
INTRODUCTION
Mitochondrial DNA (mtDNA) is a 16.6 kb circular double-stranded DNA encoding the protein subunits of mitochondrial respiratory chain complexes I, III, IV, and V and which functions in energy production [1, 2]. All of the mtDNA replication, transcription factors, and the primary components of the mitochondrial translation machinery are encoded by nuclear genes [1, 2]. Mutations in nuclear genes that function in either mitochondrial deoxyribonucleoside triphosphate (dNTP) synthesis or mtDNA replication can lead to a severe reduction in cellular mtDNA content in affected organs, which is termed mtDNA depletion syndrome (MDS) [1, 2]. Thymidine kinase 2, adenosine diphosphate-forming succinyl CoA ligase beta subunit, guanosine diphosphate-forming succinyl CoA ligase alpha subunit, ribonucleotide reductase M2 B subunit, thymidine phosphorylase, and deoxyguanosine kinase (DGUOK) encode proteins that maintain the mitochondrial dNTP pool, and mutations in any of these genes can result in mtDNA depletion [2]. DNA polymerase gamma and Twinkle are essential for mtDNA replication, and mutations in these genes can result in inadequate mtDNA synthesis [2].
Clinically, MDS is classified into myopathic, encephalomyopathic, neurogastrointestinal, and hepatocerebral forms that are associated with mutations in the above genes [2]. Of the four forms of MDS, hepatocerebral MDS is associated with DGUOK gene mutations and occurs during the neonatal period [2-31]. The common clinical features of hepatocerebral MDS include hepatic dysfunction, psychomotor delay, hypotonia, rotary nystagmus developing into opsoclonus, lactic acidosis, and hypoglycemia [2-31]. Here, we report a fatal case of MDS with a novel compound heterozygous mutation in the DGUOK gene as well as a systemic literature review.
RESULTS
Clinical findings
Physical examination at 9 months and 4 days of age revealed slight jaundice, with a head circumference, chest circumference, height, weight, temperature, heart rate, and respiration rate of 40.0 cm, 41.0 cm, 69.0 cm, 7.0 kg, 37.2°C, 134 beats per minute, and 32 times per minute, respectively (Table 1). No obvious psychomotor delay, hypotonia, or rotary nystagmus was observed. Visual inspection and palpation found no obvious abnormalities except for splenomegaly.
Table 1: Clinical and laboratory findings
Personal history |
|
Age |
9 months and 4 days |
Gravidity (G) and Parity (P) |
G3P1 |
Gestation |
37 weeks+2days |
Birth weight (g) |
2,550 |
Birth history |
Breech position caesarean section |
Family history |
|
Pregnancy history of mother |
1-0-2-1 |
Physical examination |
|
Head circumference (cm) |
40.0 |
Chest circumference (cm) |
41.0 |
Height (cm) |
69.0 |
Weight (kg) |
7.0 |
Temperature (°C) |
37.2 |
Heart rate (beats per minute) |
134.0 |
Respiration rate (times per minute) |
32.0 |
Biochemical examination |
|
Albumin (g/L) |
25.0–33.7* |
Alanine aminotransferase, ALT (IU/L) |
77.0–293.0 |
Glutamate oxalacetate transaminase, GOT (IU/L) |
203.0–623.0 |
Direct bilirubin (umol/L) |
24.4–265.0 |
Total bilirubin (umol/L) |
213.5–340.8 |
Alpha fetal protein (ng/mL) |
29,090–121,000 |
Lactic acid (mmol/L) |
2.1–5.4 |
Glucose (mmol/L) |
1.44–4.10 |
Ammonia (umol/L) |
80.0–130.0 |
Coagulation test |
|
D-dimer (mg/L) |
0.49–20.0 |
Activated partial thromboplastin time (s) |
> 180 |
Thrombin time (s) |
23.0–42.8 |
Prothrombin time (s) |
39.2–75.9 |
Fibrinogen (g/L) |
0.6 |
Blood routine examination |
|
Hemoglobin (g/L) |
111.0–115.0 |
Red blood cell count (per liter) |
3.2–3.9 × 1012 |
White blood cell count (per liter) |
14.3 × 109 |
Lymphocytes (%) |
38.7–52.5 |
Neutrophils (%) |
35.4–50.0 |
Platelets count (per liter) |
146–479 × 109 |
Blood gas analysis |
|
pH |
7.45 |
pCO2 (mmHg) |
32.80 |
pO2 (mmHg) |
43.60 |
*Multiple test results are presented as (25.0–29.7).
