
INTRODUCTION
Parkinson’s disease (PD), characterized by resting tremor, rigidity, slowness of movements and postural instability, is the second most common neurodegenerative disorder after Alzheimer’s disease [1]. Although the cause of PD is still not completely known, it is already known that both genetic susceptibility factors and environmental factors could lead to the occurrence and the development of PD. According to one recent GWAS study related with PD [2], we know that a number of gene variants such as SNCA, LRRK2, PARK2 and GBA, have been shown to modulate the risk of PD. Additionally, mutations in a number of mitochondrial related genes, such as PARK6, PARK2, 12SrRNA, PINK1, POLG and DJ, have also been confirmed to be associated with PD [3, 4]. Among these genes, 12SrRNA was located in mitochondrial and the others were located in the nucleus. These genes participate in the coding of mitochondrial proteins required to build mitochondria and maintain the function of mitochondria [5].
Human mitochondria have their own DNA, containing 37 genes in a single chromosome [6]. Some genes located in the mitochondria encode subunits of enzymes that are components of the respiratory chain or ATP synthesis, thus playing a critical role in energy production. Since the brain constantly requires high amounts of energy consumption for maintenance of its functions, an alteration in the mtDNA functions may cause changes in the brain functions and brain disorders. The genetic polymorphism A10398G, due to an amino acid substitution from Thr to Ala, is located on the ND3 gene that encodes one subunits constituting Complex I [7]. In the substantia nigra and platelets of PD patients, the biochemical activity in the electron transport chain of complex I is impaired. Therefore, the 10398G allele could have an effect on the development of PD. In recent years, the polymorphism of mtDNA A10398G has been suggested as a protective factor in some studies [8–10] but not in others [11–15], and one study even indicated that it was a risk factor [16]. To help resolve the controversy over whether the mtDNA A10398G variant is associated with PD susceptibility, we conducted a meta-analysis according to the eligible case-control data from 9 reported studies.
RESULTS
Characteristics of eligible studies
The selection flow diagram and exclusion reasons for articles are described in Figure 1. First, 498 articles were searched as potential relevant articles through database searching. After scanning titles and abstracts, articles that did not examine the correlation between the mtDNA A10398G variant and PD risk or did not have a case-control design (n = 451) were excluded. In addition, reviews or meta-analyses (n = 2) and animal studies (n = 28) were excluded, and 17 potentially relevant articles were considered worth further reviewing. Then, through screening the full text, studies with no sufficient allele data (n = 8) were excluded, and 9 potentially relevant articles were identified. Finally, after estimating the quality of the eligible studies, 9 articles (including 9 clinical case-control studies with a total of 3381 PD cases and 2810 healthy individuals) met our inclusion criteria and were included in our meta-analysis. Among them, 6 studies’ subjects were Caucasian, and 3 studies’ subjects were Asian. In these studies, they used PCR or TaqMan as their genotyping method and the genotype frequencies of the controls were in HWE. The detailed characteristics of the included studies are described in Table 1, and the NOS scores of these articles are listed in Table 2.
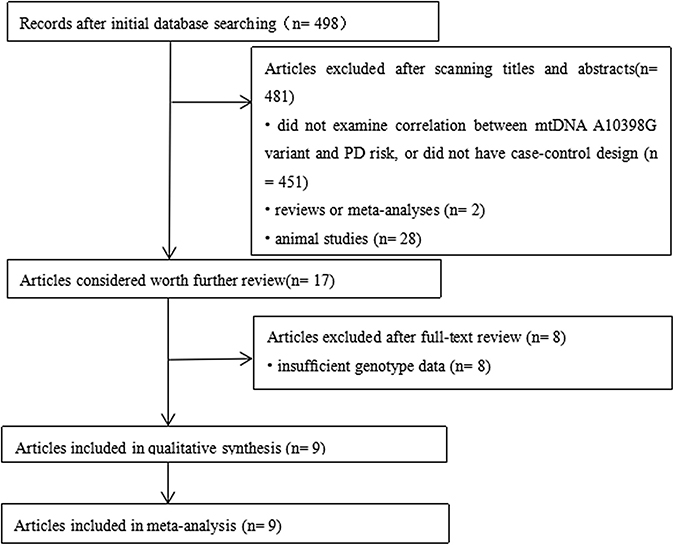
Figure 1: Flow diagram for publication selection in the present meta-analysis.
