro and in vivo [54]. Very promising results came out of two clinical trials with MK-2206 in HNSCC patients (Table 5). In a phase 1 trial two HNSCC patients showed complete and partial response and in a phase 2 study with 21 patients, 9 patients were alive and progression-free at the end of the trial [73].
Table 5: Clinical trials with the AKT inhibitor MK-2206 in head and neck cancer patients
Compound |
Combination |
Phase |
State |
NCT number |
|---|---|---|---|---|
MK-2206 |
/ |
2 |
Completed |
NCT01349933 |
/ |
2 |
Completed |
NCT01370070 |
NCT: Number clinicaltrials.gov identifier.
mTOR inhibitors
Other possibilities to attack this pathway are by focusing on the central mTOR complex. Most mTOR inhibitors are derived from Rapamycin, also known as rapalogues, for example Temsirolimus and Everolimus. Rapalogues bind to mTORC1 and stereotacticly interrupt the ability to signal to the downstream effectors [53, 54]. Temsirolimus is already approved by the FDA to treat renal cell carcinoma. In contrast to its analogue Everolimus, Temsirolimus showed very promising results in a phase 2 trial in HNSCC [58, 74, 75]. In an orthotopic HNSCC model, the combination of Temsirolimus with Cetuximab inhibited the PI3K pathway as well as the MAPK pathway, and showed antiangiogenic effects, leading to almost a complete tumor response [58]. Multiple clinical trials in HNSCC patients with Temsirolimus are completed and indicated a significant inhibition of mTOR (see Table 6) [76]. In a trial with platinum- and Cetuximab-recurrent HNSCC patients, Temsirolimus treatment induced stabilization in 58% of the patients and 39% showed tumor shrinkage. However, combination treatment with Temsirolimus can be limited due to numerous toxicities and overall the therapeutic benefit was unsatisfactory [54, 66, 77].
Table 6: Clinical trials with Temsirolimus in head and neck cancer patients
Compound |
Combination |
Phase |
State |
NCT number |
|---|---|---|---|---|
Temsirolimus |
Paclitaxel + Carboplatin |
1/2 |
Not yet recruiting |
NCT01016769 |
/ |
2 |
Completed |
NCT01172769 |
|
/ |
unknown |
Completed |
NCT00195299 |
|
Cetuximab |
2 |
Completed |
NCT01256385 |
|
Erlotinib |
2 |
Completed |
NCT01009203 |
|
Cetuximab + Cisplatin + RT |
Pilot |
Withdrawn |
NCT01326468 |
|
Cetuximab + Cisplatin |
1/2 |
Terminated |
NCT01015664 |
RT: Radiotherapy, NCT: Number clinicaltrials.gov identifier.
The effect of mTOR inhibition could even be broadened via dual pan-class 1 PI3K-mTOR inhibitors that are based on the structural similarities of the catalytic subunit of mTOR and the p110 subunit of PI3K. Thanks to this, the pathway is targeted at two levels. Examples of dual inhibitors are SF1126, PF-05212384, BEZ235, GSK1059615 and XL765. These dual inhibitors all showed radiosensitizing effects in HNSCC cells and multiple clinical trials are ongoing in the HNSCC field (see Table 7) [54, 59, 78–81]. These dual inhibitors are not discussed in detail as this is beyond the scope of this review, but can be explored in the following articles [79, 82].
Table 7: Clinical trials with dual PI3K-mTOR inhibitors in head and neck cancer patients
Compound |
Combination |
Phase |
State |
NCT number |
|---|---|---|---|---|
PF-05212384 |
PD-0332991 |
1 |
Recruiting |
NCT03065062 |
Paclitaxel, Carboplatin |
1 |
Recruiting |
NCT02069158 |
|
SF1126 |
/ |
2 |
Recruiting |
NCT02644122 |
BEZ235 |
/ |
1 |
Completed |
NCT00620594 |
NCT: Number clinicaltrials.gov identifier.
Biomarkers
To assess the level of inhibition of the PI3K pathway, the phosphorylation level of AKT or its downstream effectors gives a good indication [61, 79]. The phosphorylation status of AKT was significantly associated with the sensitivity for the dual PI3K-mTOR inhibitor PF-05212384 in HNSCC cells and can predict resistance to standard HNSCC therapies like Cetuximab and RT, regardless the PIK3CA status. Even better, this resistance can be overcome by combination of mTOR-PI3K and MEK inhibition therapy [57]. The major drawbacks to use AKT phosphorylation level as biomarker are the strict handling and sampling conditions of the tissue, which makes implementation in clinical trials difficult [59, 61].
Preclinical studies indicated that tumors with PIK3CA mutations were more sensitive to PI3K inhibition treatment in contrast to PTEN loss which indicated resistance [58, 60]. Early clinical trials have not found a clear correlation between molecular alterations in the PI3K pathway and antitumor effects of the therapy. However tumors with activating mutations in members of the MAPK pathway are potentially resistant to PI3K inhibitors [58, 59]. These conflicting results make the predictive value of PIK3CA or PTEN mutations inconclusive and unable to implement in clinical practice. However, these contrasting findings can be attributed to external factors. First, PI3K alterations can be missed with early detection methods that were based on a limited number of assays or the lack of well-defined thresholds to define PTEN loss for example. Secondly, other alterations in genes such as AKT1/2 encoding AKT, PIK3R1 encoding the regulatory subunit p85α, liver kinase B1 (LKB1) that activates AMPK and neurofibromin 1 (NF1) that encodes the negative regulator of the RAS pathway, can induce sensitivity. Furthermore, PI3K-mutant HNSCC patients could also have coexisting mutations that induce resistance to PI3K inhibitors, such as KRAS mutations. Tumor heterogeneity and incorrect or invalidated assays can also explain the variation. These tumor-based markers should be expanded so that not only genomic aberrations in PIK3CA, PTEN, ATKT1/2, etc. could be targeted, but also a ‘PI3Kness’ status is included that can serve as indicator for all activating alterations. Finally, a recent study concluded that 15% of the mutations in PI3K pathway genes are subclonal rather than truncal. These subclonal driver mutations can explain the uncertain predictive value of PI3K/AKT/mTOR mutations and suggest that the response to PI3K inhibitors should be assessed by the proportion of tumor cells in which the driver mutation is identified [61].
The complexity of the pathway and its feedback loops hypothesizes that a clear prediction of the response via genotype will be difficult. Furthermore, PI3K inhibitors can have a plethora of effects on tumors, going from angiogenesis to immune cells and other environmental interactions. Therefore it is probable that not a single biomarker can predict sensitivity but rather a molecular signature will be required [61].