Biochemical examination revealed hepatic dysfunction, lactic acidosis, and hypoglycemia. The serum levels of albumin, alanine aminotransferase, glutamate oxalacetate transaminase, direct bilirubin, total bilirubin, lactic acid, and glucose were 25.0–33.7 g/L (normal range: 60–83 g/L), 77.0–293.0 IU/L (normal range: 0–40 IU/L), 203.0–623.0 IU/L (normal range: 0–40 IU/L), 24.4–265.0 umol/L (normal range: 0–6 umol/L range), 213.5–340.8 umol/L (normal range: 5.1–17.1 umol/L), 2.1–5.4 mmol/L (normal range: <1.8 mmol/L), and 1.44–4.10 mmol/L (normal range: 3.9–5.8 mmol/L), respectively (Table 1). Biochemical tests also showed extremely high alpha fetal protein 29,090–121,000 ng/mL (normal range: 0 -77 ng/mL) and elevated ammonia 80.0–130.0 umol/L (normal range: 10–47 umol/L). Coagulation tests showed dysfunctional blood clotting. The D-dimer, activated partial thromboplastin time, thrombin time, prothrombin time, and fibrinogen were 0.49–20.0 mg/L (normal range: 0.0–0.3 mg/L), >180 s (normal range: 28.0–44.5 s), 23.0–42.8 s (normal range: 14.0–21.0), 39.2–75.9 s (normal range: 12.0–14.8 s) and 0.6 g/L (normal range: 2.0 -4.0 g/L), respectively (Table 1). Blood gas analysis showed that pH, pCO2, and pO2 were 7.45 (normal range: 7.35–7.45), 32.80 mmHg (normal range: 35.0–45.0 mmHg), and 43.60 mmHg (normal range: 80 -100), respectively (Table 1).
Abdominal ultrasonography showed multiple focal liver lesions, hepatic fibrosis, and massive ascites. Brain magnetic resonance imaging (MRI) showed higher DWI and T2WI signals in the bilateral frontal, temporal, parietal, and occipital lobes, the corpus callosum, internal and external capsules, and the brain stem (Figure 1A). In addition, a high T2WI signal also was observed in the bilateral globus pallidus (Figure 1A). Proton magnetic resonance spectroscopic imaging showed normal N-acetylaspartate, choline and creatine peaks in the basal ganglia and thalamus. The results suggested that the patient had hepatocerebral MDS and was thus given symptomatic and supportive treatment for eight days in our hospital. The girl died one and a half months after release from the hospital.
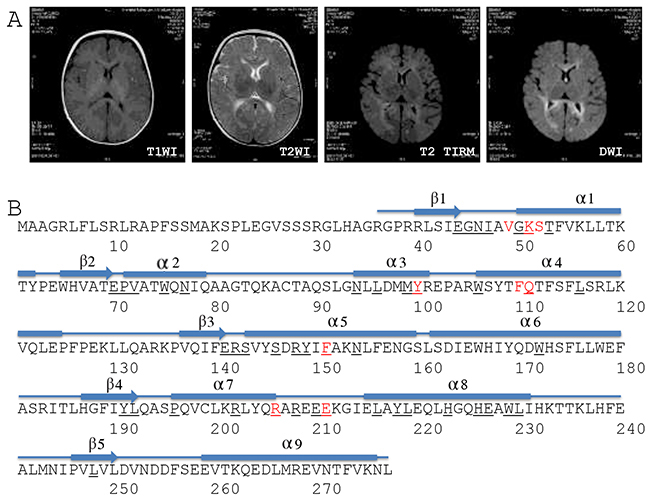
Figure 1: Brain MRI and DGUOK structure. (A) Brain MRI. T1WI, T1 weighted image; T2WI, T2 weighted image; TIRM, turbo inversion recovery magnitude; DWI, diffusion-weighted magnetic resonance imaging. (B) Structure-based sequence alignment of the human DGUOK gene (SwissProt Q16854). The numbers refer to the DGUOK amino acid sequence. Red characters refer to the conserved substrate-binding site. Black fields represent residues identical to the sequences of deoxycytidine kinase, thymidine kinase 2, and deoxyribonucleoside kinase.
Genetic findings
Genetic testing revealed that this girl was a compound heterozygote for DGUOK gene alleles c.128T>C (p.I43T) and c.313C>T (p.R105*) (Table 2). Her father was a heterozygous carrier of allele c.128T>C (p.I43T). c.128T>C (p.I43T), a novel variant located in exon 1 (National Center for Biotechnology Information ID: NM_080916) that encoded an amino acid in the first beta sheet of the DGUOK protein (Table 2 and Figure 1B). c.128T>C (p.I43T) has been included in the Human Gene Mutation Database, the NHLBI GO Exome Sequencing Project, or the 1000 Genomes Project to date, and has not been previously reported (Table 2). The patient’s mother was a heterozygous carrier of allele c.313C>T (p.R105*). c.313C>T (p.R105*) is located in exon 2 (NM_080916) and encodes amino acids located in the third and fourth alpha helixes of the DGUOK protein (Table 2 and Figure 1B). c.313C>T (p.R105*) is predicted to result in a 173 amino acid residue truncation at the C terminus of the DGUOK protein [18]. No prior cases of MDS were found in the family histories of either parent.
