
INTRODUCTION
Phosphatase with tensin homology (PTEN) is one of the most frequently inactivated tumor suppressor genes in human cancers [1]. PTEN controls phosphoinositide 3-kinase (PI3K) signaling, which has critical roles in diverse cellular functions [2, 3]. As such, PTEN is involved in cell proliferation and survival, energy metabolism, and cellular architecture [4].
In PCa, loss of PTEN has been consistently associated with more aggressive disease features and a worse prognosis [5–16]. Estimates of PTEN loss range from less than 20 percent for clinically localized prostate tumors to more than 40 percent for metastatic castrate-resistant PCa [14, 17]. PTEN loss also frequently co-occurs with the TMPRSS2:ERG gene fusion, which exists in about half of all localized prostate tumors in men of European ancestry, suggesting that both somatic events might cooperate in prostate tumorigenesis [18, 19]. Loss of PTEN is now recognized as one of the major driving events in PCa [20].
Tumors with PTEN loss have significantly altered gene expression profiles. Saal et al. previously generated a tumor transcriptomic signature of PTEN loss in breast tumors [21]. The signature included 246 genes and the most significant differentially expressed gene was PTEN itself. The PTEN loss-like mRNA expression signature was also strongly associated with PTEN status based on copy number levels in PCa [22]. Further, in independent datasets of breast, prostate, and bladder carcinoma, the signature significantly correlated with worse patient outcomes [21].
Tumor epigenomic changes, in particular changes at the DNA methylation level, may also contribute to the progression of PCa [23]. Several studies have identified individual differentially methylated CpG sites and methylation signatures (combination of CpGs) associated with PCa recurrence and metastatic progression [24–26]. Recent research on PCa also revealed that methylation patterns are more tightly associated with patient outcomes than other genomic characteristics (e.g., copy number alterations, single nucleotide variants) [27].
Therefore, differential DNA methylation profiles in prostate tumors with PTEN loss may help to better understand the mechanisms that drive cancer progression in the absence of PTEN. To gain insights on this issue, we examined the association of PTEN loss with PCa recurrence in a cohort of patients diagnosed with clinically localized disease, and used methylome data from the same cohort to profile tumors with and without PTEN loss. As far as we know, this is the first large study to investigate PTEN status according to prostate cancer recurrence and epigenome-wide changes in tumor DNA methylation profiles.
RESULTS
Patient characteristics
There were 403 patients (85.6%) with PTEN intact tumors, and 47 (10.0%) and 21 (4.5%) patients with hemi- and homozygous PTEN loss, respectively (Table 1). PTEN loss was associated with higher Gleason scores (P = 0.03) and regional pathological stage (P < 0.01) as well as occurrence of the somatic TMPRSS2:ERG gene fusion (P < 0.01). There was no significant association between PTEN loss and race (European-American vs. African American; P = 0.4), but there were only 2 African-American patients with hemizygous PTEN loss and 3 African- American patients with homozygous PTEN loss.
Table 1: Selected patient characteristics by tumor PTEN status in the radical prostatectomy patient cohorta
PTEN status |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Intact (n = 403) |
Hemizygous loss (n = 47) |
Homozygous loss (n = 21) |
||||||||
No. |
% |
Mean (SD) |
No. |
% |
Mean (SD) |
No. |
% |
Mean (SD) |
P-valueb |
|
Age at diagnosis (years) |
58.4 (7.2) |
57.3 (6.6) |
56.2 (7.2) |
0.10 |
||||||
Race |
0.36 |
|||||||||
Caucasian |
367 |
91.1% |
45 |
95.7% |
18 |
85.7% |
||||
African-American |
36 |
8.9% |
2 |
4.3% |
3 |
14.3% |
||||
Pathological stagec |
< 0.01 |
|||||||||
Local |
295 |
73.2% |
31 |
66.0% |
8 |
38.1% |
||||
Regional |
108 |
26.8% |
16 |
34.0% |
13 |
61.9% |
||||
Gleason score |
0.03 |
|||||||||
≤6 |
207 |
51.4% |
21 |
44.7% |
5 |
23.8% |
||||
7(3+4) |
141 |
35.0% |
13 |
27.7% |
12 |
57.1% |
||||
7(4+3) |
30 |
7.4% |
6 |
12.8% |
3 |
14.3% |
||||
8–10 |
25 |
6.2% |
7 |
14.9% |
1 |
4.8% |
||||
PSA at diagnosis (ng/mL) |
0.13 |
|||||||||
0–3.9 |
65 |
17.1% |
8 |
17.0% |
3 |
16.7% |
||||
4–9.9 |
248 |
65.3% |
25 |
53.2% |
11 |
61.1% |
||||
10–19.9 |
49 |
12.9% |
8 |
17.0% |
3 |
16.7% |
||||
≥20 |
18 |
4.7% |
6 |
12.8% |
1 |
5.6% |
||||
TMPRSS2:ERG fusion status |
< 0.01 |
|||||||||
Negative |
192 |
50.3% |
14 |
29.8% |
1 |
4.8% |
||||
Positive |
190 |
49.7% |
33 |
70.2% |
20 |
95.2% |
||||
Recurrence |
0.04 |
|||||||||
No |
256 |
78.8% |
26 |
70.3% |
8 |
53.3% |
||||
Yesd |
69 |
21.2% |
11 |
29.7% |
7 |
46.7% |
||||
a Ninety-three patients had missing data on PTEN status. Patient characteristics were not substantially different for these patients.
b P-value from either a T-test or chi-square test
c Local: pT2, N0/NX, M0; regional: pT3–T4 and/or N1, M0
d Of the patients with PCa recurrence, 17 had metastatic-lethal progression.