Future challenges of PI3K inhibitors
PI3K/AKT/mTOR inhibitors can effectively inhibit the PI3K pathway and are well tolerated [59]. Preclinical research with PI3K inhibitors in HNSCC has provided very promising results. Translation to clinical trials was expected to have encouraging results, which is unfortunately not the case. Different explanations could address this discrepancy. As discussed previously, PI3K inhibitors as monotherapy have limited effect due to the compensatory feedback via other pathways like RAS/RAF/MEK/ERK. This can be addressed by combining PI3K inhibitors with DNA damaging therapies or Cetuximab, which results in dramatic growth inhibition via an anti-proliferative effect rather than a pro-death action [80]. Secondly, PI3K inhibition can induce mitochondrial reprogramming that will promote tumor invasion and progression. Furthermore, preclinical research that provides optimal dosing schedules, is scarce although this is very important for clinical trial success. Most of all, inadequate patient selection can affect trial results. The correlation between PI3K pathway mutational status and responsiveness is unclear and currently patients are selected based on prior treatment failure. This lack of patient stratification can have tremendous effects on response to targeted therapies. Evidently there is room for improvement, combining PI3K inhibitors with other DNA damaging therapies or Cetuximab may have clinical benefit regardless of the mutational status [80]. To improve the success of PI3K inhibitors, pharmacological, biological and translational issues must be better identified. Valuable biomarkers, which are necessary to correctly use these targeted agents, need to be determined, but it is likely that this will depend on the tumor type, genotype and the compound. Biomarker-driven clinical trials are necessary to answer these questions together with thorough preclinical research to stratify patients and predict response [54, 61].
ATM and ATR inhibitors
The cell cycle consists of different checkpoints that are activated in case of DNA damage to ensure genomic stability [83]. The G2/M checkpoint is the last major opportunity to prevent that DNA damage is taken into mitosis which otherwise leads to mitotic catastrophe and cell death [84]. ATM and ATR are two critical components of the DDR that sense DNA damage to activate the G2/M cell cycle checkpoint. They are both members of the PIKK family, making it difficult to design specific inhibitors [83]. ATR and ATM phosphorylate CHK1 and CHK2 respectively to eventually arrest the cell cycle enabling DDR. Somatic mutations in ATR and ATM are seen in various frequencies in HNSCC with 4–10% and 1–16% for ATR and ATM respectively [30].
ATM and ATR inhibitors investigated in HNSCC
Three ATR and ATM inhibitors have already been developed: VX-970 and AZD6738 as ATR inhibitor and AZD0156 as ATM inhibitor. VX-970, previously known as VE-822, is the first-in-class ATR inhibitor which has a sensitizing effect to chemotherapeutic drugs that induce replication fork collapse like Cisplatin. VX-970 increased the antitumor activity of Cisplatin in vivo in patient derived xenograft models which is translated to an ongoing phase 1 trial in HPV-negative HNSCC (see Table 8) [19, 51]. A phase 1 trial with VX-970 as monotherapy showed good tolerability. Secondly, AZD6738 is another potent, selective, oral ATR inhibitor that demonstrated radiosensitization in a panel of human cancer cell lines independent of the p53 or BRCA2 status and is currently tested in phase 1 clinical trials as monotherapy or in combination with radiotherapy, chemotherapy or Olaparib [19, 21, 85]. Two clinical trials are currently ongoing with AZD6738 combined with Olaparib in HNSCC and these are investigating possible biomarkers (Table 8).
Table 8: Clinical trials with ATR inhibitors in HNSCC patients
Compound |
Combination |
Phase |
State |
NCT number |
|---|---|---|---|---|
AZD6738 |
Olaparib, Carboplatin or MED14736 |
1 |
Recruiting |
NCT02264678 |
Olaparib |
1 |
Recruiting |
NCT03022409 |
|
VX-970 |
Cisplatin, RT |
1 |
Recruiting |
NCT02567422 |
RT: Radiotherapy, NCT: Number clinicaltrials.gov identifier.
Biomarkers
Currently, biomarkers in ATM and ATR inhibitors are limited. Alterations in ATM/ATR and DDR deficiencies are suggested to increase reliance to cell cycle checkpoints. Also alterations that cause increased replication stress can be possible biomarkers, such as p53 mutations, Cyclin E upregulation and RAS or MYC mutations. A more advanced biomarker is alternative lengthening of telomeres (ALT), together with loss of ATP-dependent helicase (ATRX), as the loss of ATRX results in increased ALT. Therefore, the loss of ATRX and the increase in ALT may predict the response to ATR inhibitors [19]. In a study in HNSCC combining ATR and CHK1 inhibitors evoked distal loss of 11q as a possible biomarker [86]. Since research with ATM inhibitors is confined, the only suggested biomarker is H2AX foci as for other DNA repair inhibitors.
Future challenges with ATR/ATM inhibitors
ATR inhibitors VX-970 and AZD6738 showed promising preclinical results which led to ongoing clinical trials in combination with other antitumor agents. Unfortunately, with the AZD6738 inhibitor it is a tradeoff between antitumor activity and bone marrow toxicity [21]. These toxicity issues should be addressed by thorough preclinical research focused on optimal scheduling of the combined agents. To select proper HNSCC patients, the above suggested biomarkers must be validated so that profound clinical trials can be designed in the future. Furthermore, ATR inhibition activates backup pathways via DNA-PK and CHK1 as the ATR-CHK1 axis is not linear [84]. This could be circumvented by combining ATR inhibition with DNA-PK or CHK1 inhibition, which is a possible novel strategy in HNSCC research.
Cell cycle checkpoint targeted molecules
As mentioned before, the targets of ATR and ATM are the kinases CHK 1 and CHK2. After ATR activated CHK1, this will then phosphorylate CDC25. Additionally, CHK1 also activates WEE1 to phosphorylate the cyclin dependent 1 (CDK1)/CyclinB complex. The phosphorylation of CDK1 will activate the G2 cell cycle checkpoint and arrests the cell cycle so that the DNA damage can be repaired. This is normally prevented by the phosphatase CDC25 that dephosphorylates CDK1 to deactivate the checkpoint resulting in continuation of the cell cycle. The key components of the G2/M checkpoint, being CHK 1&2, WEE1 and CDC25, are interesting targets for cancer therapy as the activation of this checkpoint leads to DNA repair [6, 8, 22, 87].