Table 2: Cumulative frequencies of reported variants of DGUOK gene
Ethnicity / Nationality |
Variants |
Zygotic type |
Sex |
Consanguinity |
Source |
Death |
Reference |
|---|---|---|---|---|---|---|---|
DGUOK mutations in children with MDS |
|||||||
Chinese |
c.128T>C(p.I43T)/c.313C>T(p.R105*) |
CH |
F |
No |
HP |
Yes |
this report |
French? |
c.2T>C(p.M1T) |
Homozygote |
F |
No |
HP |
Yes |
4 |
French? |
c.2T>C(p.M1T) |
Homozygote |
F |
No |
HP |
Yes |
5 |
Italian? |
c.2T>C(p.M1T)/c.677A>G (p.H226R) |
CH |
M |
? |
? |
? |
6 |
Polish? |
c.3G>A(p.M1I)/c.813_814insTTT(p.N271_T272insF) |
CH |
F |
? |
? |
Yes |
7 |
Polish? |
c.3G>A(p.M1I) |
Homozygote |
F |
? |
? |
Yes |
7 |
Turkish |
c.34C>T(p.R12X) |
Homozygote |
? |
Yes |
? |
Yes |
8 |
Hispanic |
c.80delC/c.763_c766dupGATT |
CH |
F |
No |
? |
Yes |
9 |
Turkish? |
c.130G>A(p.E44K) |
Homozygote |
M2 |
Yes |
HP |
Yes |
11 |
French? |
c.137A>G(p.N46S) |
Homozygote |
M |
No |
? |
? |
5 |
American? |
c.137A>G(p.N46S)/c.352C>T(p.R118C) |
CH |
M |
No |
HP |
Yes |
13 |
American? |
c.137A>G(p.N46S)/c.352C>T(p.R118C) |
CH |
M2 |
No |
HP |
4y (2009) |
13 |
Hispanic |
c.137A>G(p.N46S)/c.352C>T(p.R118C) |
CH |
M |
? |
? |
Yes |
9 |
Japanese |
g.11692_12026del335(p.A48fsX90) |
Homozygote |
F2 |
No |
? |
Yes |
14 |
Japanese |
g.11692_12026del335(p.A48fsX90) |
Homozygote |
F |
No |
? |
Yes |
14 |
German |
c.155C>T(p.S52F) |
Homozygote |
M |
Yes |
? |
1y (2006) |
3 |
Russian |
c.155C>T(p.S52F)/c.681-684delGTTT |
CH |
F2 |
No |
? |
Yes |
3 |
European |
c.165G>A(p.W65X)/c.487_c490dupGACA |
CH |
M2 |
? |
? |
Yes |
9 |
Israeli-Druze |
c.204del A |
Homozygote |
M5F15 |
Yes |
HP |
19 died |
15 |
Arabs |
c.223T>A(p.W75R) |
Homozygote |
F |
? |
? |
Yes |
16 |
Lebanese |
c.235C>T(p.79*) |
Homozygote |
F2 |
Yes |
? |
Yes |
9,17 |
German |
c.313C>T (p.R105*) |
Homozygote |
M |
No |
HP |
Yes |
18 |
French? |
c.313 C>T (p.R105*) |
Homozygote |
M |
No |
? |
Yes |
5 |
Indian |
c.318 G>A (p.W106*) |
? |
F |
No |
? |
Yes |
9,17 |
European |
c.352C>T(p.R118C) |
Homozygote |
F |
No |
MUD |
Yes |
19 |
Italian |
c.444-11C>G/c.605_c.606delGA |
CH |
M |
? |
? |
Yes |
9 |
Turkish? |
c.493G>A(p.E165K) |
Homozygote |
M |
Yes |
? |
Yes |
11 |
Polish? |
c.494A>T(p.E165V) |
Homozygote |
M |
? |
? |
Yes |
7 |
French? |
c.495A>T(p.E165V)/P246R |
CH |
M |
No |
? |
? |
5 |
Turkish |
c.509A>G(p.Q170R) |
Homozygote |
F |
No |
HP |
2y (2006) |
3 |
French? |
c.591A>G/nt424_425delAG |
CH |
M |
No |
? |
? |
5 |
Hispanic |
c.533G>A(p.W178X) |
Homozygote |
F |
No |
PUI |
Yes |
9 |
American? |
c.533G4A (p.W178X) |
Homozygote |
? |
No |
PUI |
Yes |
21 |
African American |
c.572A>G(p.Y191C) |
Homozygote |
F |
No |
HP |
Yes |
22 |
Chinese |
c.572A>G(p.Y191C)/c.151A>C(p.K51Q) |
CH |
F |
? |
? |
Yes |
9 |
French |
c.4G>T(p.A2S)/c.591G>A |
CH |
M |
No |
HP |
Yes |
23 |
Caucasian/Hispanic |
c.605_c606 delGA/c.591G>A (p.Q197Q) |
CH |
F |
? |
? |
Yes |
9 |
Mexican |
c.592-4_c.592-3delTT |
Homozygote |
F |
Yes |
HP |
Yes |
24 |
Mexican |
c.592-4_c.592-3delTT |
Homozygote |
F |
Yes |
HP |
? |
24 |
Italian? |
c.delGA603_604(p.K201fs214X) |
Homozygote |
M |
? |
? |
Yes |
6 |
American? |
c.609_c.610delGT |
Homozygote |
F2 |
? |
? |
Yes |
20 |
Arabs |
c. 617G>A(p.R206K) |
Homozygote |
F |
Yes |
HP |
Yes |
16 |
Hispanic |
c.677A>G(p.H226R)/c.592-4_c.592-3delTT |
CH |
M |
? |
? |
Yes |
9 |
Caucasian |
c.677A>G (p.H226R) |
Homozygote |
M1F1 |
Yes |
? |
Yes |
25 |
American? |
c.707+417(intron 5)/c.834 (end of 3’UTR)+3416, del3127bp |