PTEN loss and prostate cancer recurrence
The association between PTEN loss and recurrence-free survival was investigated (Figure 1, Table 2). In total, 87 patients developed PCa recurrence during a mean follow-up of 8 years (Table 1). Compared to patients with PTEN intact tumors, those with homozygous PTEN deleted tumors had an increased risk of recurrence (HR = 2.84, 95% CI: 1.30, 6.19). Hemizygous loss was not significantly associated with recurrence. For any versus no PTEN loss, the HR was 1.74 (95% CI: 1.03, 2.93). The median time to recurrence in men with intact PTEN, hemizygous PTEN loss, and homozygous PTEN loss was 7.3, 7.8, and 5.7 years (P = 0.5), respectively.
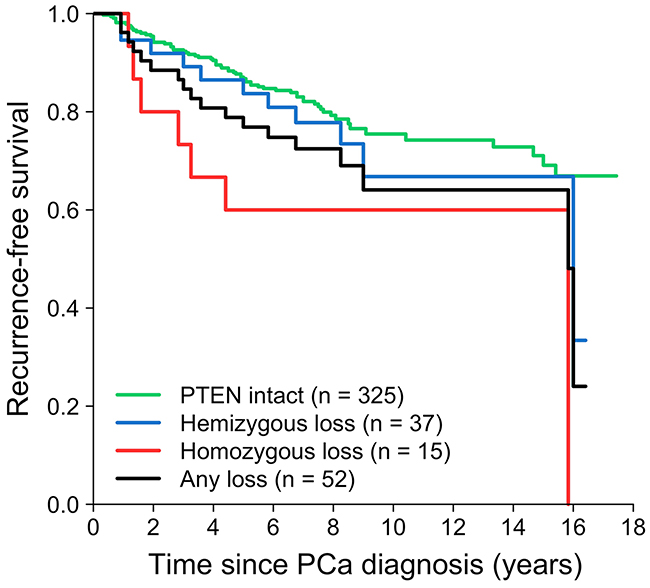
Figure 1: Loss of PTEN in relation to recurrence-free survival in the radical prostatectomy cohort.
Table 2: Age-adjusted hazard ratios and 95% confidence intervals for the association of PTEN loss with prostate cancer recurrence and by selected disease features
Patients |
No. patients |
No. events |
PTEN status |
||||||
|---|---|---|---|---|---|---|---|---|---|
Intact (ref.) |
Hemizygous loss |
Homozygous loss |
Any loss |
||||||
HR |
HR |
(95% CI) |
HR |
(95% CI) |
HR |
(95% CI) |
|||
All |
377 |
87 |
1.00 |
1.39 |
(0.73, 2.64) |
2.84 |
(1.30, 6.19) |
1.74 |
(1.03, 2.93) |
Local pathological stage |
270 |
41 |
1.00 |
1.96 |
(0.86, 4.50) |
4.30 |
(1.31, 14.18) |
2.35 |
(1.15, 4.83) |
Regional pathological stage |
107 |
46 |
1.00 |
0.87 |
(0.31, 2.45) |
1.23 |
(0.43, 3.49) |
1.02 |
(0.47, 2.21) |
Lower Gleason score (≤7) |
318 |
58 |
1.00 |
1.23 |
(0.53, 2.89) |
2.17 |
(0.78, 6.03) |
1.49 |
(0.75, 2.96) |
Higher Gleason score (8–10) |
59 |
29 |
1.00 |
0.80 |
(0.26, 2.46) |
5.95 |
(1.58, 22.50) |
1.28 |
(0.51, 3.21) |
TMPRSS2:ERG fusion-negative |
161 |
37 |
1.00 |
2.26 |
(0.78, 6.54) |
– |
– |
2.05 |
(0.70, 6.00) |
TMPRSS2:ERG fusion-positive |
198 |
45 |
1.00 |
1.20 |
(0.53, 2.72) |
3.26 |
(1.43, 7.45) |
1.75 |
(0.93, 3.29) |
Subgroup analyses revealed significant associations between homozygous PTEN loss and PCa recurrence for patients with local pathological stage (HR = 4.30), higher Gleason score (8–10) tumors (HR = 5.95), and TMPRSS2:ERG fusion-positive tumors (HR = 3.26; Table 2). The association was not studied in the subgroup of patients with TMPRSS2:ERG fusion-negative tumors because only one patient in this subgroup had homozygous PTEN loss.
An ROC analysis revealed that a clinical model based on Gleason score and pathological stage had an AUC for PCa recurrence of 0.72. Loss of PTEN only had an AUC for recurrence of 0.69. After additionally including PTEN loss in the multivariable model with Gleason score and stage, the AUC improved slightly (0.73). Larger AUC improvements were observed in patients with local pathological stage (+3%), and in patients with higher Gleason scores of 8–10 (+4%).