CHK1/2 inhibitors
As described above, CHK1 mediates the S- and G2 phase checkpoint and its function is important in DNA replication, cell cycle progression and survival [22, 40]. Furthermore, CHK1 promotes the recruitment of RAD51, involved in HR, to repair the DNA damage in the S phase [22, 83]. CHK2 promotes the G1 checkpoint regulated by p53. When p53 is mutated, the G1 checkpoint is abrogated. Therefore, p53-mutant cells rely more on the G2 checkpoint. Tp53 is mutated in 85% of HNSCC and additionally HPV causes the deregulation of p53 and retinoblastoma (pRb) via oncoproteins E6 and E7 [88, 89]. These cells are no longer protected by the G1 checkpoint, so when the S-G2 checkpoint would be abrogated by the inhibition of CHK1 and/or WEE1, the cell cycle would proceed despite the presence of DNA-damage which would lead to mitotic catastrophe and cell death [8]. Likewise, it would enhance apoptosis induced by DNA damaging agents. In contrast, p53-potent cells are protected by the intact G1 checkpoint which will stop the cell cycle and induce NHEJ [22]. Several studies have proven the sensitizing effect of CHK1/2 inhibitors to DNA damaging agents in p53-deficient cells [3, 6, 88]. The greatest potentiating effect was seen with antimetabolites like Gemcitabine, as antimetabolites temporarily redistribute cells into the S phase. This synchronization of cells maximizes the effect of CHK1 inhibitors on radiation-induced DSBs [22].
Different CHK1/2 inhibitors are already developed with UNC-01 and AZD7762 being the first that proved synergism with DNA damaging agents in humans [15]. However, lack of efficacy, pharmacokinetic and toxicity issues were revealed in early clinical trials [40, 45, 87]. LY2606368 is a strong ATP-competitive CHK1/2 inhibitor, being the improved analogue of LY2603618 that showed cardiotoxicity in phase 1 trial [40]. LY2606368 showed growth inhibition, but the poor oral bioavailability and intravenous administration are important disadvantages [84]. Next, MK-8776 also known as SCH-900776, showed good in vitro results and phase 1 trials combining MK-8776 with Gemcitabine showed clinical efficacy and moderate tolerability [21]. Other CHK1 inhibitors that recently entered clinical trials are: PF0477736, CCT245737, V158411, GDC-0425 and GDC-0575 [19, 21, 40, 83, 84].
CHK1/2 inhibitors investigated in HNSCC
Preclinical studies in HNSCC confirmed the radiosensitizing effect of CHK1 inhibitors [22, 23, 87]. When PF0477736, AZD7762 or SAR-020106 is administered to p53-deficient cells, G2 arrest is abrogated which results in radiosensitization [90]. CHK1 inhibitors can be combined with PARP inhibitors to further enhance the radiosensitizing effect in HPV-positive cells [23]. Combining CHK1/2 inhibitors with Cetuximab and RT results in a significant delay of in vivo tumor growth without increased toxicity [91]. These promising effects of CHK1 inhibitors resulted in numerous clinical trials that are conducted in solid tumors with LY2603618, AZD7762 and CCT245737 as monotherapy or in combination with other DNA damaging therapies [11, 45]. Focusing on HNSCC, two clinical trials are ongoing with CHK1 inhibitors. LY2606368, or Prexasertib, is investigated as monotherapy in a completed phase 1 trial (NCT01115790) and is combined with Cisplatin and Cetuximab in an ongoing phase 1 trial in advanced HNSCC (NCT02555644) (see Table 9).
Table 9: Clinical trials with cell cycle checkpoint inhibitors in HNSCC
Class |
Compound |
Combination |
Phase |
State |
NCT number |
|---|---|---|---|---|---|
CHK1/2 inhibitor |
LY2606368 |
/ |
1 |
Completed |
NCT01115790 |
Cisplatin, RT, Cetuximab |
1 |
Recruiting |
NCT02555644 |
||
WEE1 inhibitor |
AZD1775 |
Cisplatin, RT |
1 |
Recruiting |
NCT02585973 |
Cisplatin, RT |
1 |
Not yet recruiting |
NCT03028766 |
||
Cisplatin, Docetaxel, Surgery |
1 |
Recruiting |
NCT02508246 |
||
Cisplatin |
2 |
Terminated |
NCT02196168 |
RT: Radiotherapy, NCT: Number clinicaltrials.gov identifier.
Biomarkers
CHK1 overexpression is the most suggested biomarker for CHK1 inhibition associated with sensitivity [19, 40, 83]. Some clinical trials use p53 status together with pCDK1/2 status as CHK1 pathway biomarker [92]. Other studies suggest that high levels of DNA-PK may be a potential biomarker to stratify patients to CHK1 inhibitor therapy, as cells deficient in DNA-PK were resistant to CHK1 inhibition [93].
Future challenges of CHK1/2 inhibitors
CHK1 inhibitors showed radiosensitizing effects in p53-mutant and HPV-positive HNSCC cells. The combination of CHK1 inhibitors with Cetuximab and radiation induced significant tumor growth delay, which led to four ongoing clinical trials. However, the caveat when combining CHK1/2 inhibitors with DNA damaging agents is the schedule of administration as CHK1 is especially important for the stability of stalled replication forks, often caused by chemotherapy [15]. Chemotherapy should be given first, so that the cells accumulate in the S phase, then CHK inhibitors can be given and thereafter radiotherapy to maximize the effect [22].
WEE1 inhibitors
WEE1 is responsible for the inhibitory phosphate on CDK1 and G2/M checkpoint activation [11]. Inhibition of WEE1 results in the abrogation of the G2 checkpoint and premature enter of mitosis [84, 94]. As with CHK1 inhibitors, p53-proficient cells are protected from G2 checkpoint abrogation due to the intact G1 checkpoint. The first-in-class small molecule WEE1 inhibitor AZD1775, also known as MK-1775, has shown to potentiate multiple chemo- and radiotherapies [11]. The IC50 of AZD1775 is 20 times lower in p53-mutated HNSCC cells than wildtype. Combining WEE1 inhibitors with DNA damaging agents has proven to be a rational strategy. When AZD1775 is combined with RT in HPV-positive cells, an increase in cell death is seen regardless the p53 status [88]. In combination with chemotherapy, this induces mitotic catastrophe and senescence in HPV-negative cells that are p53-mutated, whereas in HPV-positive cells it increases apoptosis [95].