Table 1: Characteristics of studies included in this analysis
ID |
First author |
year |
Ethnicity |
Genotyping method |
Sample size (case/control) |
Allele distribution (case/control) |
HWE, |
association |
|---|---|---|---|---|---|---|---|---|
1 |
Cecilia Huerta |
2006 |
Caucasian |
PCR-sequencing |
450/200 |
58/44 392/156 |
0.382 |
Had |
2 |
Joanne Clark |
2011 |
Caucasian |
PCR-RFLP |
376/173 |
84/44 292/129 |
0.571 |
Hadn’t |
3 |
C. M. Chen |
2007 |
Asian |
PCR-RFLP |
416/372 |
257/223 159/149 |
0.132 |
Hadn’t |
4 |
Qiaohong Chu |
2015 |
Asian |
PCR-RFLP |
322/332 |
186/168 136/164 |
0.573 |
Had |
5 |
Cecilia Huerta |
2005 |
Caucasian |
PCR-RFLP |
271/230 |
34/49 237/181 |
0.191 |
Had |
6 |
D. Otaegui |
2004 |
Caucasian |
PCR-sequencing |
40/64 |
9/10 31/54 |
0.068 |
Hadn’t |
7 |
Joelle M. van der Walt |
2003 |
Caucasian |
Taqman |
557/312 |
97/81 460/231 |
0.386 |
Had |
8 |
Helen Latsoudis |
2008 |
Caucasian |
PCR-RFLP |
224/383 |
43/78 181/305 |
0.368 |
Hadn’t |
9 |
C. W. Liou |
2016 |
Asian |
SPCR |
725/744 |
397/406 328/338 |
0.526 |
Hadn’t |
PCR, polymerase chain reaction. PCR-RFLP, Polymerase Chain Reaction-Restriction Fragment Length Polymorphism.
SPCR, symmetric PCR. HWE, Hardy–Weinberg equilibrium. The P value of HWE determined by the χ2 test or Fisher’s exact test in control groups.
Table 2: Quality assessment for the eligible studies according to NOS
First author |
Selection (stars) |
Comparability (stars) |
Exposure (stars) |
Total Quality score |
Refs. |
|
|---|---|---|---|---|---|---|
1 |
Cecilia Huerta |
4 |
2 |
1 |
7 |
8 |
2 |
Joanne Clark |
3 |
2 |
1 |
6 |
12 |
3 |
C. M. Chen |
4 |
2 |
1 |
7 |
13 |
4 |
Qiaohong Chu |
4 |
2 |
1 |
7 |
16 |
5 |
Cecilia Huerta |
4 |
2 |
1 |
7 |
9 |
6 |
D. Otaegui |
4 |
2 |
1 |
7 |
11 |
7 |
Joelle M. van der Walt |
3 |
2 |
1 |
6 |
10 |
8 |
Helen Latsoudis |
4 |
2 |
1 |
7 |
14 |
9 |
C. W. Liou |
3 |
2 |
1 |
6 |
15 |
NOS: Newcastle-Ottawa-Scale [32].
Quantitative synthesis
The results of our meta-analysis are summarized in Table 3. According to the I-squared value, heterogeneity was present in the allele model (I² = 69.8%, P = 0.001), so the random effect model was selected. We also performed subgroup analysis stratified by ethnicity (in the Caucasian population: I² = 43.0%, P = 0.118; in the Asian population: I² = 10.0%, P = 0.329). According to our results, although no significant association between mtDNA A10398G polymorphism and PD was found when polling all populations overall(G vs. A: OR = 0.858, 95% CI = 0.686–1.074, P = 0.181) or in the Asian population (G vs. A: OR = 1.090, 95% CI = 0.939–1.284, P = 0.242), in the Caucasian population (G vs. A: OR = 0.699, 95% CI = 0.546–0.895, P = 0.005) the G allele of mtDNA A10398G mutations may be a potential protective factor of PD. The forest plot of mtDNA A10398G polymorphism in PD is shown in Figure 2.