PTEN loss and tumor DNA methylation levels
Tumor epigenome-wide methylation differences between PTEN deleted (any loss) and PTEN intact tumors were studied. In total, 4,208 differentially methylated CpGs were identified (False Discovery Rate [FDR] Q-value < 0.01), of which 1,924 (46%) were hypermethylated in PTEN deleted tumors (Figure 2A). Of the 4,208 differentially methylated CpGs, 485 had a mean methylation difference of more than 10%. Genome-wide methylation levels in homozygous versus hemizygous PTEN deleted tumors were also compared. This analysis, however, revealed no significant differentially methylated CpGs between the two subsets (FDR Q-value = 1). Therefore, further methylation analyses involved contrasting tumors with any PTEN loss versus those with intact PTEN. Figure 2B shows the proportion of differentially methylated CpGs by genomic region, which showed that hypermethylated CpGs were more commonly found in gene promoter regions; and hypomethylated CpGs were more commonly found in gene body and intergenic regions.
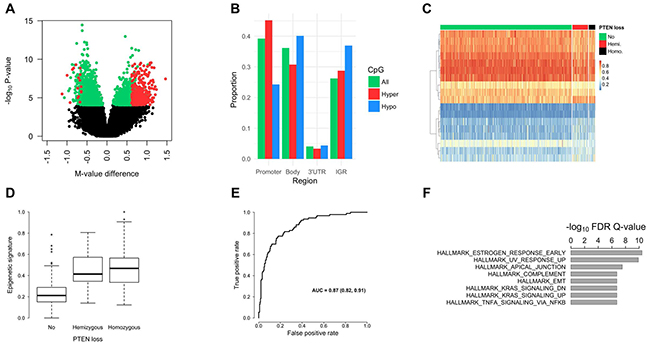
Figure 2: Prostate tumor DNA methylation profiles by PTEN status. (A) Volcano plot for the differential methylation analysis of any PTEN loss versus intact PTEN. Each point in the figure represents a CpG site. Differentially methylated CpGs are shown in green or red (FDR Q-value < 0.01; n = 4,208). The CpGs shown in red have a mean methylation difference (PTEN loss vs. intact PTEN) of more than 10% (n = 485). CpGs with a higher mean methylation level in PTEN deleted tumors (i.e., hypermethylated) have a positive methylation M-value (logit transformation of β-value) difference, and hypomethylated CpGs have a negative M-value difference. (B) Proportion of significantly hypermethylated (red bars) and hypomethylated CpGs (blue bars) by genomic region. As a comparison, the proportion of all measured CpGs (n = 480K) by genomic region is shown (green bars). (C) Heat map (supervised) of the 18 CpG sites selected using Elastic Net in our cohort. This panel of 18 CpGs optimally distinguished PTEN deleted from PTEN intact tumors. The rows of the heatmap are the CpG sites and the columns are the tumor samples, which were grouped based on PTEN status. Methylation β-values (figure legend; range 0−1) were used and the highest methylation levels are shown in red. The number of patients with intact PTEN, hemizygous PTEN loss, and homozygous PTEN loss was 388, 46, and 19, respectively. The rows were clustered based on Euclidean distance. (D) Epigenetic signature of PTEN loss in TCGA. The 18 differentially methylated CpGs, identified in our cohort, were combined into a single epigenetic signature, which was then tested in the TCGA dataset. As expected, tumors with PTEN loss had significantly higher levels of the signature compared to PTEN intact tumors. (E) ROC curve for classifying any PTEN loss versus intact PTEN using the methylation signature in TCGA. Values for the AUC and associated 95% confidence interval are shown in the figure. (F) Top-ranked GSEA hallmark gene sets, which showed enrichment for the genes with differentially methylated CpGs (1,908 genes).
The Elastic Net method was used to identify a panel of CpGs that, in combination, distinguished PTEN deleted from PTEN intact tumors in our cohort. In total, eighteen CpGs were identified (Table 3; Figure 2C). These CpGs were in 13 genes: ATP11A, BAT4, CALD1, CSNK2B, GDNF, GNB1, JAK1, JAM3, RHOBTB1, RNF144A, SEZ6, VAPA, and YPEL3; several of which have previously been implicated in cancer development and progression. The 18 CpGs were combined into a signature, as described in the methods. This signature was tested in the PCa TCGA dataset, which showed that both hemi- and homozygous deleted tumors have significantly higher levels of the signature compared to PTEN intact tumors (Figure 2D; P < 0.001). An ROC analysis showed an AUC of 0.87 for any PTEN loss versus intact PTEN (Figure 2E). The classification performance was similar in patient subsets based on tumor TMPRSS2:ERG fusion status.