Inhibition of WEE1 evokes an upregulation of CHK1 due to S phase accumulation and replication stress. This feedback mechanism reduces the efficacy of WEE1 inhibition, by combining CHK1 and WEE1 inhibitors this effect can be antagonized. Different studies have proven the synergistic effects of CHK1 and WEE1 inhibitors resulting in tumor growth reduction regardless the p53 status [88, 96]. Combining AZD1775 and LY2603618 showed more efficient radiosensitization with lower drug concentrations in HPV-positive cells while sparing normal cells [87]. Adding PARP inhibitors to the combination of WEE1 and CHK1 inhibitors increases the sensitivity to RT even further and is translated to a phase 2 clinical trial (NCT02576444) [28]. CHK1 and WEE1 inhibitors combined with RT and chemotherapy induced high levels of sensitivity due to G2 checkpoint and HR abrogation [22].
The promising chemo- and radiopotentiating effect of WEE1 inhibitors in HNSCC cells are translated to four ongoing clinical trials. Here, Cisplatin and RT are combined with AZD1775 while two other clinical trials are performing laboratory biomarker analyses next to the effect of the treatment (Table 9). Preliminary clinical trial results show good tolerability and minimal collateral cytotoxicity [95].
Biomarkers
As indicated above, the status of p53 as biomarker remains a matter of active debate. The phase 1 trial that combined AZD1775 with chemotherapy showed superior results in p53-mutants in contrast to the phase 2 trial in non-small cell lung cancer that observed no association between p53 status and response [97, 98]. Further research is necessary to unravel the significance of the p53 mutational status on CHK1 and WEE1 inhibitors.
Possible biomarkers for combining AZD1775 with DNA damaging agents are not known yet. However, different studies are suggesting some markers like WEE1 and PAX-interacting protein 1 (PAXIP1) levels as mechanism-based biomarkers. PAXIP1 is a PAX protein essential for cells to progress through mitosis and regulates WEE1 activity [99]. Together with low PKMYT1 expression, a kinase functionally related to WEE1, this could serve as an enrichment biomarker for AZD1775 sensitivity [100]. Another study suggested WEE1 inhibition gene signature as pharmacodynamic biomarker based on mRNA expression in tissue biopsies [101]. According to a very recent study focused on the use of mitotic inhibitors in HNSCC, mutations in AJUBA, SMAD4 and RAS predict the sensitivity to CHK1 and WEE1 inhibitors [102]. However, these suggested biomarkers are not validated yet, nor implemented in clinical trials.
Partial response of a head and neck cancer patient with a BRCA1 mutation was seen in a phase 1 study, indicating that DNA repair pathways may be a therapeutic target in HNSCC patient selection [30]. A large-scale study in 59 HNSCC cell lines suggested that insulin receptor substrate 4 (IRS4) and SMAD4 mutations could predict sensitivity to CHK1/2 and WEE1 targeted agents [102]. As described in this article, HNSCC cell lines have a wide range of sensitivity indicating the need for thorough preclinical research to enable in vivo translation. Personalized treatment will be necessary for such specialized treatment strategies, therefore biomarkers are urgently awaited.
Future challenges of WEE1 inhibitors
The combination of adduct-forming chemotherapies and radiotherapy with WEE1 inhibition is a reasonable strategy that should be further investigated in the future. For now the focus was laid on the combination with antimetabolites seen the synchronization of cells in the S phase, where WEE1 is most active. The combination schedule of CHK1 and WEE1 inhibitors with chemo- and radiotherapy is based on early preclinical studies, where a logical order of administration is the inhibitor given after chemotherapy but just before RT so that early repair is inhibited and sustained till extended time thereafter so that late repair and G2-S checkpoints are inhibited [22].
WEE1 inhibition forces the cells through the replication cycle resulting in genomic instability, apoptosis and mitotic catastrophe. A concern therefore is that cancer cells that survived this forced mitosis would increase their proliferation rate and become resistant to chemotherapy, as was seen in proliferation assays. Another important aspect for future research is to determine off-target effects of AZD1775 and improve the selectivity of WEE1 inhibitors [103].
Furthermore, CHK1 inhibitors combined with WEE1 inhibitors showed synergistic effects to result in growth reduction in HNSCC cells regardless the p53 status. Preclinical studies that investigate biomarkers are restricted, luckily ongoing clinical trials are making an attempt to do laboratory biomarkers analyses. However, biomarkers for the discussed inhibitors are unclear and invalidated, despite the fact that they are needed for appropriate patient selection. Another critical point in developing reasonable clinical trials, is the administration schedule. This should be based on the mechanism of action and must be tested in preclinical studies first.
Conclusion
Combining DDR inhibitors with DNA damaging agents can overcome cancer defense mechanisms and improve therapy significantly. A plethora of preclinical studies proved the effect of DDR inhibitors in HNSCC. This review indicates that there is great potential for DNA repair targeted agents combined with antiproliferative therapies in HNSCC. This potential together with the high need for novel targeted therapies to improve the treatment of HNSCC stresses the need for good preclinical research to support rational clinical trial design.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
REFERENCES
1. Bann DV, Deschler DG, Goyal N. Novel immunotherapeutic approaches for head and neck squamous cell carcinoma. Cancers (Basel). 2016; 8:1–20. https://doi.org/10.3390/cancers8100087.
2. Li H, Wawrose JS, Gooding WE, Garraway LA, Lui VW, Peyser ND, Grandis JR. Genomic analysis of head and neck squamous cell carcinoma cell lines and human tumors: a rational approach to preclinical model selection. Mol Cancer Res. 2014; 12:571–82. https://doi.org/10.1158/1541-7786.MCR-13-0396.
3. Suh Y, Amelio I, Guerrero Urbano T, Tavassoli M. Clinical update on cancer: molecular oncology of head and neck cancer. Cell Death Dis. 2014; 5:e1018. https://doi.org/10.1038/cddis.2013.548.
4. Kalu NN, Johnson FM. Do CDK4/6 inhibitors have potential as targeted therapeutics for squamous cell cancers? Expert Opin Investig Drugs. 2017; 26: 207-17. https://doi.org/10.1080/13543784.2017.1274731.
5. Sharma RA, Plummer R, Stock JK, Greenhalgh TA, Ataman O, Kelly S, Clay R, Adams RA, Baird RD, Billingham L, Brown SR, Buckland S, Bulbeck H, et al, and NCRI CTRad Academia-Pharma Joint Working Group. Clinical development of new drug-radiotherapy combinations. Nat Rev Clin Oncol. 2016; 13:627–42. https://doi.org/10.1038/nrclinonc.2016.79.
6. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–78. https://doi.org/10.1038/nature08467.
7. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008; 8:193–204. https://doi.org/10.1038/nrc2342.
8. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011; 11:239–53. https://doi.org/10.1038/nrc3007.
9. Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014; 30:326–39. https://doi.org/10.1016/j.tig.2014.06.003.
10. Wagner LM. Profile of veliparib and its potential in the treatment of solid tumors. Onco Targets Ther. 2015; 8:1931–39. https://doi.org/10.2147/OTT.S69935.
11. Cerrato A, Morra F, Celetti A, Hoeijmakers J, Friedberg E, Aguilera A, Gellert M, Hanawalt P, Hays J, Lehmann A, Lindahl T, Lowndes N, Sarasin A, et al. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: the rationale for their inclusion in the clinic. J Exp Clin Cancer Res. 2016; 35:179. https://doi.org/10.1186/s13046-016-0456-2.
12. Cseh AM, Fábián Z, Sümegi B, Scorrano L. Poly(adenosine diphosphate-ribose) polymerase as therapeutic target: lessons learned from its inhibitors. Oncotarget. 2017; 8:50221–39. https://doi.org/10.18632/oncotarget.16859.
13. Rajawat J, Shukla N, Mishra DP. Therapeutic Targeting of Poly(ADP-Ribose) Polymerase-1 (PARP1) in Cancer: Current Developments, Therapeutic Strategies, and Future Opportunities. Med Res Rev. 2017 May 16. https://doi.org/10.1002/med.21442. [Epub ahead of print]
14. Tangutoori S, Baldwin P, Sridhar S. PARP inhibitors: A new era of targeted therapy. Maturitas. 2015; 81:5–9. https://doi.org/10.1016/j.maturitas.2015.01.015.
15. Shaheen M, Allen C, Nickoloff JA, Hromas R. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood. 2011; 117:6074–82. https://doi.org/10.1182/blood-2011-01-313734.
16. Helleday T, Bryant HE, Schultz N. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle. 2005; 4:1176–78. https://doi.org/10.4161/cc.4.9.2031.
17. Group NP. FDA approves PARP inhibitor for ovarian cancer. Nat Biotechnol. 2017; 35:398. https://doi.org/10.1038/nbt0517-398.
18. Wurster S, Hennes F, Parplys AC, Seelbach JI, Mansour WY, Zielinski A, Petersen C, Clauditz TS, Münscher A, Friedl AA, Borgmann K. PARP1 inhibition radiosensitizes HNSCC cells deficient in homologous recombination by disabling the DNA replication fork elongation response. Oncotarget. 2016; 7:9732–41. https://doi.org/10.18632/oncotarget.6947.
19. Stover EH, Konstantinopoulos PA, Matulonis UA, Swisher EM. Biomarkers of Response and Resistance to DNA Repair Targeted Therapies. Clin Cancer Res. 2016; 22: 5651-60. https://doi.org/10.1158/1078-0432.CCR-16-0247.
20. McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006; 66:8109–15. https://doi.org/10.1158/0008-5472.CAN-06-0140.
21. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA Repair in Cancer: beyond PARP Inhibitors. Cancer Discov. 2017; 7:20–37. https://doi.org/10.1158/2159-8290.CD-16-0860.
22. Morgan MA, Parsels LA, Maybaum J, Lawrence TS. Improving the efficacy of chemoradiation with targeted agents. Cancer Discov. 2014; 4:280–91. https://doi.org/10.1158/2159-8290.CD-13-0337.
23. Güster JD, Weissleder SV, Busch CJ, Kriegs M, Petersen C, Knecht R, Dikomey E, Rieckmann T. The inhibition of PARP but not EGFR results in the radiosensitization of HPV/p16-positive HNSCC cell lines. Radiother Oncol. 2014; 113:345–51. https://doi.org/10.1016/j.radonc.2014.10.011.
24. Verhagen CV, de Haan R, Hageman F, Oostendorp TP, Carli AL, O’Connor MJ, Jonkers J, Verheij M, van den Brekel MW, Vens C. Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother Oncol. 2015; 116:358–65. https://doi.org/10.1016/j.radonc.2015.03.028.
25. De Haan R, Verhagen CV, Hageman F, Oostendorp T, Di Carli A, O’Connor MJ, Jonkers J, Van Triest B, Van den Brekel MW, Verheij M, Vens C. OC-0617: the PARP inhibitor olaparib is effective as radiosensitizer at 10-fold lower doses than as single agent. Radiother Oncol. 2015; 115:S298–99. https://doi.org/10.1016/S0167-8140(15)40609-7.
26. Weaver AN, Cooper TS, Rodriguez M, Trummell HQ, Bonner JA, Rosenthal EL, Yang ES. DNA double strand break repair defect and sensitivity to poly ADP-ribose polymerase (PARP) inhibition in human papillomavirus 16-positive head and neck squamous cell carcinoma. Oncotarget. 2015; 6:26995–7007. https://doi.org/10.18632/oncotarget.4863.
27. Dok R, Kalev P, Van Limbergen EJ, Asbagh LA, Vázquez I, Hauben E, Sablina A, Nuyts S. p16INK4a impairs homologous recombination-mediated DNA repair in human papillomavirus-positive head and neck tumors. Cancer Res. 2014; 74:1739–51. https://doi.org/10.1158/0008-5472.CAN-13-2479.
28. Morgan MA, Lawrence TS. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin Cancer Res. 2015; 21:2898–904. https://doi.org/10.1158/1078-0432.CCR-13-3229.
29. Yasukawa M, Fujihara H, Fujimori H, Kawaguchi K, Yamada H, Nakayama R, Yamamoto N, Kishi Y, Hamada Y, Masutani M. Synergetic effects of PARP inhibitor AZD2281 and cisplatin in oral squamous cell carcinoma in vitro and in vivo. Int J Mol Sci. 2016; 17:272. https://doi.org/10.3390/ijms17030272.
30. Aung KL, Siu LL. Genomically personalized therapy in head and neck cancer. Cancers Head Neck. BioMed Central. 2016; 1:1–10. https://doi.org/10.1186/s41199-016-0004-y.
31. Heitmann J, Geeleher P, Zuo Z, Weichselbaum RR, Vokes EE, Fetscher S, Seiwert TY. Poly (ADP-ribose) polymerase inhibitor efficacy in head and neck cancer. Oral Oncol. 2014; 50:825–31. https://doi.org/10.1016/j.oraloncology.2014.06.004.