Table 3: Meta-analysis of the association between the A10398G gene mutation and PD
Ethnicity |
Fix-effect model |
Random-effect model |
Heteogeneity |
Begg’s test |
|||||
|---|---|---|---|---|---|---|---|---|---|
OR |
95% CI |
P |
OR |
95% CI |
P |
I² |
Z |
P |
|
Overall population |
0.910 |
0.812–1.019 |
0.102 |
0.858 |
0.686–1.074 |
0.181 |
69.8% |
0.52 |
0.602 |
Caucasian population |
0.689 |
0.576–0.824 |
< 0.001 |
0.699 |
0.546–0.895 |
0.005 |
43.0% |
< 0.01 |
1.000 |
Chinese population |
1.094 |
0.945–1.267 |
0.231 |
1.098 |
0.939–1.284 |
0.242 |
10.1% |
1.04 |
0.296 |
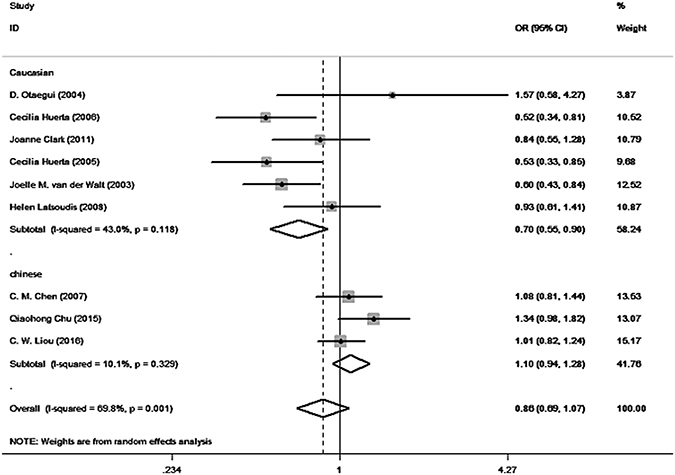
Figure 2: Forest plot for the relationship of mtDNA A10398G polymorphism and PD (G allele vs. A allele).
Publication bias
Begg’s funnel plots and tests were used to estimate the publication bias. No publication bias was identified for the association between A10389G polymorphism and PD susceptibility (P = 0.602, Figure 3).
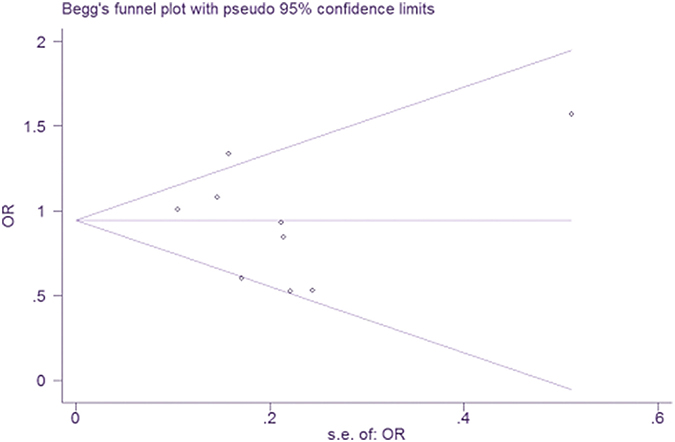
Figure 3: Begg’s funnel plot for publication bias analysis for mtDNA A10398G polymorphisms.
Sensitivity analysis
We removed each study one by one every time the results showed no significant changes in the pooled OR and 95% CI in the allele model. This demonstrates that our meta-analysis is of high stability (Figure 4).
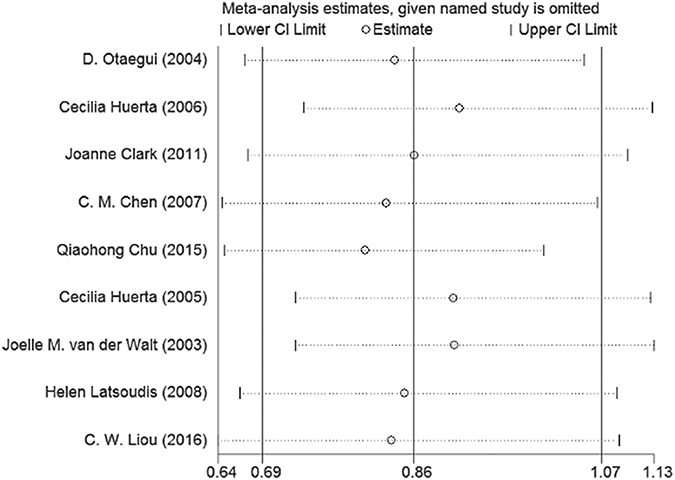
Figure 4: Sensitivity analysis of the the summary odds ratio coefficients on the association between mtDNA A10398G polymorphism and PD.