Table 3: Eighteen top-ranked CpG sites for classifying prostate tumors with any PTEN loss versus intact PTEN
CpG ID |
Chr. |
Gene name |
Genetic location |
Epigenetic location |
Mean β PTEN intact |
Mean β PTEN deleted |
Mean β difference |
Elastic Net coefficient |
|---|---|---|---|---|---|---|---|---|
cg05877648 |
6 |
Island |
0.09 |
0.12 |
0.03 |
2.30 |
||
cg12150066 |
1 |
GNB1 |
TSS1500 |
S_Shore |
0.09 |
0.14 |
0.05 |
1.16 |
cg17422460 |
6 |
BAT4;CSNK2B |
Body;TSS1500 |
N_Shore |
0.23 |
0.32 |
0.08 |
0.90 |
cg04121624 |
10 |
RHOBTB1 |
Body;TSS200 |
N_Shore |
0.27 |
0.36 |
0.08 |
0.32 |
cg12444684 |
1 |
JAK1 |
5’UTR |
N_Shore |
0.17 |
0.27 |
0.10 |
0.13 |
cg27106909 |
16 |
YPEL3 |
1stExon;5’UTR |
N_Shore |
0.18 |
0.27 |
0.09 |
0.06 |
cg16166160 |
6 |
Island |
0.18 |
0.26 |
0.08 |
0.05 |
||
cg03640071 |
11 |
JAM3 |
3’UTR |
0.69 |
0.76 |
0.07 |
0.01 |
|
cg12930882 |
5 |
GDNF |
Body |
0.69 |
0.62 |
0.08 |
-0.10 |
|
cg20554353 |
7 |
S_Shore |
0.79 |
0.72 |
0.07 |
-0.14 |
||
cg02072532 |
14 |
0.87 |
0.79 |
0.07 |
-0.31 |
|||
cg13657981 |
7 |
CALD1 |
Body |
0.79 |
0.73 |
0.06 |
-0.31 |
|
cg16937410 |
3 |
0.60 |
0.51 |
0.09 |
-0.73 |
|||
cg20670923 |
18 |
VAPA |
Body |
S_Shore |
0.41 |
0.34 |
0.07 |
-0.85 |
cg10162251 |
2 |
RNF144A |
5’UTR |
0.89 |
0.84 |
0.04 |
-0.90 |
|
cg04838191 |
2 |
0.87 |
0.81 |
0.06 |
-0.90 |
|||
cg20708856 |
13 |
ATP11A |
Body |
S_Shore |
0.84 |
0.77 |
0.07 |
-0.92 |
cg24742298 |
17 |
SEZ6 |
Body |
N_Shelf |
0.80 |
0.72 |
0.08 |
-1.47 |
Pathway analysis
The 4,208 differentially methylated CpGs in PTEN deleted versus PTEN intact tumors were in 1,908 genes. Gene Set Enrichment Analysis (GSEA) showed that this gene list was enriched for genes in different pathways related to signaling, DNA repair, immune functions, and developmental processes (Figure 2F). Previously, Vivanco and colleagues identified gene expression differences in PTEN wild-type versus PTEN knockdown cell lines [28]. Epidermoid carcinoma, non-small-cell lung carcinoma, and mammary adenocarcinoma cells were used to generate the PTEN knockdown cell lines by performing retroviral transduction with a small hairpin RNA targeting PTEN. Comparing our findings to the sets of differentially expressed genes after PTEN knockdown in vitro (Molecular Signatures Database gene sets: PTEN_DN.V1_DN, PTEN_DN.V1_DN) showed significant gene set enrichment (GSEA, FDR Q-value < 0.0001). As such, these findings provide further evidence that the differentially methylated genes found in our study are at least partially regulated by PTEN. One of the upregulated genes after PTEN knockdown in vitro was JAM3, which was also identified in our methylation signature of PTEN loss. Further, Ouyang et al. studied gene expression differences in prostate tissue from PTEN mutant mice (Nkx3.1; Pten) [29]. One of the 20 upregulated genes in that study (gene set: OUYANG_PROSTATE_CANCER_PROGRESSION_UP) was JAK1, which is also one of the 18 genes included in the methylation signature of PTEN loss.
DISCUSSION
This prospective study showed a positive association between homozygous PTEN loss and PCa recurrence after radical prostatectomy for clinically localized PCa. To gain further insights into the mechanisms that contribute to tumor progression in PCa patients with genomic deletion of PTEN, tumor DNA methylation profiles were investigated. The study revealed significantly different genome-wide methylation profiles in tumors classified by PTEN status, and identified a methylation signature that was uniquely associated with PTEN loss. The differentially methylated CpG sites were in biological pathways related to cell signaling (e.g., estrogen), DNA repair, and immune processes.
Several studies have shown that PTEN loss is associated with worse recurrence-free survival [5–16]. While some studies found that both homozygous and hemizygous loss increase the risk of adverse outcomes, two large, recent studies suggest a stronger association for homozygous loss. A study by Lotan et al. showed that patients with hemizygous and homozygous deleted tumors had a relative risk for recurrence of 1.24 (95% CI: 0.93, 1.65) and 1.66 (95% CI: 1.22, 2.24), respectively [12]. A study by Ahearn et al. evaluated the association of PTEN loss with PCa mortality and found that homozygous (HR = 1.9), but not hemizygous PTEN loss was significantly associated with a worse prognosis [5]. This suggests that tumors with a higher mass of PTEN-null cells have a higher propensity for metastatic spread [12].
The study by Lotan et al. also compared PTEN status to standard clinical-pathological parameters for predicting PCa recurrence (e.g., Gleason score, tumor stage) [12]. This showed that adding data on PTEN loss to the standard clinical model only modestly improved the AUC for recurrence (0.72 vs. 0.74). A similar result was seen in our study (0.72 vs. 0.73). However, predictors that result in small shifts in the AUC may be clinically useful and can improve clinical decision making for individual patients. Further, molecular tumor markers such as PTEN status might be more important and result in larger AUC improvements when detected in biopsy specimens from patients for whom data on pathological stage are unavailable.