32. Chalmers AJ, Lakshman M, Chan N, Bristow RG. Poly(ADP-Ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin Radiat Oncol. 2010; 20: 274–81. https://doi.org/10.1016/j.semradonc.2010.06.001.
33. Gachechiladze M, Škarda J, Soltermann A, Joerger M. RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies. Int J Cancer. 2017; 141:1286–94. https://doi.org/10.1002/ijc.30764.
34. Talens F, Jalving M, Gietema JA, Van Vugt MA. Therapeutic targeting and patient selection for cancers with homologous recombination defects. Expert Opin Drug Discov. 2017; 12:565–81. https://doi.org/10.1080/17460441.2017.1322061.
35. Yang Z, Liao XM, Chen Y, Shen YY, Yang XY, Su Y, Sun YM, Gao YL, Ding J, Zhang A, He JX, Miao ZH. Combining 53BP1 with BRCA1 as a biomarker to predict the sensitivity of poly(ADP-ribose) polymerase (PARP) inhibitors. Acta Pharmacol Sin. 2017; 38:1038-1047. https://doi.org/10.1038/aps.2017.8.
36. del Rivero J, Kohn EC. PARP Inhibitors: The Cornerstone of DNA Repair-Targeted Therapies. Oncology (Williston Park). 2017; 31:265–73.
37. Konecny GE, Kristeleit RS. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions. Br J Cancer. 2016; 115:1157–73. https://doi.org/10.1038/bjc.2016.311.
38. Nickson CM, Moori P, Carter RJ, Rubbi CP, Parsons JL. Misregulation of DNA damage repair pathways in HPV-positive head and neck squamous cell carcinoma contributes to cellular radiosensitivity. Oncotarget. 2017; 8:29963–75. https://doi.org/10.18632/oncotarget.16265.
39. Lempiäinen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009; 28:3067–73. https://doi.org/10.1038/emboj.2009.281.
40. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, Turchi JJ. DNA repair targeted therapy: the past or future of cancer treatment? Pharmacol Ther. 2016; 160:65–83. https://doi.org/10.1016/j.pharmthera.2016.02.003.
41. Yu L, Shang ZF, Hsu FM, Zhang Z, Tumati V, Lin YF, Chen BP, Saha D. NSCLC cells demonstrate differential mode of cell death in response to the combined treatment of radiation and a DNA-PKcs inhibitor. Oncotarget. 2015; 6:3848–60. https://doi.org/10.18632/oncotarget.2975.
42. Goodwin JF, Knudsen KE. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014; 4:1126–39. https://doi.org/10.1158/2159-8290.CD-14-0358.
43. Mathews LA, Cabarcas SM, Hurt EM, Editors. DNA Repair of Cancer Stem Cells. Dordrecht, Netherlands: Springer, Dordrecht; 2013. 180 p.
44. Davidson D, Amrein L, Panasci L, Aloyz R. Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front Pharmacol. 2013; 4:5. https://doi.org/10.3389/fphar.2013.00005.
45. Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014; 105:370–88. https://doi.org/10.1111/cas.12366.
46. Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, Calvert AH, Newell DR, Smith GC, Curtin NJ. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006; 66:5354–62. https://doi.org/10.1158/0008-5472.CAN-05-4275.
47. Sunada S, Kanai H, Lee Y, Yasuda T, Hirakawa H, Liu C, Fujimori A, Uesaka M, Okayasu R. Nontoxic concentration of DNA-PK inhibitor NU7441 radio-sensitizes lung tumor cells with little effect on double strand break repair. Cancer Sci. 2016; 107:1250–55. https://doi.org/10.1111/cas.12998.
48. Yang C, Wang Q, Liu X, Cheng X, Jiang X, Zhang Y, Feng Z, Zhou P. NU7441 Enhances the Radiosensitivity of Liver Cancer Cells. Cell Physiol Biochem. 2016; 38:1897–905. https://doi.org/10.1159/000445551.
49. Ciszewski WM, Tavecchio M, Dastych J, Curtin NJ. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res Treat. 2014; 143:47–55. https://doi.org/10.1007/s10549-013-2785-6.
50. Nowsheen S, Bonner JA, Lobuglio AF, Trummell H, Whitley AC, Dobelbower MC, Yang ES. Cetuximab augments cytotoxicity with poly (adp-ribose) polymerase inhibition in head and neck cancer. PLoS One. 2011; 6:e24148. https://doi.org/10.1371/journal.pone.0024148.
51. Yamamoto VN, Thylur DS, Bauschard M, Schmale I, Sinha UK. Overcoming radioresistance in head and neck squamous cell carcinoma. Oral Oncol. 2016; 63:44–51. https://doi.org/10.1016/j.oraloncology.2016.11.002.
52. Cowell IG, Durkacz BW, Tilby MJ. Sensitization of breast carcinoma cells to ionizing radiation by small molecule inhibitors of DNA-dependent protein kinase and ataxia telangiectsia mutated. Biochem Pharmacol. 2005; 71:13–20. https://doi.org/10.1016/j.bcp.2005.09.029.
53. Holmes D. PI3K pathway inhibitors approach junction. Nat Rev Drug Discov. 2011; 10:563–64. https://doi.org/10.1038/nrd3527.
54. Cai Y, Dodhia S, Su GH. Dysregulations in the PI3K pathway and targeted therapies for head and neck squamous cell carcinoma. Oncotarget. 2017; 8:22203–17. https://doi.org/10.18632/oncotarget.14729.
55. Simpson DR, Mell LK, Cohen EE. Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2015; 51:291–98. https://doi.org/10.1016/j.oraloncology.2014.11.012.
56. Wang CC, Cirit M, Haugh JM. PI3K-dependent cross-talk interactions converge with Ras as quantifiable inputs integrated by Erk. Mol Syst Biol. 2009; 5:246. https://doi.org/10.1038/msb.2009.4.
57. Mohan S, Vander Broek R, Shah S, Eytan DF, Pierce ML, Carlson SG, Coupar JF, Zhang J, Cheng H, Chen Z, Van Waes C. MEK Inhibitor PD-0325901 overcomes resistance to PI3K/mTOR inhibitor PF-5212384 and potentiates antitumor effects in human head and neck squamous cell carcinoma. Clin Cancer Res. 2015; 21:3946–56. https://doi.org/10.1158/1078-0432.CCR-14-3377.
58. Bozec A, Ebran N, Radosevic-Robin N, Sudaka A, Monteverde M, Toussan N, Etienne-Grimaldi MC, Nigro CL, Merlano M, Penault-Llorca F, Milano G. Combination of mTOR and EGFR targeting in an orthotopic xenograft model of head and neck cancer. Laryngoscope. 2016; 126:E156–63. https://doi.org/10.1002/lary.25754.
59. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013; 10:143–53. https://doi.org/10.1038/nrclinonc.2013.10.
60. Michmerhuizen NL, Leonard E, Kulkarni A, Brenner JC. Differential compensation mechanisms define resistance to PI3K inhibitors in PIK3CA amplified HNSCC. Med Image Anal. 2016; 116:1477–90. https://doi.org/10.1161/CIRCRESAHA.116.303790.
61. Josephs DH, Sarker D. Pharmacodynamic biomarker development for PI3K pathway therapeutics. Transl Oncogenomics. 2016 (Suppl 1); 7:33–49. https://doi.org/10.4137/TOG.S30529.
62. Smolensky D, Rathore K, Bourn J, Cekanova M. Inhibition of the PI3K/AKT Pathway Sensitizes Oral Squamous Cell Carcinoma Cells to Anthracycline-Based Chemotherapy In Vitro. J Cell Biochem. 2017; 118:2615–24. https://doi.org/10.1002/jcb.25747.
63. Zimmerman PM, Zhang Z, Musgrave CB. Singlet fission in pentacene through multi-exciton quantum states. Nat Chem. 2010; 2:648-652. doi: 10.1038/nchem.694.
64. Estévez LG, García E, Hidalgo M. Inhibiting the PI3K signaling pathway: buparlisib as a new targeted option in breast carcinoma. Clin Transl Oncol. 2016; 18:541-9. https://doi.org/10.1007/s12094-015-1410-z.
65. Lattanzio L, Tonissi F, Monteverde M, Vivenza D, Russi E, Milano G, Merlano M, Lo Nigro C. Treatment effect of buparlisib, cetuximab and irradiation in wild-type or PI3KCA-mutated head and neck cancer cell lines. Invest New Drugs. 2015; 33:310–20. https://doi.org/10.1007/s10637-015-0210-1.
66. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014; 13:140–56. https://doi.org/10.1038/nrd4204.
67. Echarri MJ, Lopez-Martin A, Hitt R. Targeted therapy in locally advanced and recurrent/metastatic head and neck squamous cell carcinoma (LA-R/M HNSCC). Cancers (Basel). 2016; 8:E27. https://doi.org/10.3390/cancers8030027.
68. Soulières D, Faivre S, Mesía R, Remenár É, Li SH, Karpenko A, Dechaphunkul A, Ochsenreither S, Kiss LA, Lin JC, Nagarkar R, Tamás L, Kim SB, et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017; 18:323–35. https://doi.org/10.1016/S1470-2045(17)30064-5.
69. Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R, Hirawat S. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016; 9:203–10. https://doi.org/10.2147/OTT.S89967.
70. Doi T, Fuse N, Yoshino T, Kojima T, Bando H, Miyamoto H, Kaneko M, Osada M, Ohtsu A. A Phase I study of intravenous PI3K inhibitor copanlisib in Japanese patients with advanced or refractory solid tumors. Cancer Chemother Pharmacol. 2017; 79:89–98. https://doi.org/10.1007/s00280-016-3198-0.
71. Keam B, Kim S, Ahn YO, Kim TM, Lee SH, Kim DW, Heo DS. In vitro anticancer activity of PI3K alpha selective inhibitor BYL719 in head and neck cancer. Anticancer Res. 2015; 35:175–82.
72. Zhao W, Qiu Y, Kong D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm Sin B. 2017; 7:27–37. https://doi.org/10.1016/j.apsb.2016.07.006.
73. Akt Inhibitor MK2206 in Treating Patients With Recurrent or Metastatic Head and Neck Cancer-Study Results-ClinicalTrials. gov [Internet]. [cited 2017 Mar 15]. Available 2017 Mar 15, from https://clinicaltrials.gov/ct2/show/results/NCT01349933.
74. Massarelli E, Lin H, Ginsberg LE, Tran HT, Lee JJ, Canales JR, Williams MD, Blumenschein GR Jr, Lu C, Heymach JV, Kies MS, Papadimitrakopoulou V. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2015; 26:1476-80. https://doi.org/10.1093/annonc/mdv194.
75. Geiger JL, Bauman JE, Gibson MK, Gooding WE, Varadarajan P, Kotsakis A, Martin D, Gutkind JS, Hedberg ML, Grandis JR, Argiris A. Phase II trial of everolimus in patients with previously treated recurrent or metastatic head and neck squamous cell carcinoma. Head Neck. 2016; 38:1759–64. https://doi.org/10.1002/hed.24501.
76. Isaacsson Velho PH, Castro G Jr, Chung CH. Targeting the PI3K Pathway in Head and Neck Squamous Cell Carcinoma. Am Soc Clin Oncol Educ Book. 2015:123-8. https://doi.org/10.14694/EdBook_AM.2015.35.123.
77. Bossi P, Alfieri S. Investigational drugs for head and neck cancer. Expert Opin Investig Drugs. 2016; 25:797–810. https://doi.org/10.1080/13543784.2016.1175435.
78. SF1126 in Recurrent or Progressive SCCHN and Mutations in PIK3CA Gene and/or PI-3 Kinase Pathway Genes. Available 2017 Mar 15, from https://clinicaltrials.gov/ct2/show/NCT02644122?term=SF1126+head&rank=1.
79. Leiker AJ, DeGraff W, Choudhuri R, Sowers AL, Thetford A, Cook JA, Van Waes C, Mitchell JB. Radiation Enhancement of Head and Neck Squamous Cell Carcinoma by the Dual PI3K/mTOR Inhibitor PF-05212384. Clin Cancer Res. 2015; 21:2792–801. https://doi.org/10.1158/1078-0432.CCR-14-3279.
80. Swick AD, Prabakaran PJ, Miller MC, Javaid AM, Fisher MM, Sampene E, Ong IM, Hu R, Iida M, Nickel KP, Bruce JY, Wheeler DL, Kimple RJ. Cotargeting mTORC and EGFR Signaling as a Therapeutic Strategy in HNSCC. Mol Cancer Ther. 2017; 16:1257–1268. https://doi.org/10.1158/1535-7163.MCT-17-0115.
81. Xie J, Li Q, Ding X, Gao Y. GSK1059615 kills head and neck squamous cell carcinoma cells possibly via activating mitochondrial programmed necrosis pathway. Oncotarget. 2017; 8:50814-23. https://doi.org/10.18632/oncotarget.15135.