Homozygote |
F |
? |
? |
Yes |
26 |
French? |
c.721_724insTGAT+ |
Homozygote |
M |
? |
? |
Yes |
5 |
Italian? |
c.749T>C(p.L250S) |
Homozygote |
F |
? |
? |
Yes |
6 |
French? |
c.749T>C(p.L250S) |
Homozygote |
M1F1 |
Yes |
HP |
Yes |
4 |
Italian |
c.749T>C(p.L250S) |
Homozygote |
F |
No |
? |
Yes |
27 |
French? |
c.749T>C(p.L250S) |
Homozygote |
F |
Yes |
? |
? |
5 |
Portuguese? |
c.749T>C(p.L250S) |
Homozygote |
M |
Yes |
? |
Yes |
25 |
Portuguese |
c.749T>C(p.L250S)/c.1A>G(p.M1T) |
CH |
M/F |
No |
HP |
Yes |
3 |
French? |
IVS2nt-2/c.749T>C(p.L250S) |
CH |
F |
? |
? |
Yes |
5 |
American? |
c.763_766dupGATT |
Homozygote |
F2 |
Yes |
HP |
Yes |
20 |
Moroccan |
c.763_766dupGATT |
Homozygote |
F |
Yes |
? |
Yes |
28 |
Moroccan |
c.763_766dupGATT |
Homozygote |
? |
? |
? |
Yes |
23 |
Moroccan |
c.763_766dupGATT |
Homozygote |
M2F1 |
Yes |
HP |
Yes |
29 |
Algerian |
c.763_766dupGATT |
Homozygote |
M2 |
No |
HP |
Yes |
29 |
Moroccan |
c.763_766dupGATT |
Homozygote |
F |
Yes |
HP |
? |
29 |
Italian? |
c.763_766dupGATT/c.130g>A(p.E44K) |
CH |
F |
? |
? |
10mo (2002) |
6 |
Mennonite |
c.763G>T (p.D255Y) |
Homozygote |
M1F1 |
? |
HP |
Yes |
30 |
Arabs |
c.766_767insGATT(p.F256*) |
Homozygote |
M2 |
? |
? |
Yes |
16 |
Polish? |
c.766_767insGATT(p.F256X) |
Homozygote |
F |
? |
? |
Yes |
7 |
French? |
c.633A>G(p.E211G)/c.797T>G(p.L266R) |
CH |
M |
No |
? |
Yes |
4 |
French? |
c.495A>T(p.E165V)/c.797T>G(p.L266R) |
CH |
M |
No |
? |
Yes |
4 |
French? |
c.495A>T(p.E165V)/c.797T>G(p.L266R) |
CH |
M |
No |
? |
Yes |
5 |
Tunisian |
c.444-62C>A |
Homozygote |
M |
Yes |
HP |
Yes |
31 |
Moroccan |
c.444-62C>A |
Homozygote |
M2 |
Yes |
HP |
Yes |
31 |
Turkish? |
c.707+3_6delTAAG |
Homozygote |
F |
Yes |
HP |
Yes |
11 |
Cases of reversible MDS |
|||||||
French |
c.137A>G (N46S)/c.797T>G(p.L266R) |
CH |
M |
No |
HP |
10y (2007) |
23 |
American? |
c.81_c.82insCC(p.S28P)/c.4G>T (p.A2S) |
CH |
M |
? |
? |
21y (2012) |
10 |
Italian |
c.319T>C (p.S107P) |
Homozygote |
M |
? |
? |
3.5y (2007) |
9 |
Hispanic |
c.137A>G(p.N46S)/c.352C>T(p.R118C) |
CH |
M |
? |
? |
3.5y (2007) |
9 |
Cases of MDS underwent liver transplantation |
|||||||
American? |
c.425G>A(p.R142K)/c.679G>A(p.E227K) |
CH |
M |
No |
HP |
5y (2002) |
20 |
Polish |
c.1A>G(p.M1V)/c.3G>A(p.M1I) |
CH |
F |
No |
? |
1y (2006) |
3 |
Mennonite |
c.763G>T (p.D255Y) |
Homozygote |
F |
? |
HP |
3y (2005) |
30 |
DGUOK mutations in adult patients with mitochondrial DNA multiple deletions |
|||||||
Italian? |
c.130G>A(p.E44K)/c.462 T>A(p.N154K) |
CH |
F |
? |
? |
72y |
12 |
Italian? |
c.186 C>A(p.Y62*)/c.509A>G(p.Q170R) |
CH |
M |
? |
? |
80y |
12 |
Italian? |
c.444-11C>G/c.509A>G(p.Q170R) |
CH |
F1M1 |
? |
? |
46y,48y |
12 |
Italian? |
c.605_606delGA(p.R202YfsX12)/c.462T>A(p.N154K) |
CH |
F |
? |
? |
69y |
12 |
Italian? |
c.605_606delGA(p.R202YfsX12)/c.137A4G p.N46S |
CH |
F |
? |
? |
23y |
12 |
?, unspecified; CH, compound heterozygote; HP, heterozygous parents; PUI, paternal uniparental isodisomy; MUD, maternal uniparental disomy; M5F15, 5 males and 15 females in a single kindred; 72y, 72 years old when data collected; 10mo, 10 months old when data collected; 4y (2009), 4 years old in the published year. Cases with confirmed improvement in the pathological manifestation of MDS were sorted in “Cases of reversible MDS”. Three cases of MDS with liver transplantation were collected together in section of “Cases of MDS underwent liver transplantation”.