Experimental studies have shown that tumor somatic PTEN loss in combination with the TMPRSS2:ERG gene fusion may result in accelerated tumor progression [18, 19]. The gene fusion exists in about half of all localized tumors in Caucasian men and is therefore the most common somatic alteration in PCa [6]; but presence of the fusion alone is not associated with adverse patient outcomes [30]. Several epidemiological and clinical investigations have studied the association between PTEN loss and adverse PCa outcomes in subgroups stratified by TMPRSS2:ERG fusion status, and the results are mixed. While some studies, including our study, found a stronger association of PTEN loss with recurrence among patients with TMPRSS2:ERG fusion-positive tumors [12, 15, 16], other studies reported a stronger association with prognosis in the TMPRSS2:ERG fusion-negative subgroup [5, 8, 10]. Importantly, one of the largest studies on PTEN loss and PCa recurrence to date by Lotan and coworkers [12], showed that PTEN loss was more strongly associated with recurrence-free survival among patients that harbored the gene fusion; but the authors also noted that there was no statistically significant interaction between PTEN loss and TMPRSS2:ERG fusion status. Thus, further research on this topic in larger datasets is needed.
Tumor DNA methylation profiling in our study revealed that tumors with hemi- and homozygous PTEN loss harbor significant genome-wide methylation alterations compared to PTEN intact tumors. These differentially methylated CpGs were enriched in genes involved in different biological processes such as signaling, DNA repair, immune functions, and developmental processes. The study also showed that many of the significant CpGs were in genes known to be differentially expressed after PTEN knockdown with RNAi, suggesting that these genes might be epigenetically regulated in PCa. One of the most significantly enriched pathways was related to estrogen signaling. Interestingly, previous research found that somatic PTEN mutations occur more frequently in tumors with estrogen receptor overexpression [31], and that estrogen receptor β (ERβ) is targeted for repression in PCa caused by PTEN deletion [32]. Our study also showed that DNA methylation profiles were similar in homozygous versus hemizygous deleted tumors. Thus, although some epidemiological, studies including our study, showed that patients with homozygous loss have a worse prognosis than patients with hemizygous loss, these prognostic differences appear to be unrelated to any substantial methylomic changes.
Using feature selection, we identified an 18-CpG methylation signature that classified tumors with PTEN loss. Importantly, this molecular classifier was validated using TCGA data where it accurately distinguished PTEN deleted from PTEN intact tumors. As the methylation signature is a genomic correlate of PTEN loss, the CpGs/genes included in the signature may provide mechanistic insights into the pathways altered in PTEN deleted tumors that contribute to PCa progression. The 13 genes (18 CpGs) in the PTEN signature have roles in various pathways, including cell signaling; and some of the genes have known roles in cancer development (e.g., JAK1, GDNF). Several of the genes have also been implicated in PCa or PTEN biology. For example, VAPA is an endogenous RNA that regulates PTEN levels in a microRNA-dependent manner [33].
Other noteworthy genes with CpGs in the epigenetic signature include ATP11A, JAM3, and GDNF. A previous study from our group identified a CpG biomarker in ATP11A for predicting metastatic-lethal PCa [26]. The gene encodes a membrane ATPase. JAM3 was also included in the tumor mRNA expression signature of PTEN loss in breast cancer generated by Saal et al. [21], thereby providing further evidence of a link between this gene and PTEN activity. Aberrant methylation of JAM3 has also been associated with cervical cancer [34]. Finally, GDNF has been shown to be elevated in PCa reactive tumor stroma and, as such, may contribute to tumor growth and invasion [35]. Therefore, for several of the genes that encompass CpGs in the signature there is plausible evidence for a role in prostate tumorigenesis.
Important strengths of the present study include the relatively large sample size and long-term follow-up for recurrence. Our methylation findings were confirmed using data from TCGA. A potential limitation of the study is that only a small subset of patients with PCa recurrence progressed to metastatic-lethal PCa so this critical endpoint could not be analyzed separately.
In conclusion, PTEN loss in PCa was associated with significantly altered epigenome-wide tumor methylation profiles. As PCa with PTEN loss has a more aggressive phenotype with shorter relapse-free survival, our findings suggest that aberrant DNA methylation may mediate tumor progression when PTEN is deleted.
MATERIALS AND METHODS
Study population
The cohort includes 566 PCa patients who underwent radical prostatectomy as primary therapy for clinically localized adenocarcinoma of the prostate. These patients were previously enrolled in population-based studies of PCa among residents of King County, WA (diagnosed in 1993–1996 or 2002–2005) [36, 37]. Clinical information and survival data were collected from the Seattle-Puget Sound Surveillance, Epidemiology, and End Results (SEER) Program cancer registry. Prostate cancer recurrence status was determined from two detailed follow-up surveys that were completed by patients in 2004–2005 and in 2010–2011, with review of medical records or physician follow-up as needed. A patient was considered to have disease recurrence based on: 1) a post-surgery PSA value of 0.2 ng/mL or greater; 2) metastatic progression on a bone scan, MRI, CT or biopsy; or 3) PCa-specific death. The mean follow-up time for biochemical recurrence was 8 years. The Fred Hutchinson Cancer Research Center Institutional Review Board approved the study and all participants signed informed consent statements.