DISCUSSION
As the second most common neurodegenerative disorder, PD is characterized by the loss of dopaminergic neurons in the substantia nigra (SN). We all know that although the aetiological factor of PD is still obscure, the genetic factor has been well confirmed to have a great contribution to the development of PD. Many studies have suggested that the dysfunction of mitochondria could be involved in the development of PD [17–19]. Additionally, mtDNA mutations have been reported to contribute to some neurodegenerative disorders, including Lewy body disease (LBD, mainly PD), Alzheimer’s disease, amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) [20–22]. However, the relationship between mtDNA A10398G and PD is still unclear. In the past several years, many studies have researched the relationship between the mtDNA A10398Gpolymorphism and the susceptibility of PD in different areas around the world, but their results are conflicting. Considering the contradictory conclusions that they reached and that most of these studies’ sample sizes were relatively small, a meta-analysis should be applied to further verify the association between mitochondrial DNA A10398G and PD.
In this meta-analysis, we included a total of 9 eligible case-control studies above. Among the 9 included studies, six were in the Caucasian population and three were in the Asian population. In a study based on a regional Caucasian population from Spain, with a total of 450 cases and 200 controls, the 10398G allele was found to decrease the risk of PD with an OR (95% CI) of 0.52 (0.34–0.81) [8]. In the study by Cecilia Huerta [9] with a total of 271 cases and 230 controls, the 10398G allele was also shown to decrease the risk of PD in the Spanish population with an OR (95% CI) of 0.53 (0.33–0.86) [9]. In the study by Joelle M. van der Walt [10], 10398G is strongly associated with a protective effect of PD in European with an OR (95% CI) of 0.53 (0.39–0.73). However, these results were not always consistent. Qiaohong Chu [16] detected an inverse association between A10398G polymorphism and the risk of PD. In a northern Chinese population,carrying the 10398G allele had a significantly increased risk of PD (OR = 1.30; 95% CI = 0.95–1.76; P = 0.013). After stratification by gender, the increased risk appeared to be more significant in females (OR = 1.91; 95% CI = 1.16–3.16; P = 0.001). Of note, such a statistically significant association between A10398G polymorphism and the risk of PD was not always duplicated in other studies. D. Otaegui [11] reported that no statistically significant correlation was identified in the general Caucasian population of Spain. However, Joanne Clark [12] observed that G10398G was not significantly associated with PD risk among the Caucasian population. Similarly, no association was detected in C. M. Chen’s [13] case-control study comprising 416 cases and 372 controls. The frequencies of the G allele among cases and controls from both Helen Latsoudis’s [14] case-control study (including 224 cases and 383 controls) and C. W. Liou’s [15] study (including 725 cases and 744 controls) were similar to each other. For our meta-analysis, although the results support that individuals carrying the G allele did not exhibit an increased risk of PD as per the eligible studies, when compared with individuals carrying the A allele in the Asian population (G vs. A: OR = 1.090, 95% CI = 0.939–1.284, P = 0.242), the G allele of mtDNA A10398G polymorphism seems to be a potential protective factor of PD in the Caucasian population (G vs. A: OR = 0.699, 95% CI = 0.546–0.895, P = 0.005).
Mutation of mtDNA A10398G (A to G transition due to an amino acid substitution from Thr to Ala) was located in the ND3 gene that encodes one subunit constituting Complex I [7]. Complex I was the first enzyme in the mitochondrial respiratory chain and a key enzyme for energy production and a site for ROS production [23, 24]. Therefore, it is possible that 10398G may increase the performance of complex I. Thus, carriers of the 10398G allele would have enhanced capability for energy metabolism when the cell was either in a normal state or under abnormal oxidative stress conditions. An increase in complex I activity can result in an increase in ATP synthesis and protect against a range of PD-relevant biotoxins [25]. Thus, the carriers of the 10398G allele are at a lower risk for developing PD. In addition, the 10398G allele may induce more ROS production compared with 10398A. Complex I normally produces ROS during cellular activity [26]. However, when the complex I is enhanced, generation of ROS is decreased, alleviating oxidative stress [27]. SN tissues are susceptible to oxidative damage, and excessive oxidative stress over time may lead to the degeneration of SN neurons [28]. Therefore, the 10398G allele could play a role in determining neuronal protection to reduce the risk of developing PD. Further study is needed to verify the mechanism for its regulation on complex I.