Demographic and genetic characteristics of cumulative variants of the DGUOK gene
To map the DGUOK gene mutations to MDS, we collected all reports published in the NCBI database (Table 2). In total, 112 infantile MDS cases had been reported worldwide by the end of April 2017. The reports consisted of 60 (53.6%) females and 49 (43.8%) males; 92 (82.1%) died before 1 year of age; 13 (11.6%) subjects were alive when the study was published [3, 6, 9, 10, 13, 15, 20, 23, 30]; 47 (42.0%) and 35 (31.3%) cases were consanguineous offspring and non-consanguineous offspring, respectively (Table 2); 81 (72.3%) and 31 (27.7%) were homozygotes and compound heterozygotes, respectively (Table 2); and the parents of 55 (49.1%) cases were heterozygous carriers of related DGUOK variants (Table 2).
Three reviewed MDS cases underwent liver transplantation at age 5, 1, and 3 years old in 2002, 2006, and 2005, respectively (Table 2) [3, 20, 30]. There were four reversible MDS cases, and the pathological manifestation of the four cases was confirmed to be improved [9, 10, 23]. The ages of the patients with reversible cases were 10, 21, 3.5, and 3.5 years old in 2007, 2012, 2007, and 2007, respectively (Table 2). It is difficult to find reversible MDS specific variants in the existing data. Although c.319T>C (p.S107P) was found in an Italian reversible MDS case [9], other variants including c.137A>G (N46S), c.797T>G(p.L266R), c.4G>T (p.A2S), c.137A>G(p.N46S), and c.352C>T(p.R118C) were also found in fatal cases (Table 2).
One previous study has reported DGUOK mutations in adults [12]. The subjects ranged in age from 23 to 80 years old. Clinical presentations were variable, and included mitochondrial myopathy with or without progressive external ophthalmoplegia, recurrent rhabdomyolysis, and adult-onset lower motor neuron syndrome with mild cognitive impairment [12]. The genotypes of six individuals in that study were c.130G>A/c.462T>A, c.186C>A/c.509A>G, c.444–11C>G/c.509A>G, c.605_606delGA/c.462T>A, and c.605_606delGA/c.137A>G (Table 2). As the authors indicated, the mutations identified in adults were associated with an infantile hepatocerebral form of MDS [12]. It is also difficult to find adult mtDNA multiple deletion-specific variants.
Taken together, these studies suggest that DGUOK variants can occur anywhere in the DGUOK gene, and that it remains difficult to find reversible MDS-specific or adult mtDNA multiple deletion-specific variants.
DISCUSSION
MDS is an autosomal recessive disorder. In this report, we describe a fatal MDS case with novel compound heterozygous variants of the DGUOK gene, c.128T>C(p.I43T)/c.313C>T(p.R105*). To date, two additional reports have described the c.313C>T(p.R105*) variant [5, 18]. The patients in which these mutations were found were both boys, and one was from Germany and one was suspected to be French. Both boys died before 1 year of age. c.313C>T is predicted to result in a truncated DGUOK protein lacking the 173 amino acid residue at the C terminus. c.128T>C (p.I43T) is a novel variant. Because the girl died before reaching 1 year of age, this allele is likely also lethal. Thus, this finding should be considered during pre-pregnancy checkups.
During the data-collection process, we found that variants of the DGUOK gene were varied. As shown in Table 2, all types of genetic variants could be found in the DGUOK gene. Missense mutations [3-13, 15-23, 25, 27, 30, 31] can change highly conserved amino acids [3, 20, 23], affect start codon recognition by the ribosome [7, 10, 23], substitute a nonpolar neutral amino acid with a polar one [16], result in a premature stop codon [5, 12, 17, 18], affect the initial methionine [3], or induce splicing anomalies [23]. Nucleotide deletions [3, 5, 6, 9, 11, 12, 14, 15, 20, 24, 26] can lead to frameshift mutations, which could result in either a premature termination of translation [3, 12, 15, 20, 26] or aberrant splicing [24]. Nucleotide insertions [5, 7, 10, 16] can either change the polarity of an amino acid [10] or lead to a premature termination [10, 16]. Finally, nucleotide duplications [6, 9, 20, 23, 28-30] can produce a frameshift mutation, a premature stop codon [9, 20, 28], or induce mRNA instability due to nonsense mutations [15, 30].
The DGUOK protein consists of five beta sheets and nine alpha helixes (Figure 1B) [32]. Nine amino acids make up a conserved substrate-binding site, and 45 amino acids are identical to deoxycytidine kinase, thymidine kinase 2, and deoxyribonucleoside kinase (Figure 1B) [32]. Although previous reports have shown that some variants change highly conserved amino acids [3, 4, 9, 16, 20, 23], integrated data show that mutations are not confined to these conserved sites. Many variants occur outside of the conserved region [3-9, 11-13, 19, 21, 25, 27, 30].