Fluorescence in situ hybridization (FISH)
Loss of PTEN was assessed using a FISH assay as described previously [38]. Hemizygous PTEN loss was defined as a ratio of the total number of PTEN signals divided by the total number of signals from the chromosome 10 centromere (CEP10) ≤ 0.75. Homozygous PTEN loss was defined as PTEN/CEP10 ≤ 0.2. In total, 71 patients had missing data on PTEN status. An additional 24 patients had PTEN gain; and these patients were not considered in the present analyses. FISH was also used to determine TMPRSS2:ERG gene fusion status, as described previously [39].
DNA isolation, methylation profiling, and data preprocessing
Formalin-fixed paraffin-embedded prostate tumor tissue blocks were obtained from radical prostatectomy specimens and used to make hematoxylin and eosin stained slides, which were reviewed by a PCa pathologist to confirm the presence and location of prostate adenocarcinoma. For each patient two 1-mm tumor tissue cores from the dominant lesion enriched with ≥ 75% tumor cells were taken for DNA purification. The RecoverAll Total Nucleic Acid Isolation Kit (Ambion/Applied Biosciences, Austin, TX) was used to extract DNA, which was then shipped to Illumina (Illumina, Inc., San Diego, CA) for methylation profiling.
Tumor DNA was bisulfite converted. The Infinium HumanMethylation450 BeadChip array (Illumina) was used for methylation profiling. Methylation data were normalized using subset-quantile within array normalization (minfi in Bioconductor) [40], and batch effects were removed using ComBat [41]. Methylation β-values were calculated, which represent the percentage of DNA methylation at a CpG site. Methylation M-values were also calculated, which are a logit transformation of the β-values [42].
Genome annotation was based on the Illumina protocol. A gene promoter region was defined as: TSS1500, TSS200, 5’UTR, and 1stExon. Across the 96-well plates, we incorporated blind duplicate (n = 16) and replicate (n = 2) samples. A sample was excluded if less than 95% of the CpG sites for that sample on the array were detected with a detection P-value (probability of a CpG being detected above the background level defined by negative control probes) < 0.05. Further, CpG sites with a detection P-value of > 0.01 were excluded. Correlation coefficients for duplicate samples were 0.96–0.99. The correlation coefficient for the replicate samples was 0.99. After data preprocessing, there were 523 patients in the radical prostatectomy cohort with DNA methylation data.
The cancer genome atlas (TCGA)
The TCGA PCa dataset included 333 patients, with oversampling of men with higher Gleason score tumors [43]. Allelic copy number derived from ABSOLUTE was used [44], along with relative copy number to determine hemizygous and homozygous PTEN deletions, as described previously [43]. In total, there were 50 homozygous PTEN deleted tumors, 43 hemizygous PTEN deleted tumors, and 240 PTEN intact tumors. Level 1 Infinium HumanMethylation450 data from TCGA (https://gdc.cancer.gov) were preprocessed as described above.
Statistical data analysis
Cox proportional hazards regression and Kaplan-Meier analyses (survival in R) were used to evaluate the association of PTEN loss with PCa recurrence. In addition to the overall association, analyses were stratified by pathological stage, Gleason score, and TMPRSS2:ERG fusion status. Hazard ratios (HRs) and 95% confidence intervals (CIs) were computed. A ROC (Receiver Operating Characteristic) analysis was performed (pROC in R) to compare the prognostic classification performance (recurrence vs. no recurrence) of PTEN loss versus Gleason score (6, 7[3+4], 7[4+3], or 8–10) and pathological stage (local: pT2, N0/NX, M0; regional: pT3–T4 and/or N1, M0).
Epigenome-wide tumor methylation data (478,998 CpG sites) were analyzed to find differential methylation profiles between PTEN deleted (any PTEN loss) and PTEN intact tumors, and between hemi- and homozygous deleted tumors. Differentially methylated CpGs were identified using linear models (limma in Bioconductor).
Elastic Net regularization (glmnet in R; [45]) was used to identify a reduced panel of CpGs that, in combination, distinguished prostate tumors based on PTEN status. All measured CpG sites were used as input for the limma and glmnet analyses except CpGs in 10q22.1–10q25.1 (n = 7,604), which were excluded because this genomic region is deleted in tumors with PTEN loss [46]. Five-fold cross-validation and the AUC (Area Under the Curve) criterion were used to determine the optimal tuning parameter for classification. After variable selection using Elastic Net, the selected CpGs were combined into an epigenetic signature as follows: signaturei = , where g is the CpG site; n is the number of CpGs; βg is the Elastic Net coefficient for CpG g; and Xgi is the methylation value for CpG g in the tumor of patient i.
A heatmap of the data was generated using pheatmap in R. Gene Set Enrichment Analysis (GSEA) was done using the Molecular Signatures Database (http://software.broadinstitute.org/gsea) by comparing our findings to the hallmark [47], and oncogenic signatures (C6) gene sets. All other analyses were done using R.