Several reasons may help account for why the results between the Caucasian population and the Asian population are different. First, the A10398G polymorphism is differently distributed in the Caucasian population compared to the Asian population: the frequency of 10398G allele of mtDNA, which is found only 26% in Caucasian, is much higher in Asian population [29]. The negative result of the mtDNA A10398G polymorphism in PD in the Asian population may be due to the very high frequency of the 10398G allele in the Asian population. Second, because gene polymorphisms could influence the risk of developing PD through interaction with other genes or with some environmental toxins [30], an environmental difference between the Caucasian population and the Asian population may lead to the inconsistent results. Therefore, further well-designed studies with larger samples, particularly studies stratified by ethnic and gene-environmental interactions, should be performed to validate these results.
We are convinced that our meta-analysis has some special characteristics. Most importantly, compared with previous research, a larger sample size was used to estimate the effect, so our results are more reliable than the previous research results. In addition, according to a recent review on Parkinson’s disease pharmacogenetics included all the meta-analysis published in PD [31], and A10398G was not included. Therefore this is apparently the first meta-analysis on this issue and this study is the first meta-analysis to demonstrate that the G allele of the mtDNA A10398G polymorphism does not appear to associated with PD risk in the Asian population, and it may be a potential protective factor of PD in the Caucasian population, which may encourage more research focused on the mtDNA A10398G polymorphism in different races. Last but certainly not least, we conducted a sensitivity analysis to estimate the effect of each study on the overall estimate, and the results suggested that our meta-analysis results were stable.
There are also some limitations of our meta-analysis. First, as PD is a multifactorial disease, the potential interactions of genetic-genetic or genetic-environmental factors may influence the process of PD. Second, in view of the relatively small sample size and the limited study number, especially in the subgroup of the Asian population where only three studies were included, the power used to detect the real difference between cases and controls may not be very strong. Finally, as a result of insufficient data, other possible variables, including age of onset, gender or smoking, for the association between A10398G and PD could not be evaluated.
In conclusion, our meta-analysis indicated that although the G allele of the mtDNA A10398G polymorphism has no significant relationship with PD in the Asian populations, it seems to be a potential protective factor of PD in the Caucasian population. Although a systematic investigation of the relationship between A10398G polymorphisms and the risk of PD could not be conducted because of the aforementioned limitations, it is important to gain a better understanding of the effect of A10398G polymorphisms on PD. Considering the limitations, investigations involving large case-control studies of different ethnicities should be conducted to clarify the possible roles of the mtDNA A10398G polymorphism in the aetiology of PD.
MATERIALS AND METHODS
Literature search
Two investigators independently searched the related literature from PubMed, Web of Science, Springer Link, EMBASE and EBSCO databases published up to March 2017. We used the following key words: (“mitochondrial DNA” or “A10398G”) and (“gene polymorphism” or “variant”) and (“Parkinson’s disease” or “PD”). References of the eligible articles were also searched. Searching was restricted to human case-control studies.
Inclusion and exclusion criteria
To be included, the studies must have met the following inclusion criteria: (1) case-control studies that studied the relationship between mitochondrial DNA A10398G and PD, (2) studies that included sufficient allele data for both patients and controls to calculate the odds ratio (OR) and 95% confidence interval (CI), (3) studies in which cases and controls were based on the same population, (4) studies that included participants diagnosed with PD according to the UK Parkinson’s Disease Society Brain Bank criteria, and (5) full text studies written in English.
The excluded criteria included the following: (1) case reports, editorials, meta-analysis or reviews; (2) studies with insufficient information about the mitochondrial DNA A10398G; (3) family-based studies; (4) studies whose subjects were ethnic minorities; and (5) studies in which the allele distribution in controls did not meet Hardy-Weinberg equilibrium (HWE).
Extraction
Two investigators independently selected the eligible articles and extracted the needed data. Inconsistencies between the two investigators were resolved by discussing with a third reviewer. The information extracted from each study included the first author’s name, year of publication, ethnicity of the study population, and number of cases and controls for each allele.