Hepatocerebral MDS always leads to hepatic failure and a variable neurological phenotype [1-3]. Our patient manifested jaundice, hepatosplenomegaly, abnormal hepatic function, and multiple abnormal signals in the brain. It is worth mentioning that the alpha-fetoprotein levels in all reports was extremely high [7, 11, 16]. In our patient, levels were as high as 29,090 ng/mL, which may occur due to the liver regeneration process or the early onset of hepatocellular carcinoma.
Since the first report of MDS by Moraes et al. in 1991 [1], the number of cases has increased, and now stand at more than 100. There is confusion in the position and nomenclature of MDS-related DGUOK variants, and many studies have either not provided a reference sequence or have adopted different sequence nomenclature. In Table 2, we list the original expression of variants. A pathogenic map of the DGUOK gene needs to be created. Therefore, it is important to follow Human Genome Variation Society (http://varnomen.hgvs.org) recommendations to standardize the location and nomenclature of MDS-related DGUOK variants going forward.
The main limitation to this study is that the patient was critically ill, which prevented us from performing biopsies for complete respiratory chain analysis and mtDNA quantification. Therefore, we were unable to provide any molecular evidence regarding the influence of the c.128T>C(p.I43T) variant on DGUOK expression or activity.
In conclusion, our report shows that diversified DGUOK variants can occur anywhere in the DGUOK gene, and that mutations affect both conserved and non-conserved DGUOK amino acids are associated with fatalities.
MATERIALS AND METHODS
This study was approved by the Research Ethics Committee of Children’s Hospital of Fudan University and was conducted under the Declaration of Helsinki ethical principles for medical research involving human subjects. Informed consent was obtained from the child’s parents.
Patient
A Chinese girl, aged 9 months and 4 days, visited our hospital on February 17, 2016, due to jaundice. The patient was a gravidity 3 and parity 1 (G3P1) infant of unrelated parents. The mother was 31 years old and had three pregnancies, with pregnancy terminations occurring at 1 and 3 months due to fetal growth arrest. The father was 32 years old. The patient was delivered by cesarean section at 37 weeks and 2 days due to a breech position, and had a birth weight of 2,550 g. Eight days after birth, she was hospitalized due to recurrent hypoglycemia (1.0–4.1 mmol/L). Physical and laboratory examinations found jaundice, coagulation disorders, and abnormal liver function. After her release from the hospital, the patient visited multiple hospitals for jaundice, weight stagnation, hepatomegaly, and abnormal liver function. Clinical laboratory and imaging examinations were performed in the corresponding clinical departments in our hospital.
DGUOK gene detection
During multiple hospitalizations, the patient was screened by whole exome sequencing (WES) and multi-gene panels on several occasions to find risk gene variants. WES was performed by an authorized independent gene testing company (Jinyu, Changsha, China). The testing process is briefly described as follows: WES was initiated by preparing a library using 3 μg purified DNA; enrichment was achieved using the SOLiDoptimized Sure Select All Human Exon Kit (Agilent, Technologies, Santa Clara, CA); sequencing was performed using 5500XL sequencers (Life Technologies, Carlsbad, CA). Quality control parameters were strictly observed throughout the WES workflow, and samples failing quality testing were either restarted completely or restarted from an earlier point. Sequence reads were aligned to hg19 using Lifescope v2.1 software (Life Technologies, Shanghai, China) followed by variant analysis of the aligned sequence. Variants were annotated using a custom analysis pipeline. A minimum of 30×median coverage per sample was required for sufficient data quality.
In our hospital, rare diseases in pediatrics are further screened using an in-house multi-gene panel [33, 34]. The panel included the following 40 genes: ATPase phospholipid transporting 8B1, ATP binding cassette subfamily B member 11, ATP binding cassette subfamily B member 4, tight junction protein 2, bile acid-CoA: amino acid N-acyltransferase, claudin 1, hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 7, Aldo-Keto reductase family 1 member D1, cytochrome P450 family 7 subfamily B member 1, alpha-methylacyl-CoA racemase, cytochrome P450 family 27 subfamily A member 1, 7-dehydrocholesterol reductase, jagged 1, Notch 2, solute carrier family 25 member 13, DGUOK, MPV17, mitochondrial inner membrane protein, fumarylacetoacetate hydrolase, ATP binding cassette subfamily C member 2, UDP glucuronosyltransferase family 1 member A1, NPC intracellular cholesterol transporter 1, NPC intracellular cholesterol transporter 2, galactose-1-phosphate uridylyltransferase, UDP-galactose-4-epimerase, aldolase fructose-bisphosphate A, aldolase fructose-bisphosphate B, keratin 18, keratin 8, UTP4 small subunit processome component, cystic fibrosis transmembrane conductance regulator, glutamyl-TRNA synthetase 2 mitochondrial, hydroxysteroid 17-beta dehydrogenase 4, lipase A lysosomal acid type, peroxisomal biogenesis factor 1, peroxisomal biogenesis factor 5, POU class 1 homeobox 1, HESX homeobox 1, serpin family A member 1, VPS33B interacting protein, apical-basolateral polarity regulator, Spe-39 homolog, and late endosome and lysosome associated VPS33B. Other than DGUOK, no variants in any of the above genes were found. The DGUOK gene variants were further confirmed by Sanger sequencing using DNA from peripheral blood leukocytes.