Author contributions
MSG performed the data analysis and drafted the manuscript. JLS initiated the patient cohort study and helped draft the manuscript. BK carried out the methylation assays. MF and XQ performed the PTEN FISH assays. All authors read the manuscript, revised it critically for important intellectual content, and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Drs. Beatrice Knudson, Antonio Hurtado-Coll, and Xiaotun Zhou for their assistance with the pathology; Lena Glaskova and Sarah Schroeder for assisting with the PTEN FISH assay; and Manuel Luedeke, Antje Rinckleb, and Christiane Maier for performing the TMPRSS2:ERG FISH assays. We also thank all the men who participated in these studies.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
GRANT SUPPORT
This work was supported by the National Cancer Institute, NIH (R01 CA056678, R01 CA092579, and K05 CA175147 to JLS, and P50 CA097186); the Intramural Research Program of the National Human Genome Research Institute; and the Fred Hutchinson Cancer Research Center. Illumina, Inc. provided the DNA methylation arrays and performed the methylation assays. MSG is the recipient of a Dutch Cancer Society Fellowship (BUIT 2014-6645).
REFERENCES
1. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011; 11:289-301.
2. Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999; 96:4240-4245.
3. Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012; 13:195-203.
4. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012; 13:283-296.
5. Ahearn TU, Pettersson A, Ebot EM, Gerke T, Graff RE, Morais CL, Hicks JL, Wilson KM, Rider JR, Sesso HD, Fiorentino M, Flavin R, Finn S, et al. A prospective investigation of PTEN loss and ERG expression in lethal prostate cancer. J Natl Cancer Inst. 2016; 108.
6. Bostrom PJ, Bjartell AS, Catto JW, Eggener SE, Lilja H, Loeb S, Schalken J, Schlomm T, Cooperberg MR. Genomic predictors of outcome in prostate cancer. Eur Urol. 2015; 68:1033-1044.
7. Chaux A, Peskoe SB, Gonzalez-Roibon N, Schultz L, Albadine R, Hicks J, De Marzo AM, Platz EA, Netto GJ. Loss of PTEN expression is associated with increased risk of recurrence after prostatectomy for clinically localized prostate cancer. Mod Pathol. 2012; 25:1543-1549.
8. Kim SH, Kim SH, Joung JY, Lee GK, Hong EK, Kang KM, Yu A, Nam BH, Chung J, Seo HK, Park WS, Lee KH. Overexpression of ERG and wild-type PTEN are associated with favorable clinical prognosis and low biochemical recurrence in prostate cancer. PLoS One. 2015; 10:e0122498.
9. Krohn A, Diedler T, Burkhardt L, Mayer PS, De Silva C, Meyer-Kornblum M, Kotschau D, Tennstedt P, Huang J, Gerhauser C, Mader M, Kurtz S, Sirma H, et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am J Pathol. 2012; 181:401-412.
10. Lahdensuo K, Erickson A, Saarinen I, Seikkula H, Lundin J, Lundin M, Nordling S, Butzow A, Vasarainen H, Bostrom PJ, Taimen P, Rannikko A, Mirtti T. Loss of PTEN expression in ERG-negative prostate cancer predicts secondary therapies and leads to shorter disease-specific survival time after radical prostatectomy. Mod Pathol. 2016; 29:1565-1574.
11. Lotan TL, Carvalho FL, Peskoe SB, Hicks JL, Good J, Fedor HL, Humphreys E, Han M, Platz EA, Squire JA, De Marzo AM, Berman DM. PTEN loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy. Mod Pathol. 2015; 28:128-137.
12. Lotan TL, Wei W, Morais CL, Hawley ST, Fazli L, Hurtado-Coll A, Troyer D, McKenney JK, Simko J, Carroll PR, Gleave M, Lance R, Lin DW, et al. PTEN loss as determined by clinical-grade immunohistochemistry assay is associated with worse recurrence-free survival in prostate cancer. Eur Urol Focus. 2016; 2:180-188.
13. Tosoian JJ, Almutairi F, Morais CL, Glavaris S, Hicks J, Sundi D, Humphreys E, Han M, De Marzo AM, Ross AE, Tomlins SA, Schaeffer EM, Trock BJ. Prevalence and prognostic significance of PTEN loss in African-American and European-American men undergoing radical prostatectomy. Eur Urol. 2017; 71:697-700.
14. Troyer DA, Jamaspishvili T, Wei W, Feng Z, Good J, Hawley S, Fazli L, McKenney JK, Simko J, Hurtado-Coll A, Carroll PR, Gleave M, Lance R, et al. A multicenter study shows PTEN deletion is strongly associated with seminal vesicle involvement and extracapsular extension in localized prostate cancer. Prostate. 2015; 75:1206-1215.
15. Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, Ludkovski O, Zielenska M, Soares FA, Squire JA. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol. 2008; 21:1451-1460.
16. Leinonen KA, Saramaki OR, Furusato B, Kimura T, Takahashi H, Egawa S, Suzuki H, Keiger K, Ho Hahm S, Isaacs WB, Tolonen TT, Stenman UH, Tammela TL. Loss of PTEN is associated with aggressive behavior in ERG-positive prostate cancer. Cancer Epidemiol Biomarkers Prev. 2013; 22:2333-2344.
17. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215-1228.
18. King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, Gerald WL, Sawyers CL. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009; 41:524-526.
19. Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino PT, Cordon-Cardo C, Gerald W, Pandolfi PP. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009; 41:619-624.
20. Wise HM, Hermida MA, Leslie NR. Prostate cancer, PI3K, PTEN and prognosis. Clin Sci (Lond). 2017; 131:197-210.
21. Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, Koujak S, Ferrando AA, Malmstrom P, Memeo L, Isola J, Bendahl PO, Rosen N, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007; 104:7564-7569.
22. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011; 19:575-586.
23. Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, Esopi D, Irizarry RA, Getzenberg RH, Nelson WG, Luo J, Xu J, Isaacs WB, et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med. 2013; 5:169ra110.
24. Chao C, Chi M, Preciado M, Black MH. Methylation markers for prostate cancer prognosis: a systematic review. Cancer Causes Control. 2013; 24:1615-1641.
25. Geybels MS, Wright JL, Bibikova M, Klotzle B, Fan JB, Zhao S, Feng Z, Ostrander EA, Lin DW, Nelson PS, Stanford JL. Epigenetic signature of Gleason score and prostate cancer recurrence after radical prostatectomy. Clin Epigenetics. 2016; 8:97.
26. Zhao S, Geybels MS, Leonardson A, Rubicz R, Kolb S, Yan Q, Klotzle B, Bibikova M, Hurtado-Coll A, Troyer D, Lance R, Lin DW, Wright JL, et al. Epigenome-wide tumor DNA methylation profiling identifies novel prognostic biomarkers of metastatic-lethal progression in men diagnosed with clinically localized prostate cancer. Clin Cancer Res. 2017; 23:311-319.
27. Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, Shiah YJ, Yousif F, Lin X, Masella AP, Fox NS, Xie M, Prokopec SD, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017; 541:359-364.
28. Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, Jiao J, Rose J, Xie W, Loda M, Golub T, Mellinghoff IK, Davis RJ, et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell. 2007; 11:555-569.
29. Ouyang X, Jessen WJ, Al-Ahmadie H, Serio AM, Lin Y, Shih WJ, Reuter VE, Scardino PT, Shen MM, Aronow BJ, Vickers AJ, Gerald WL, Abate-Shen C. Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 2008; 68:2132-2144.
30. Pettersson A, Graff RE, Bauer SR, Pitt MJ, Lis RT, Stack EC, Martin NE, Kunz L, Penney KL, Ligon AH, Suppan C, Flavin R, Sesso HD, et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: a cohort study and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2012; 21:1497-1509.
31. Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19784 diverse solid tumors. JAMA Oncol. 2016; 2:1565-1573.
32. Mak P, Li J, Samanta S, Chang C, Jerry DJ, Davis RJ, Leav I, Mercurio AM. Prostate tumorigenesis induced by PTEN deletion involves estrogen receptor beta repression. Cell Rep. 2015; 10:1982-1991.
33. Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, Lieberman J, Rigoutsos I, Pandolfi PP. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011; 147:344-357.
34. Yin A, Zhang Q, Kong X, Jia L, Yang Z, Meng L, Li L, Wang X, Qiao Y, Lu N, Yang Q, Shen K, Kong B. JAM3 methylation status as a biomarker for diagnosis of preneoplastic and neoplastic lesions of the cervix. Oncotarget. 2015; 6:44373-44387. https://doi.org/10.18632/oncotarget.6250.
35. Huber RM, Lucas JM, Gomez-Sarosi LA, Coleman I, Zhao S, Coleman R, Nelson PS. DNA damage induces GDNF secretion in the tumor microenvironment with paracrine effects promoting prostate cancer treatment resistance. Oncotarget. 2015; 6:2134-2147. https://doi.org/10.18632/oncotarget.3040.
36. Stanford JL, Wicklund KG, McKnight B, Daling JR, Brawer MK. Vasectomy and risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 1999; 8:881-886.
37. Agalliu I, Salinas CA, Hansten PD, Ostrander EA, Stanford JL. Statin use and risk of prostate cancer: results from a population-based epidemiologic study. Am J Epidemiol. 2008; 168:250-260.
38. Qu X, Jeldres C, Glaskova L, Friedman C, Schroeder S, Nelson PS, Porter C, Fang M. Identification of combinatorial genomic abnormalities associated with prostate cancer early recurrence. J Mol Diagn. 2016; 18:215-224.
39. Egbers L, Luedeke M, Rinckleb A, Kolb S, Wright JL, Maier C, Neuhouser ML, Stanford JL. Obesity and prostate cancer risk according to tumor TMPRSS2:ERG gene fusion status. Am J Epidemiol. 2015; 181:706-713.
40. Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012; 13:R44.
41. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007; 8:118-127.
42. Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010; 11:587.
43. Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015; 163:1011-1025.
44. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012; 30:413-421.
45. Zou H, Hastie T. Regularization and variable selection via the elastic net. J Roy Stat Soc B. 2005; 67:301-320.
46. Yoshimoto M, Ludkovski O, DeGrace D, Williams JL, Evans A, Sircar K, Bismar TA, Nuin P, Squire JA. PTEN genomic deletions that characterize aggressive prostate cancer originate close to segmental duplications. Genes Chromosomes Cancer. 2012; 51:149-160.
47. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417-425.