Quality assessment
The Newcastle-Ottawa Scale (NOS) [32] was used to estimate the quality of the eligible studies by two reviewers independently based on the following aspects: selection, comparability and exposure. The NOS scores of each article equal or greater than 6 stars indicated high quality.
Statistical analysis
We used the Stata 12.0 software (Stata Corporation, College Station, TX, USA) to calculate all data analyses. In addition, we used the allele model to evaluate the association between mtDNA A10398G variant and PD risk. The OR and CI were used to assess the strength of the association between the variant and PD susceptibility. The pooled OR was measured using the Z-test. P < 0.05 was considered to demonstrate a statistically significant difference. Heterogeneity between studies was examined by the Q-test (meaningful value of P < 0.05) and I-square statistic tests (I² < 50% shows little difference) [33]. The fixed effects model or the random effects model was selected according to the heterogeneity. If the P-value of the Q-test > 0.05 or I² < 50%, the fixed effects model was selected. Inversely, the random effects model was applied [34]. We also performed subgroup analysis to seek reasons for heterogeneity. Stratified analysis was performed according to ethnicity and source of controls. Sensitivity analysis was conducted to assess the stability of the results. Potential publication bias was assessed by Begg’s test and funnel plots. A P-value of Begg’s test greater than 0.05 and articles with symmetrical distribution in the Begg’s funnel plot were considered to have no significant publication bias [35].
ACKNOWLEDGMENTS AND FUNDING
We would like to thank all the investigators for their helpful comments on this article. This work was supported by a grant from the Natural Science Foundation of China (81571225).
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
REFERENCES
1. Jiang H, Wang J, Rogers J, Xie J. Brain Iron Metabolism Dysfunction in Parkinson's Disease. Mol Neurobiol. 2017; 54:3078–3101.
2. Benitez BA, Davis AA, Jin SC, Ibanez L, Ortega-Cubero S, Pastor P, Choi J, Cooper B, Perlmutter JS, Cruchaga C. Resequencing analysis of five Mendelian genes and the top genes from genome-wide association studies in Parkinson's Disease. Mol Neurodegener. 2016; 11:29.
3. Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006; 7:207–19.
4. Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC, Brice A, van Duijn CM, Oostra B, Meco G, Heutink P. DJ-1( PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol Sci. 2003; 24:159–60.
5. Muqit MM, Gandhi S, Wood NW. Mitochondria in Parkinson disease: back in fashion with a little help from genetics. Arch Neurol. 2006; 63:649–54.
6. Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999; 23:147.
7. Oliver NA, Greenberg BD, Wallace DC. Assignment of a polymorphic polypeptide to the human mitochondrial DNA unidentified reading frame 3 gene by a new peptide mapping strategy. J Biol Chem. 1983; 258:5834–9.
8. Huerta C, Sanchez-Ferrero E, Coto E, Blazquez M, Ribacoba R, Guisasola LM, Salvador C, Alvarez V. No association between Parkinson's disease and three polymorphisms in the eNOS, nNOS, and iNOS genes. Neurosci Lett. 2007; 413:202–5.
9. Huerta C, Castro MG, Coto E, Blázquez M, Ribacoba R, Guisasola LM, Salvador C, Martínez C, Lahoz CH, Alvarez V. Mitochondrial DNA polymorphisms and risk of Parkinson's disease in Spanish population. Journal of the Neurological Sciences. 2005; 236:49–54.
10. van der Walt JM, Nicodemus KK, Martin ER, Scott WK, Nance MA, Watts RL, Hubble JP, Haines JL, Koller WC, Lyons K, Pahwa R, Stern MB, Colcher A, et al. Mitochondrial Polymorphisms Significantly Reduce the Risk of Parkinson Disease. American Journal of Human Genetics. 2003; 72:804–11.
11. Otaegui D, Paisan C, Saenz A, Marti I, Ribate M, Marti-Masso JF, Perez-Tur J, Lopez de Munain A. Mitochondrial polymporphisms in Parkinson's Disease. Neurosci Lett. 2004; 370:171–4.
12. Clark J, Reddy S, Zheng K, Betensky RA, Simon DK. Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson's disease. BMC Medical Genetics. 2011; 12:69.
13. Chen CM, Kuan CC, Lee-Chen GJ, Wu YR. Mitochondrial DNA polymorphisms and the risk of Parkinson’s disease in Taiwan. Journal of Neural Transmission. 2007; 114:1017.