Literature search
To understand the relationships between MDS and DGUOK gene variants, we systematically searched all published studies in PubMed. The latest published data were updated in April 2017. The following terms were used to search titles and abstracts: (mitochondrial DNA depletion syndrome or MDS or OMIM 251880) and (deoxyguanosine kinase or DGUOK or dGK). In total, 31 multi-case and single-case reports were reviewed in this study.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
1. Moraes CT, Shanske S, Tritschler HJ, Aprille JR, Andreetta F, Bonilla E, Schon EA, DiMauro S. mtDNA depletion with variable tissue expression: A novel genetic abnormality in mitochondrial diseases. Am J Hum Genet. 1991; 48:492–501.
2. El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013; 10:186–198.
3. Freisinger P, Fütterer N, Lankes E, Gempel K, Berger TM, Spalinger J, Hoerbe A, Schwantes C, Lindner M, Santer R, Burdelski M, Schaefer H, Setzer B, et al. Hepatocerebral mitochondrial DNA depletion syndrome caused by deoxyguanosine kinase (DGUOK) mutations. Arch Neurol. 2006; 63:1129–1134.
4. Slama A, Giurgea I, Debrey D, Bridoux D, de Lonlay P, Levy P, Chretien D, Brivet M, Legrand A, Rustin P, Munnich A, Rötig A. Deoxyguanosine kinase mutations and combined deficiencies of the mitochondrial respiratory chain in patients with hepatic involvement. Mol Genet Metab. 2005; 86:462–465.
5. Sarzi E, Bourdon A, Chrétien D, Zarhrate M, Corcos J, Slama A, Cormier-Daire VD, de Lonlay P, Munnich A, Rötig A. Mitochondrial DNA depletion is a prevalent cause of multiple respiratory chain deficiency in childhood. J Pediatr. 2007; 150:531–534.
6. Spinazzola A, Invernizzi F, Carrara F, Lamantea E, Donati A, Dirocco M, Giordano I, Meznaric-Petrusa M, Baruffini E, Ferrero I, Zeviani M. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis. 2009; 32:143–158.
7. Pronicka E, Węglewska-Jurkiewicz A, Taybert J, Pronicki M, Szymańska-Dębińska T, Karkucińska-Więckowska A, Jakóbkiewicz-Banecka J, Kowalski P, Piekutowska-Abramczuk D, Pajdowska M, Socha P, Sykut-Cegielska J, Węgrzyn G. Post mortem identification of deoxyguanosine kinase (DGUOK) gene mutations combined with impaired glucose homeostasis and iron overload features in four infants with severe progressive liver failure. J Appl Genet. 2011; 52:61–66.
8. Kiliç M, Sivri HS, Dursun A, Tokatli A, De Meirleir L, Seneca S, Akçören Z, Yiğit S, Topaloğlu H, Coşkun T. A novel mutation in the DGUOK gene in a Turkish newborn with mitochondrial depletion syndrome. Turk J Pediatr. 2011; 53:79–82.
9. Dimmock DP, Zhang Q, Dionisi-Vici C, Carrozzo R, Shieh J, Tang LY, Truong C, Schmitt E, Sifry-Platt M, Lucioli S, Santorelli FM, Ficicioglu CH, Rodriguez M, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat. 2008; 29:330–331.
10. Buchaklian AH, Helbling D, Ware SM, Dimmock DP. Recessive deoxyguanosine kinase deficiency causes juvenile onset mitochondrial myopathy. Mol Genet Metab. 2012; 107:92–94.
11. Sezer T, Ozçay F, Balci O, Alehan F. Novel deoxyguanosine kinase gene mutations in the hepatocerebral form of mitochondrial DNA depletion syndrome. J Child Neurol. 2015; 30:124–128.
12. Ronchi D, Garone C, Bordoni A, Gutierrez Rios P, Calvo SE, Ripolone M, Ranieri M, Rizzuti M, Villa L, Magri F, Corti S, Bresolin N, Mootha VK, et al. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain. 2012; 135:3404–3415.
13. Shieh JT, Berquist WE, Zhang Q, Chou PC, Wong LJ, Enns GM. Novel deoxyguanosine kinase gene mutations and viral infection predispose apparently healthy children to fulminant liver failure. J Pediatr Gastroenterol Nutr. 2009; 49:130–132.
14. Yamazaki T, Murayama K, Compton AG, Sugiana C, Harashima H, Amemiya S, Ajima M, Tsuruoka T, Fujinami A, Kawachi E, Kurashige Y, Matsushita K, Wakiguchi H, et al. Molecular diagnosis of mitochondrial respiratory chain disorders in Japan: focusing on mitochondrial DNA depletion syndrome. Pediatr Int. 2014; 56:180–187.
15. Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M, Eriksson S, Cohen N. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001; 29:337–341.
16. Al-Hussaini A, Faqeih E, El-Hattab AW, Alfadhel M, Asery A, Alsaleem B, Bakhsh E, Ali A, Alasmari A, Lone K, Nahari A, Eyaid W, Al Balwi M, et al. Clinical and molecular characteristics of mitochondrial DNA depletion syndrome associated with neonatal cholestasis and liver failure. J Pediatr. 2014; 164:553–559.