82. Bian Y, Han J, Kannabiran V, Mohan S, Cheng H, Friedman J, Zhang L, VanWaes C, Chen Z. MEK inhibitor PD-0325901 overcomes resistance to CK2 inhibitor CX-4945 and exhibits anti-tumor activity in head and neck cancer. Int J Biol Sci. 2015; 11:411–22. https://doi.org/10.7150/ijbs.10745.
83. Velic D, Couturier AM, Ferreira MT, Rodrigue A, Poirier GG, Fleury F, Masson JY. DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer. Biomolecules. 2015; 5:3204–59. https://doi.org/10.3390/biom5043204.
84. O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell. 2015; 60:547–60. https://doi.org/10.1016/j.molcel.2015.10.040.
85. Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, Nutting CM, McLaughlin M, Harrington KJ. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther. 2017; 16:25–34. https://doi.org/10.1158/1535-7163.MCT-16-0239.
86. Sankunny M, Parikh RA, Lewis DW, Gooding WE, Saunders WS, Gollin SM. Targeted inhibition of ATR or CHEK1 reverses radioresistance in oral squamous cell carcinoma cells with distal chromosome arm 11q loss. Genes Chromosomes Cancer. 2014; 53:129–43. https://doi.org/10.1002/gcc.22125.
87. Busch CJ, Kröger MS, Jensen J, Kriegs M, Gatzemeier F, Petersen C, Münscher A, Rothkamm K, Rieckmann T. G2-checkpoint targeting and radiosensitization of HPV/p16-positive HNSCC cells through the inhibition of Chk1 and Wee1. Radiother Oncol. 2017; 122:260-266. https://doi.org/10.1016/j.radonc.2016.11.017.
88. Aung KL, Siu LL. Genomically personalized therapy in head and neck cancer. BMC Surg. 2016; 1:1–10. https://doi.org/10.1186/s41199-016-0004-y.
89. Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, Neskey DM, Frederick MJ, Woo SH, Caulin C, Hsu TK, McDonald TO, Kimmel M, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther. 2015; 14:608–19. https://doi.org/10.1158/1535-7163.MCT-14-0735-T.
90. Busch CJ, Kriegs M, Laban S, Tribius S, Knecht R, Petersen C, Dikomey E, Rieckmann T. HPV-positive HNSCC cell lines but not primary human fibroblasts are radiosensitized by the inhibition of Chk1. Radiother Oncol. 2013; 108:495–99. https://doi.org/10.1016/j.radonc.2013.06.035.
91. Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. 2017; 16:591–600. https://doi.org/10.1158/1535-7163.MCT-16-0352.
92. Infante JR, Hollebecque A, Postel-Vinay S, Bauer TM, Blackwood EM, Evangelista M, Mahrus S, Peale FV, Lu X, Sahasranaman S, Zhu R, Chen Y, Ding X, et al. Phase I Study of GDC-0425, a Checkpoint Kinase 1 Inhibitor, in Combination with Gemcitabine in Patients with Refractory Solid Tumors. Clin Cancer Res. 2017; 23:2423–2432.
93. Massey AJ, Stephens P, Rawlinson R, McGurk L, Plummer R, Curtin NJ. mTORC1 and DNA-PKcs as novel molecular determinants of sensitivity to Chk1 inhibition. Mol Oncol. 2016; 10:101–12. https://doi.org/10.1016/j.molonc.2015.08.004.
94. Kao M, Green C, Sidorova J, Méndez E. Strategies for Targeted Therapy in Head and Neck Squamous Cell Carcinoma Using WEE1 Inhibitor AZD1775. JAMA Otolaryngol Head Neck Surg. 2017; 143:631–633. https://doi.org/10.1001/jamaoto.2016.4563.
95. Tanaka N, Patel AA, Wang J, Frederick MJ, Kalu NN, Zhao M, Fitzgerald AL, Xie TX, Silver NL, Caulin C, Zhou G, Skinner HD, Johnson FM, et al. Wee-1 Kinase Inhibition Sensitizes High-Risk HPV+ HNSCC to Apoptosis Accompanied by Downregulation of MCl-1 and XIAP Antiapoptotic Proteins. Clin Cancer Res. 2015; 21:4831–44. https://doi.org/10.1158/1078-0432.CCR-15-0279.
96. Hauge S, Naucke C, Hasvold G, Joel M, Rødland GR, Juzenas P, Stokke T, Syljuåsen RG. Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage in S-phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget. 2017; 8:10966-79. https://doi.org/10.18632/oncotarget.14089.
97. Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, Reigan P, Porter CC. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther. 2013; 12:2675–84. https://doi.org/10.1158/1535-7163.MCT-13-0424.
98. Scagliotti G, Kang JH, Smith D, Rosenberg R, Park K, Kim SW, Su WC, Boyd TE, Richards DA, Novello S, Hynes SM, Myrand SP, Lin J, et al. Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer. Invest New Drugs. 2016; 34:625–35. https://doi.org/10.1007/s10637-016-0368-1.
99. Jhuraney A, Woods NT, Wright G, Rix L, Kinose F, Kroeger JL, Remily-Wood E, Cress WD, Koomen JM, Brantley SG, Gray JE, Haura EB, Rix U, Monteiro AN. PAXIP1 Potentiates the Combination of WEE1 Inhibitor AZD1775 and Platinum Agents in Lung Cancer. Mol Cancer Ther. 2016; 15:1669–81. https://doi.org/10.1158/1535-7163.MCT-15-0182.
100. Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, Hirsch HA, Feldman I, Benita Y, Toniatti C, Zawel L, Fawell SE, Gilliland DG, Shumway SD. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. 2013; 12:1442–52. https://doi.org/10.1158/1535-7163.MCT-13-0025.
101. Mizuarai S, Yamanaka K, Itadani H, Arai T, Nishibata T, Hirai H, Kotani H. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol Cancer. BioMed Central. 2009; 8:1–12. https://doi.org/10.1186/1476-4598-8-34.
102. Zhang M, Singh R, Peng S, Mazumdar T, Sambandam V, Shen L, Tong P, Li L, Kalu NN, Pickering CR, Frederick M, Myers JN, Wang J, Johnson FM. Mutations of the LIM protein AJUBA mediate sensitivity of head and neck squamous cell carcinoma to treatment with cell-cycle inhibitors. Cancer Lett. 2017; 392:71–82. https://doi.org/10.1016/j.canlet.2017.01.024.
103. Matheson CJ, Backos DS, Reigan P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol Sci. 2016; 37:872–81. https://doi.org/10.1016/j.tips.2016.06.006.