14. Latsoudis H, Spanaki C, Chlouverakis G, Plaitakis A. Mitochondrial DNA polymorphisms and haplogroups in Parkinson's disease and control individuals with a similar genetic background. J Hum Genet. 2008; 53:349–56.
15. Liou CW, Chuang JH, Chen JB, Tiao MM, Wang PW, Huang ST, Huang TL, Lee WC, Weng SW, Huang PH, Chen SD, Chen RS, Lu CS, et al. Mitochondrial DNA variants as genetic risk factors for Parkinson disease. European Journal of Neurology. 2016; 23:1289–300.
16. Chu Q, Luo X, Zhan X, Ren Y, Pang H. Female genetic distribution bias in mitochondrial genome observed in Parkinson’s Disease patients in northern China. Scientific Reports. 2015; 5:17170.
17. Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A. Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. The Lancet. 2004; 364:875–82.
18. Abeliovich A. Parkinson's disease: Mitochondrial damage control. Nature. 2010; 463:744–5.
19. Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B, Greenamyre JT. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiology of disease. 2014; 70:214–23.
20. Keogh MJ, Chinnery PF. Mitochondrial DNA mutations in neurodegeneration. Biochim Biophys Acta. 2015; 1847:1401–11.
21. van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, Kroner CC, Welsh-Bohmer KA, Saunders AM, Roses AD, Small GW, Schmechel DE, Murali Doraiswamy P, Gilbert JR, et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neuroscience Letters. 2004; 365:28–32.
22. Mancuso M, Conforti FL, Rocchi A, Tessitore A, Muglia M, Tedeschi G, Panza D, Monsurrò M, Sola P, Mandrioli J, Choub A, DelCorona A, Manca ML, et al. Could mitochondrial haplogroups play a role in sporadic amyotrophic lateral sclerosis? Neuroscience Letters. 2004; 371:158–62.
23. Schapira AH. Mitochondrial complex I deficiency in Parkinson's disease. Adv Neurol. 1993; 60:288–91.
24. Kosel S, Hofhaus G, Maassen A, Vieregge P, Graeber MB. Role of mitochondria in Parkinson disease. Biol Chem. 1999; 380:865–70.
25. Parsons RB, Aravindan S, Kadampeswaran A, Evans EA, Sandhu KK, Levy ER, Thomas MG, Austen BM, Ramsden DB. The expression of nicotinamide N-methyltransferase increases ATP synthesis and protects SH-SY5Y neuroblastoma cells against the toxicity of Complex I inhibitors. Biochem J. 2011; 436:145–55.
26. Kazuno AA, Munakata K, Nagai T, Shimozono S, Tanaka M, Yoneda M, Kato N, Miyawaki A, Kato T. Identification of Mitochondrial DNA Polymorphisms That Alter Mitochondrial Matrix pH and Intracellular Calcium Dynamics. PLoS Genetics. 2006; 2:e128.
27. Orth M, Schapira AHV. Mitochondrial involvement in Parkinson’s disease. Neurochemistry International. 2002; 40:533–41.
28. Shoffner JM, Watts RL, Juncos JL, Torroni A, Wallace DC. Mitochondrial oxidative phosphorylation defects in parkinson's disease. Annals of Neurology. 1991; 30:332–9.
29. Torroni A, Wallace DC. Mitochondrial DNA variation in human populations and implications for detection of mitochondrial DNA mutations of pathological significance. J Bioenerg Biomembr. 1994; 26:261–71.
30. Autere J, Moilanen JS, Finnilä S, Soininen H, Mannermaa A, Hartikainen P, Hallikainen M, Majamaa K. Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Human Genetics. 2004; 115:29–35.
31. Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Agúndez JA. Advances in understanding genomic markers and pharmacogenetics of Parkinson's disease. Expert Opin Drug Metab Toxicol. 2016; 12:433–48.
32. Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. European Journal of Epidemiology. 2010; 25:603–5.
33. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003; 327:557–60.
34. Zintzaras E, Ioannidis JP. Heterogeneity testing in meta-analysis of genome searches. Genet Epidemiol. 2005; 28:123–37.
35. Peters JL, Sutton AJ, Jones DR, Abrams KR, Rushton L. Comparison of two methods to detect publication bias in meta-analysis. JAMA. 2006; 295:676–80.