17. Mancuso M, Ferraris S, Pancrudo J, Feigenbaum A, Raiman J, Christodoulou J, Thorburn DR, DiMauro S. New DGK gene mutations in the hepatocerebral form of mitochondrial DNA depletion syndrome. Arch Neurol. 2005; 62:745–747.
18. Taanman JW, Kateeb I, Muntau AC, Jaksch M, Cohen N, Mandel H. A novel mutation in the deoxyguanosine kinase gene causing depletion of mitochondrial DNA. Ann Neurol. 2002; 52:237–239.
19. Haudry C, de Lonlay P, Malan V, Bole-Feysot C, Assouline Z, Pruvost S, Brassier A, Bonnefont JP, Munnich A, Rötig A, Lebre AS. Maternal uniparental disomy of chromosome 2 in a patient with a DGUOK mutation associated with hepatocerebral mitochondrial DNA depletion syndrome. Mol Genet Metab. 2012; 107:700–704.
20. Salviati L, Sacconi S, Mancuso M, Otaegui D, Camaño P, Marina A, Rabinowitz S, Shiffman R, Thompson K, Wilson CM, Feigenbaum A, Naini AB, Hirano M, et al. Mitochondrial DNA depletion and dGK gene mutations. Ann Neurol. 2002; 52:311–317.
21. Douglas GV, Wiszniewska J, Lipson MH, Witt DR, McDowell T, Sifry-Platt M, Hirano M, Craigen WJ, Wong LJ. Detection of uniparental isodisomy in autosomal recessive mitochondrial DNA depletion syndrome by high-density SNP array analysis. J Hum Genet. 2011; 56:834–839.
22. Hanchard NA, Shchelochkov OA, Roy A, Wiszniewska J, Wang J, Popek EJ, Karpen S, Wong LJ, Scaglia F. Deoxyguanosine kinase deficiency presenting as neonatal hemochromatosis. Mol Genet Metab. 2011; 103:262–267.
23. Mousson de Camaret B, Taanman JW, Padet S, Chassagne M, Mayençon M, Clerc-Renaud P, Mandon G, Zabot MT, Lachaux A, Bozon D. Kinetic properties of mutant deoxyguanosine kinase in a case of reversible hepatic mtDNA depletion. Biochem J. 2007; 402:377–385.
24. Ji JQ, Dimmock D, Tang LY, Descartes M, Gomez R, Rutledge SL, Schmitt ES, Wong LJ. A novel c.592-4_c.592-3delTT mutation in DGUOK gene causes exon skipping. Mitochondrion. 2010; 10:188–191.
25. Nobre S, Grazina M, Silva F, Pinto C, Gonçalves I, Diogo L. Neonatal liver failure due to deoxyguanosine kinase deficiency. BMJ Case Rep. 2012; 2012.
26. Mudd SH, Wagner C, Luka Z, Stabler SP, Allen RH, Schroer R, Wood T, Wang J, Wong LJ. Two patients with hepatic mtDNA depletion syndromes and marked elevations of S-adenosylmethionine and methionine. Mol Genet Metab. 2012; 105:228–236.
27. Wang L, Limongelli A, Vila MR, Carrara F, Zeviani M, Eriksson S. Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes. Mol Genet Metab. 2005; 84:75–82.
28. Mancuso M, Filosto M, Tsujino S, Lamperti C, Shanske S, Coquet M, Desnuelle C, DiMauro S. Muscle glycogenosis and mitochondrial hepatopathy in an infant with mutations in both the myophosphorylase and deoxyguanosine kinase genes. Arch Neurol. 2003; 60:1445–1447.
29. Labarthe F, Dobbelaere D, Devisme L, De Muret A, Jardel C, Taanman JW, Gottrand F, Lombès A. Clinical, biochemical and morphological features of hepatocerebral syndrome with mitochondrial DNA depletion due to deoxyguanosine kinase deficiency. J Hepatol. 2005; 43:333–341.
30. Tadiboyina VT, Rupar A, Atkison P, Feigenbaum A, Kronick J, Wang J, Hegele RA. Novel mutation in DGUOK in hepatocerebral mitochondrial DNA depletion syndrome associated with cystathioninuria. Am J Med Genet A. 2005; 135:289–291.
31. Brahimi N, Jambou M, Sarzi E, Serre V, Boddaert N, Romano S, de Lonlay P, Slama A, Munnich A, Rötig A, Bonnefont JP, Lebre AS. The first founder DGUOK mutation associated with hepatocerebral mitochondrial DNA depletion syndrome. Mol Genet Metab. 2009; 97:221–226.
32. Johansson K, Ramaswamy S, Ljungcrantz C, Knecht W, Piskur J, Munch-Petersen B, Eriksson S, Eklund H. Structural basis for substrate specificities of cellular deoxyribonucleoside kinases. Nat Struct Biol. 2001; 8:616–620.
33. Li L, Dong J, Wang X, Guo H, Wang H, Zhao J, Qiu Y, Abuduxikuer K, Wang J. JAG1 mutation spectrum and origin in Chinese children with clinical features of Alagille syndrome. PLoS One. 2015; 10:e0130355.
34. Xie X, Lu Y, Wang X, Wu B, Yu H. JAGGED1 gene variations in Chinese twin sisters with Alagille syndrome. Int J Clin Exp Pathol. 2015; 8:8506–8511.